

Abstract
Objectives
Phenothiazines have been shown to exhibit in vitro and in vivo activity against Mycobacterium tuberculosis (Mtb) and multidrug-resistant Mtb. They are predicted to target the genetically validated respiratory chain component type II NADH:quinone oxidoreductase (Ndh). Using a set of compounds containing the phenothiazine pharmacophore, we have (i) investigated whether chemical validation data support the molecular target and (ii) evaluated pharmacophore tractability for further drug development.
Methods
Recombinant Mtb Ndh was generated and its functionality confirmed by steady-state kinetics. Pharmacodynamic profiling of the phenothiazines, including antitubercular efficacy in aerobic and O2-limited conditions, time–kill assays and isobole analyses against first-line antituberculars, was performed. Potential mitochondrial toxicity was assessed in a modified HepG2 cell-line assay and against bovine cytochrome bc1.
Results
Steady-state kinetic analyses revealed a substrate preference for coenzyme Q2 and an inability to utilize NADPH. A positive correlation between recombinant Ndh inhibition and kill of aerobically cultured Mtb was observed, whilst enhanced potency was demonstrated in a hypoxic model. Time–kill studies revealed the phenothiazines to be bactericidal whilst isobolograms exposed antagonism with isoniazid, indicative of intracellular NADH/NAD+ couple perturbation. At therapeutic levels, phenothiazine-mediated toxicity was appreciable; however, specific mitochondrial targeting was excluded.
Conclusions
Data generated support the hypothesis that Ndh is the molecular target of phenothiazines. The favourable pharmacodynamic properties of the phenothiazines are consistent with a target product profile that includes activity against dormant/persistent bacilli, rapid bactericidal activity and activity against drug-resistant Mtb by a previously unexploited mode of action. These properties warrant further medicinal chemistry to improve potency and safety.
Keywords: tuberculosis, thioridazine, Ndh, latency, toxicity
Introduction
Tuberculosis (TB) is a global public health emergency, accounting for some 1.4 million deaths per annum. Whilst the WHO has recently reported that TB prevalence and associated mortality are in decline, it is clear that this progress could be jeopardized if the increasing number of multi- (MDR), extensively (XDR) and totally drug-resistant TB cases are not tackled.1
No new antitubercular drugs, exhibiting novel modes of action, have been developed for almost half a century.2 Recent additions to the antitubercular armoury have largely been derivatives of existing therapies or repurposing of antimicrobial agents (e.g. oxazolidinones and carbapenem/β-lactamase inhibitor combinations). In order to circumvent current resistance mechanisms, novel compounds focused against new targets must be developed. The current development portfolio contains several compounds with novel modes of action exhibiting promising results in initial clinical studies.3 For example, the diarylquinolone TMC207 targets the ATP synthase of the Mycobacterium tuberculosis (Mtb) electron transfer chain (ETC), resulting in disruption of ATP synthesis. It has been shown to be highly potent against both replicating and dormant Mtb, with encouraging results in murine models and clinical studies.4–6 TMC207 highlights the potential of other ETC components as novel drug targets with utility in treating drug-persistent Mtb.7
The type II NADH:quinone oxidoreductase (Ndh) of Mtb is one such ETC component showing promise as a drug target.8 Alongside a homologous single subunit protein (NdhA) and a conventional complex I, Ndh is postulated to contribute to the menaquinol/menaquinone and NADH/NAD+ redox pools of Mtb; however, it is not expected to translocate protons.9,10 This may be of particular benefit under non-replicating conditions, where cellular energy requirements are low, as it may mitigate against the accumulation of H+ that results from reduced ATPase activity.9,10 Further endorsement of the importance of Ndh comes from a recent study demonstrating that the antitubercular activity of clofazimine, including against MDR Mtb, is due to the generation of reactive oxygen species formed as a consequence of Ndh-mediated reduction of clofazimine.11 The novelty of the target and the potential role of Ndh during Mtb dormancy and persistence therefore offer an advantage over many current antituberculars.10 If successfully developed, Ndh inhibitors could circumvent current resistance mechanisms and have utility in shortening treatment times.
The essentiality of Mtb Ndh, which has no human homologue, could not be confirmed initially.12,13 However, a recent deep sequencing study of genes required for Mtb growth by Griffin et al.14 suggests that of the three Mtb NADH dehydrogenases, only ndh is unable to tolerate insertional mutations, thus signalling its potential essentiality and viability as a novel target. Further evidence for the essentiality of Mtb Ndh is reported in the study of Rao et al.,10 wherein the killing of hypoxic, non-growing Mtb was elicited by an Ndh inhibitor but not a complex I inhibitor.
It has long been recognized that the phenothiazine-derived antipsychotic agent thioridazine has antitubercular properties15,16 and studies of inhibitors containing the phenothiazine pharmacophore identified significant activity against Mtb.17,18 Furthermore, recent clinical studies indicate that 200 mg/day doses of thioridazine, in combination with moxifloxacin and linezolid, have been successfully employed to treat patients with XDR Mtb.19 Additionally, thioridazine demonstrates significant activity against MDR TB in a murine model of infection.20 Although data intimate that phenothiazines effect their antitubercular activity via Ndh in aerobic and anaerobic in vitro models, these studies are fragmentary and isolated mainly to thioridazine and trifluoperazine.8,10,15,21 It has been established that Ndh indirectly contributes to the membrane potential (ΔΨm) in hypoxic Mtb and that thioridazine treatment causes a collapse of the ΔΨm, resulting in a concomitant reduction of ATP levels in anoxic (non-replicating) Mtb as well an increase in the intracellular NADH/NAD+ ratio.10 Furthermore, Boshoff et al.22 demonstrated that thioridazine and chlorpromazine elicit a transcriptional phenotype consistent with a respiratory poison, whilst Weinstein et al.8 showed that, in addition to the inhibition of recombinant Ndh, trifluoperazine impedes Mtb membrane respiration and acts upstream of cytochrome c within the ETC.
Whilst phenothiazines are some of the most widely used antipsychotic agents, they demonstrate numerous toxicities, including QTc interval prolongation.16,23 Mitochondrial toxicity has also been reported, particularly in the human brain where diminished oxidative phosphorylation may result in extrapyramidal effects, such as tardive dyskinesia.24 As such, the development of phenothiazine derivatives as antituberculars must be approached with appropriate caution.
Here, we describe the pharmacodynamic features of a set of phenothiazines to (i) generate supportive chemical validation data for the molecular target and (ii) evaluate the potential of this class for further antitubercular drug development.
Materials and methods
Molecular biology
The Mtb ndh gene (Rv1854) was amplified from plasmid ndh pET16b (provided by Professor H. Rubin, University of Pennsylvania). The forward and reverse primers for the PCR were 5′-GGGCATGCCCATCATCATCATCATCACAGC-3′ (Fw, SphI site underlined) and 5′-CCGGATCCTAGCTGGCCACCTTAGC-3′ (Rev, BamHI site underlined), respectively. The resultant N-terminal His-tagged 1431 bp fragment was ligated to pUC19 to generate the expression plasmid pAW1. Following sequence verification by automated DNA sequencing, pAW1 was transformed into the Escherichia coli NADH dehydrogenase knockout strain ANN0222 (nuoB::nptI-sacRB, ndh::tet, a gift from Professor T. Friedrich, University of Freiburg) for heterologous expression. All molecular biology was performed according to the standard protocols of Sambrook and Russell.25
Membrane preparations
Membrane preparations were carried out as described by Fisher et al.26 The resultant membrane pellet was resuspended in buffer containing 10% (v/v) glycerol, stored at −80°C and typically contained 10–20 mg/mL protein.
Steady-state assays and enzyme inhibition studies
Mtb Ndh-catalysed oxidation of NADH and concomitant reduction of quinone were measured spectrophotometrically at 340 and 283 nm, respectively, using a Cary 300 Bio UV/visible spectrophotometer (Varian) as per Fisher et al.26 The Km and Vmax values were determined by fitting with a Michaelis–Menten rectangular hyperbola using Origin 8.5 (OriginLab Corp., USA). IC50 (50% inhibitory concentration) determinations for the phenothiazines against recombinant Mtb Ndh were performed using the steady-state assay. The IC50 values were calculated from plots of log dose versus quinone reduction rate, fitted with a four-parameter logistic function.
Chemoinformatics compound selection
Compounds were selected by searching the Zinc database of purchasable compounds (http://zinc.docking.org) for similarity to trifluoperazine. A Tanimoto coefficient of >0.7 was used.
bc1 inhibition studies
Activities of phenothiazine compounds against bc1 were determined spectrophotometrically as a function of cytochrome c reduction, as per Fisher et al.27 using Keilin–Hartree particles.28 Inhibitors were added prior to reaction initiation with 50 μM decylubiquinol and the IC50 values determined from plots of log dose versus cytochrome c reduction.
Culture of Mtb, drug susceptibility assays, isobole analyses and time–kill studies
For drug susceptibility assays, aerobic cultures of Mtb H37Rv were grown to mid-log phase at 37°C in 10 mL growth media [Middlebrook 7H9 broth supplemented with 10% albumin–dextrose–catalase solution (Becton Dickinson), 0.2% (v/v) glycerol and 0.05% (v/v) Tween 80]. Hypoxic cultures were grown similarly, but the head-space ratio was reduced to 0.5 and cultures were stirred at 120 rpm as per Wayne and Hayes.29
Mtb drug sensitivities were determined using a microplate Alamar blue assay (MABA) described by Hartkoorn et al.30 Measurements of well absorbance at 570 and 600 nm recorded using an Opsys MR plate reader were utilized to calculate IC50 values for the inhibitors. For anaerobic cultures, plates were sealed within GasPak EZ pouches containing an indicator to ensure anaerobic conditions were maintained. The plates were subsequently incubated anaerobically at 37°C for 7 days before being moved to an aerobic environment for a further 7 days. The IC50 values were calculated as described for aerobic cultures.
Drug competition assays were performed using the method of Berenbaum.31 The IC50 values for each compound to be tested were determined by the MABA technique described above.
Time–kill studies were carried out using an adaptation of previous methods.32,33 Mid-log phase Mtb H37Rv was diluted to 1 × 107 cfu/mL and subsequently added to equal volumes of drug-containing growth media. Drugs were present at final concentrations of 5× IC90 (90% inhibitory concentration), as determined from MABA studies, whilst a drug-free control was included in order to monitor normal culture growth. Aliquots of culture were withdrawn over 7 days and pelleted material washed in drug-free Middlebrook 7H9 media before cfu/mL determination by colony counting on solid growth media [Middlebrook 7H11 agar plates supplemented with 10% oleic acid–albumin–dextrose–catalase solution (Becton Dickinson), 0.2% (v/v) glycerol and 0.05% (v/v) Tween 80].
HepG2 toxicity studies
Cellular toxicities were determined using a modification of the method of Marroquin et al.34 Briefly, HepG2 cells cultured in glucose media [high-glucose Dulbecco's modified Eagle's medium (DMEM) containing 25 mM glucose, 1 mM sodium pyruvate, 5 mM HEPES, 10% fetal bovine serum and 100 μg/mL penicillin/streptomycin] or galactose media (as per glucose media but using glucose-free DMEM) were added to 96-well plates (100 μL, 1 × 104 cells/well) and incubated for 24 h at 37°C. The test compounds were added and incubation continued for a further 24 h. The plates were subsequently incubated for 2 h in the presence of 1 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution. Solubilization buffer, consisting of 50% (v/v) N,N-dimethylformamide and 20% (w/v) SDS, was added before well absorbance at 560 nm was measured using a Varioskan plate reader (ThermoScientific). The IC50 values were determined using a four-parameter logistic function.
Results
Steady-state kinetic characterization of recombinant Mtb Ndh
Initial kinetic characterization of crude membranes containing recombinant Ndh revealed typical biphasic activity with a fast linear phase followed by a second, slower phase. Unlike previous studies of Mtb Ndh,8,21 no detergent was added to the membrane preparation, as this has been shown to elicit non-enzymatic oxidation of NADH.26
Ndh activity was not diminished by the addition of the traditional complex I inhibitors rotenone and piercidin A at concentrations >50 μM. Ndh demonstrates a preference for NADH and shows no discernible activity in the presence of NADPH or deamino-NADH. In replication of the work of Yano et al.,21 apparent Km data for substrate analogues, selected for the variations in the structures of their aliphatic chains, were determined. The data (Figure 1a) identify a rank-order preference for coenzyme Q2 (Q2, C19H26O4) > coenzyme Q1 (Q1, C14H18O4) > decylubiquinone (dQ, C19H30O4) (Table 1). Vmax data indicate a ∼2-fold activity decrease for dQ compared with Q1 and Q2. As per Yano et al.,21 the determination of kinetic parameters using coenzyme Q4 and the endogenous substrate menaquinone were not possible under the described assay conditions.
Figure 1.
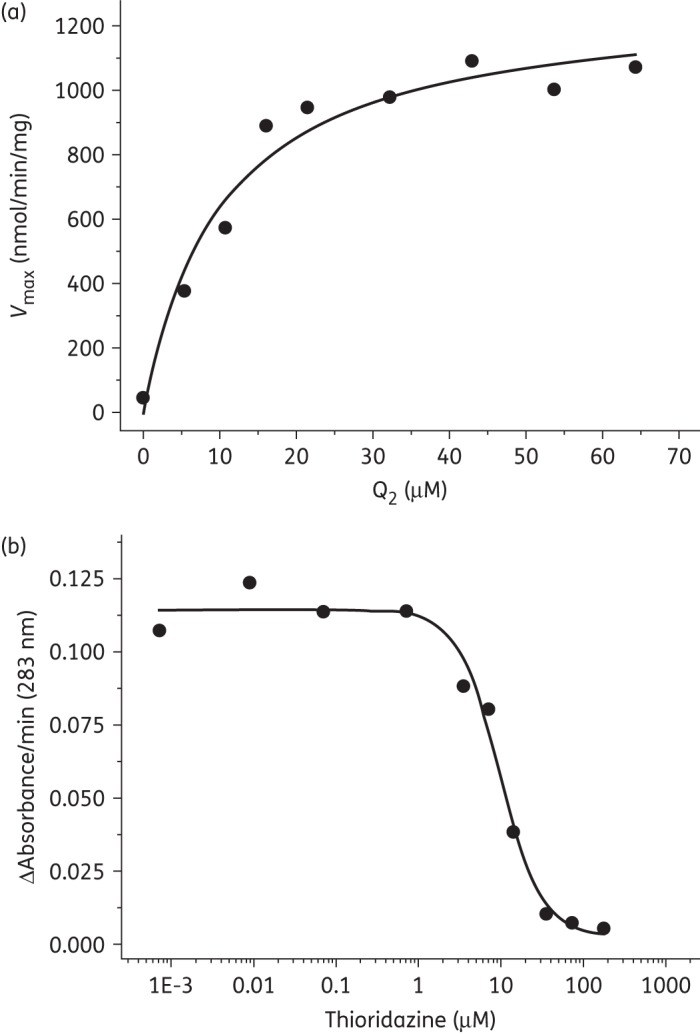
(a) Plot of coenzyme Q2 concentration dependence of recombinant Mtb Ndh, as determined by steady-state kinetic assays. The reduction of coenzyme Q2 was monitored at 283 nm and resultant data fitted with a rectangular hyperbolic function to determine the apparent Km of Q2 as 10.4 ± 3.4 μM and the Vmax as 1080.9 ± 59.4 nmol/min/mg. (b) Plot of thioridazine concentration against recombinant Mtb Ndh activity for IC50 determination. The IC50 for thioridazine was calculated as 11.4 μM ± 1.8 using a four-parameter logistic function (Origin 8.5 software). All assays contained 10–15 mg/mL protein in 50 mM potassium phosphate, 2 mM EDTA (pH 7.4) with potassium cyanide and NADH present at final concentrations of 1 mM and 200 μM, respectively. Inhibitor was introduced prior to reaction initiation by the addition of 50 μM coenzyme Q2.
Table 1.
Apparent Km and Vmax values for recombinant Mtb Ndh
| Coenzyme | Km (μM) | Vmax (nmol/min/mg) | Kmin (Vmax/Km) (/mM/s/mg) | Km (NADH) (μM) | Vmax (NADH) (nmol/min/mg) |
|---|---|---|---|---|---|
| dQ | 21.5 ± 5.0 | 673.3 ± 59.1 | 0.5 | 33.6 ± 7.9 | 441.6 ± 23.8 |
| Q1 | 18.6 ± 2.4 | 1291.2 ± 65.8 | 1.2 | 54.9 ± 12.2 | 1236.7 ± 77.1 |
| Q2 | 10.4 ± 3.4 | 1140.5 ± 106.9 | 1.8 | 69.3 ± 12.9 | 1080.9 ± 59.4 |
Values are expressed as the mean of multiple replicates ± SD and were calculated using a rectangular hyperbola (Origin 8.5 software). Km values for the quinones were determined in the presence of 200 μM NADH, whilst NADH Km values were determined in the presence of 50 μM of the appropriate quinone.
The apparent Km and Vmax values for NADH in the presence of the three quinone analogues detailed in Table 1 show that NADH binding is dependent upon the electron acceptor present, with the tightest binding observed for dQ. Vmax data show a similar trend to those of the quinone analogue study (Table 1).
Inhibition of Ndh by phenothiazines is correlated to Mtb killing
Inhibition studies for a range of commercially available phenothiazines, selected due to diversity of the side chain at the thiazine nitrogen, against recombinant Ndh identified concentration-dependent attenuation of the conversion of quinone to quinol (Figure 1b). The IC50 values ranged between 10 and 160 μM, with a rank order of potency of thioridazine > trifluoperazine > flupenthixol > perphenazine > fluphenazine > chlorpromazine > promethazine > promazine (Table 2). Phenothiazine did not exhibit an inhibitory effect.
Table 2.
IC50 values of phenothiazine-like compounds
| Compound | Structure | IC50 ± SEM (μM) |
||
|---|---|---|---|---|
| recombinant Ndh | Mtb H37Rv (aerobic) | Mtb H37Rv (Wayne model) | ||
| Chlorpromazine | 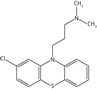 |
70.1 ± 9.6 | 23.7 ± 4.2 | 11.7 ± 2.5 |
| Flupenthixol | 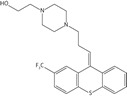 |
28.6 ± 1.7 | 13.2 ± 1.2 | 10.0 ± 0.3 |
| Fluphenazine | 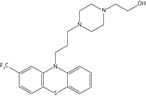 |
47.8 ± 3.2 | 14.5 ± 2.3 | 3.4 ± 0.9 |
| Perphenazine | 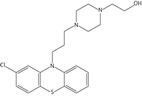 |
47.5 ± 6.6 | 18.8 ± 3.6 | 6.8 ± 1.4 |
| Phenothiazine | >200 | >100 | >100 | |
| Promazine | 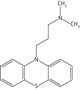 |
155.1 ± 17.3 | 62.1 ± 5.7 | 10.0 ± 3.8 |
| Promethazine | 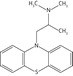 |
85.3 ± 16.1 | 50.8 ± 1.6 | 22.6 ± 4.8 |
| Thioridazine | 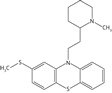 |
11.4 ± 1.8 | 13.4 ± 2.1 | 3.9 ± 0.8 |
| Trifluoperazine |  |
15.6 ± 2.2 | 10.5 ± 1.6 | 3.6 ± 0.7 |
Values were calculated spectrophotometrically (recombinant Ndh) or using the MABA assay (Mtb), as described in the Materials and methods section, and are the mean of at least two replicates.
Against Mtb under aerobic and anaerobic growth conditions (validated by an idiosyncratic improvement in metronidazole activity35), the phenothiazines exhibited significant concentration-dependent activity and the IC50 values were determined for all compounds except phenothiazine (Table 2). A plot of recombinant Mtb Ndh IC50 against Mtb IC50 (aerobically cultured, Figure 2) indicates a positive correlation; an observation echoed for anaerobic studies.
Figure 2.
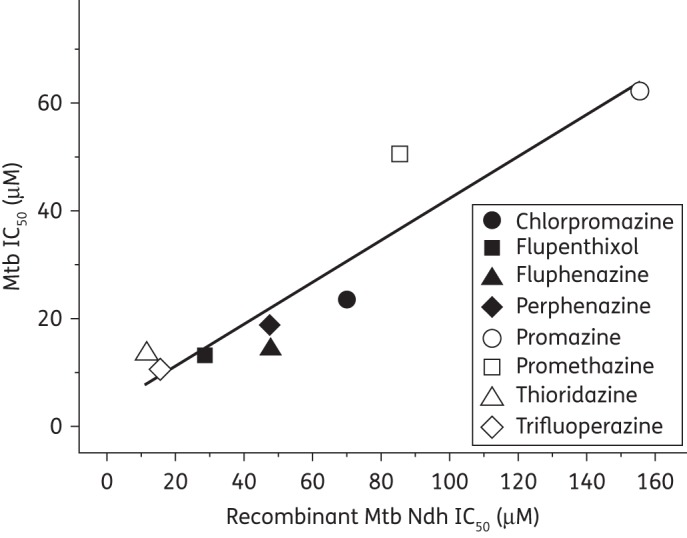
Scatterplot of the IC50s of eight phenothiazine-like compounds against recombinant Mtb Ndh and aerobically cultured Mtb, as detailed in Table 2. The data demonstrate a strong positive correlation between enzyme inhibition and in vitro antitubercular activity. Data for phenothiazine are not included, as accurate IC50 determinations could not be made.
Phenothiazines exhibit rapid bactericidal activity in time–kill studies
The bactericidal/static nature of eight phenothiazines was determined by time-dependent kill kinetic studies under aerobic conditions (Figure 3). All of the phenothiazines caused significant reductions in cfu/mL within the first 48 h and by day 4 no viable organisms remained. The order in which the phenothiazines reduced cfu/mL to zero was established as chlorpromazine = perphenazine (1 day) > trifluoperazine = thioridazine (2 days) > promethazine = promazine = flupenthixol = fluphenazine (4 days). Streptomycin (not shown) and isoniazid reduced colony counts to zero within a similar time frame, whilst ethambutol failed to completely attenuate growth.
Figure 3.

Time–kill curves for untreated cells (control), thioridazine, trifluoperazine, fluphenazine, flupenthixol, promazine, promethazine, chlorpromazine, perphenazine, isoniazid and ethambutol. Compounds were present at 5× IC90 (established from MABA assays) and cfu/mL were determined at the appropriate timepoints (as described in the Materials and methods section). Data are the mean of two experiments.
Phenothiazines display a degree of antagonism with isoniazid
The isobolograms (Figure 4) demonstrate significantly varied responses dependent upon the drug combination tested. In combinations of thioridazine or trifluoperazine with rifampicin, isoboles remained very close to the additive line. Similar responses were observed with streptomycin and ethambutol and indicate that the three Mtb therapies act in an additive manner with the tested phenothiazines. The isoniazid isoboles show considerable convexity with fractional inhibitory concentration values >2, indicating a degree of antagonism.31
Figure 4.


Isobolograms for thioridazine in combination with (a) rifampicin, (b) streptomycin, (c) ethambutol and (d) isoniazid. Panels (e–h) show isobolograms for trifluoperazine in combination with the aforementioned antitubercular compounds, respectively. Fixed ratios of each pair of compounds were prepared and used to determine IC50 values for each compound. Subsequently, the 50% fractional inhibitory concentration (FIC) for each compound in combination was calculated (see the Materials and methods section) and plotted as an isobologram. Compounds demonstrating an additive effect have a summed FIC ≈ 1 (dashed line), whilst antagonistic and synergistic combinations have values >4 and <0.5, respectively.57
Cellular toxicity of phenothiazines appears not to be associated with mitochondrial dysfunction
Mitochondrial toxicity can be identified by the differing responses observed for HepG2 cells grown in glucose-containing media, which favours glycolysis, or galactose-containing media, which promotes reliance upon oxidative phosphorylation.34 The toxicity of tamoxifen (Figure 5a) is observed to be unaltered by a change of growth media, as is expected for a compound with no known mitochondrial toxicity. Conversely, the mitochondrial complex I inhibitor rotenone is not significantly toxic in glucose media but demonstrates severe toxicity in galactose media (Figure 5b), thus indicting disruption of oxidative phosphorylation. The majority of the phenothiazines (Figure 5c) demonstrated comparable toxicity to the non-mitochondrial toxicant tamoxifen (IC50 = 19.88 μM) for HepG2 cells cultured in glucose-containing media. The toxicities of promazine (not shown) and promethazine were ∼2-fold less (IC50 = 39.15 and 41.85 μM, respectively) than other phenothiazine-like compounds, whilst minimal toxicity was observed for phenothiazine and rotenone at 100 and 1 μM, respectively. In galactose-containing media, toxicities of the phenothiazines and tamoxifen remained largely unchanged. For example, the IC50 values for fluphenazine in glucose and galactose media were 20.76 and 23.15 μM, respectively, (not shown). Phenothiazine showed little or no toxicity at 100 μM, whilst a significant increase in rotenone toxicity was observed (0.07 μM).
Figure 5.
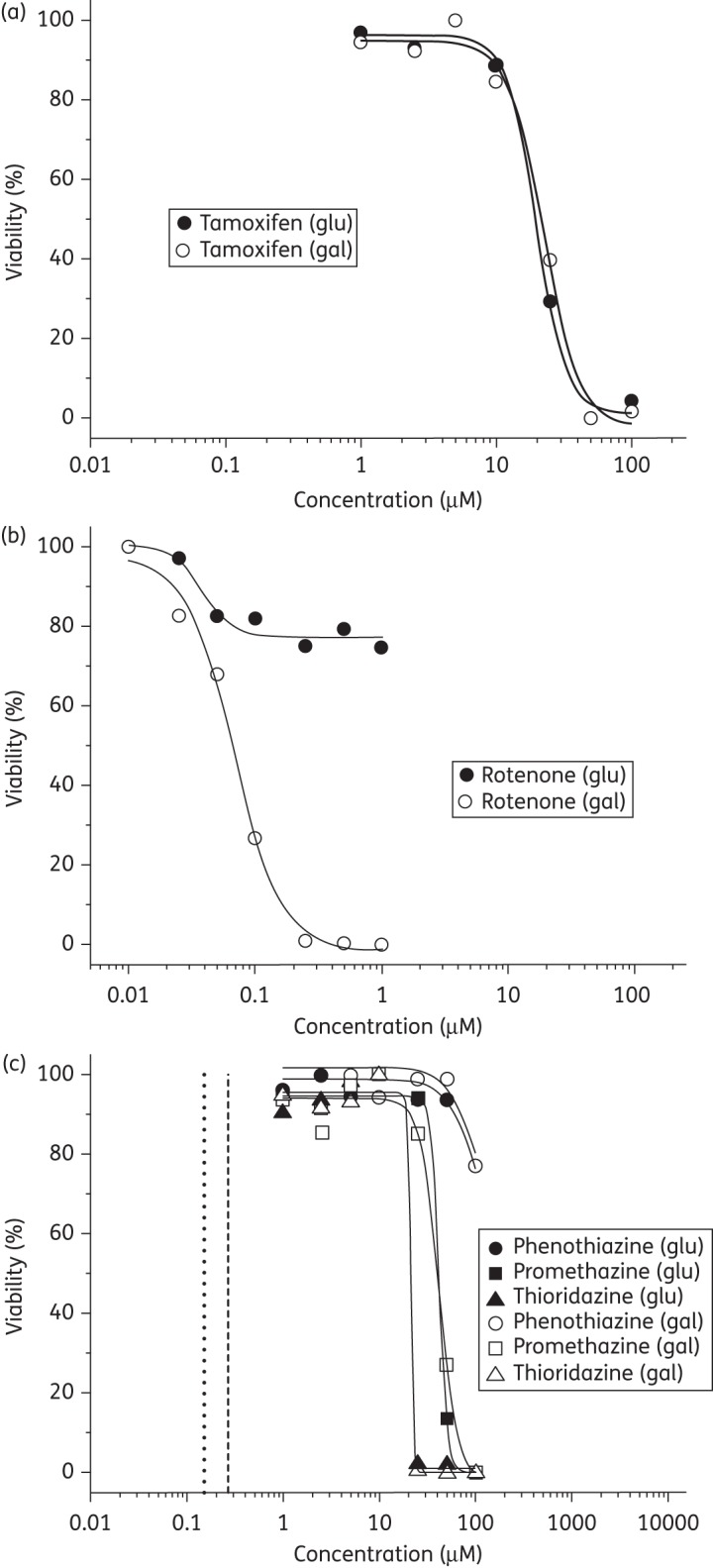
Percentage viability of HepG2 cell versus drug concentration (μM) cultured in glucose (glu) and galactose (gal) media. Panels (a) and (b) illustrate the responses elicited by a switch in media for the non-mitochondrial toxicant tamoxifen and the respiratory poison rotenone, respectively. (c) The responses for phenothiazine, promethazine and thioridazine in glucose and galactose media. In (c) the dashed vertical line represents the therapeutic maximum plasma concentration of thioridazine, whilst the dotted line indicates the concentration of thioridazine required to kill phagocytosed Mtb.52,53
The phenothiazines were counterscreened against bovine cytochrome bc1 due to concerns of complex III-mediated cardiotoxicity; however, only minimal inhibitory activity of these compounds was observed.36 IC50 values of 58.96 and 90.45 μM were recorded for thioridazine and trifluoperazine, respectively, whilst chlorpromazine and flupenthixol showed insignificant levels of inhibition at concentrations of 150 μM (data not shown).
Discussion
The findings of this kinetic characterization are consistent with previous studies wherein Mtb Ndh catalyses the conversion of quinone to quinol with concomitant NADH oxidation, complex I inhibitors demonstrate no efficacy and NADH is the preferred coenzyme over NADPH.8,21 Mtb Ndh appears somewhat unusual in this respect as other organisms, including Saccharomyces cerevisiae and Corynebacterium glutamicum, demonstrate an ability to utilize NADPH and deamino-NADH in addition to NADH.37,38 Whilst the physiological reason for these differences is not clear, structural differences in the NAD(P)H binding site are the most likely cause for a lack of activity with NADPH. The inability of deamino-NADH, which is only subtly structurally distinct from NADH, to supply reducing equivalents to Mtb Ndh is unlikely to be a consequence of an unfavourable redox potential, as it is expected to have a potential identical to that of NADH.39
The preference of Q2 > Q1 > dQ in this study is also difficult to attribute to redox potential differences. The redox potentials of the three coenzymes are not significantly different (+40, +113 and +90 mV for Q2, Q1 and dQ, respectively40–42) and without a detailed understanding of the electron transfer mechanism in operation (i.e. whether or not a semiquinone intermediate is involved), the significance of quinone redox potentials cannot be clearly ascertained. Whilst the extended hydrophobic tail of Q2 may be an important determiner of selectivity in the binding site, the Kmin values determined (Table 1) suggest that solubility is significant. They indicate that catalytic efficiency is less with dQ, which has the least favourable clogP for assays performed in aqueous media, although the differing activities of the quinones may also be a consequence of critical micelle concentrations and, hence, variability in the availability of monomeric forms in solution.
The orders of coenzyme preference in this study (Q2 > Q1 > dQ) and the study of Yano et al.21 (dQ > Q2 > Q1) are discordant. The differences most likely derive from performing assays using Ndh purified to differing degrees and that this study directly monitors the conversion of quinone to quinol rather than NADH oxidation. The latter may have additional electron acceptors (including oxygen) and may be adversely influenced by the presence of detergent.26
Alternative modes of action, including the inhibition of efflux pumps and/or calcium-binding proteins, have been suggested for the antitubercular properties of the phenothiazines.43,44 Whilst this study cannot rule out the possibility of multiple cellular targets, the drug susceptibility data presented herein demonstrate that the inhibition of Mtb Ndh by the phenothiazine class of compounds directly correlates to the antitubercular activity under aerobic and anaerobic growth conditions (Figures 1b and 2 and Table 2); thus, further supporting the hypothesis that a key molecular target for this class is Ndh.
The diversity of potency amongst these compounds is manifold and may in part be explained by structural variations. The lack of activity demonstrated by phenothiazine is likely due to the absence of aromatic ring substitution and/or the absence of a side chain at the thiazine nitrogen. As noted by Bate et al.,17 the 2-position of the phenothiazine structure appears significant to inhibitor binding, as adornments at this position appreciably increase potency (e.g. chlorpromazine compared with promazine). It should also be noted that such seemingly trivial structural modifications have been shown to be responsible for the differing modes of action of the antitubercular pyrazinamide (uncertain mode of action) and its derivative 5-chloropyrazinamide (a confirmed fatty acid synthase I inhibitor).45 Activity is further improved by the addition of an aliphatic chain at the 10-position nitrogen (promazine and promethazine) and the subsequent addition of a piperazine ring (flupenthixol, fluphenazine and trifluoperazine) or a piperidine ring (thioridazine) to this chain.
Interestingly, a slight enhancement in potency is observed for the phenothiazines in the hypoxia model in comparison with aerobic conditions. It has been previously noted that a number of phenothiazine analogues are equally potent in both models10,17 and, as such, these two datasets further validate the hypothesis that Ndh is the primary NADH dehydrogenase in non-replicating Mtb. This is highly encouraging, as the ability to target non-replicating Mtb is a key aspect of the target product profile (TPP).
The time–kill data show that unlike ethambutol (which is bacteriostatic), the tested phenothiazines are bactericidal, rapidly eliminating all Mtb organisms from liquid culture within 4 days (a time period comparable to that of streptomycin and isoniazid). This finding, which confirms previous data for trifluoperazine and chlorpromazine,8,46 is significant, as current TPPs for novel therapies include a desire for compounds to be bactericidal to minimize resistance development and to act rapidly in order that treatment regimens may be shortened.2 The data presented here clearly illustrate that the phenothiazines fulfil both these criteria.
The isobole data generated for rifampicin, ethambutol and streptomycin with the phenothiazines demonstrate non-antagonistic effects. Considering the differing modes of actions for these compounds, it is unsurprising that there is no synergism or antagonism with the respiratory chain-inhibiting phenothiazines. The antagonism observed between isoniazid and the phenothiazines may at first sight appear curious; however, prior to the inhibition of the target enzyme InhA, isoniazid is required to form an adduct with NAD+, catalysed by the catalase peroxidase KatG.47,48 Inhibition of Mtb Ndh would be expected to modulate intracellular NADH/NAD+ ratios, resulting in the accumulation of NADH, which may directly compete with the isoniazid-NAD+ adduct at the InhA active site or, as NADH is a peroxidase substrate, competitively inhibit the adduct formation catalysed by KatG.47–50 However, it should be noted that this finding may contraindicate the use of isoniazid in conjunction with a phenothiazine derivative, as it may result in a suboptimal therapeutic response.
Due to unexpected cardiotoxicity, promising compounds (e.g. the Plasmodium falciparum cytochrome bc1 inhibitor GW844520) have subsequently been withdrawn from development51 and therefore early assessment of potential toxicity is paramount when designing novel drugs against respiratory chain targets. The data presented herein identify that the inhibitory effects against bovine bc1 mediated by the phenothiazine compounds are ≥5–10-fold lower than for Mtb Ndh and are, hence, unlikely to elicit the levels of cardiotoxicity witnessed for bc1-specific compounds.
The response of HepG2 cells exposed to the phenothiazines (Figure 5c) is similar in magnitude to that observed for tamoxifen and is not appreciably altered by the respiratory pathway in operation. Significantly, the therapeutic maximum plasma concentration for thioridazine, when used as an antipsychotic, is ∼1.2–2.5 μM,52 which is ≥10-fold less than the concentrations required to elicit significant toxicity in HepG2 cells.
Whilst the concentrations of phenothiazines determined in this study that effect a complete in vitro Mtb kill (e.g. >50 μM for thioridazine) will elicit significant toxicity in humans, concentrations required to clear Mtb from macrophages may be appreciably lower. Indeed, Ordway et al.53 demonstrated accumulation of thioridazine and chlorpromazine within macrophages and established that concentrations of these drugs in the order of 0.25 μM were sufficient to kill phagocytosed antibiotic-susceptible and MDR Mtb in vitro. It is noteworthy that the drug penetration of anti-infectives into the pulmonary epithelial lining fluid is often not consistent with plasma drug levels.54,55 For some drugs it is therefore difficult to predict in vivo drug efficacy from in vitro data; a case exemplified by the recent clinical study of linezolid, which demonstrated efficacy for the treatment of chronic XDR Mtb despite having modest in vitro and in vivo (rodent models) activity.56
In conclusion, this work further validates the respiratory chain of Mtb and, more specifically, its type II NADH:quinone oxidoreductase as a viable target for the development of novel antituberculars. Furthermore, our data support the hypothesis that Ndh is a key molecular drug target for phenothiazine-based derivatives. The favourable pharmacodynamic properties of the phenothiazines, as shown here, are consistent with a TPP that includes activity against dormant/persistent bacilli, rapid bactericidal activity and activity against drug-resistant Mtb by virtue of a novel mode of action.
Funding
This work was supported by grants from the National Institute of Health Research (NIHR, BRC Liverpool), Leverhulme Trust and the Medical Research Council.
Transparency declarations
None to declare.
Acknowledgements
We are grateful to Professor H. Rubin (University of Pennsylvania) and Professor T. Friedrich (University of Freiburg) for their generous donations of biological materials.
References
- 1.WHO. Global Tuberculosis Control 2011. http://www.who.int/tb/publications/global_report/2011/gtbr11_full.pdf. (17 September 2012, date last accessed) [Google Scholar]
- 2.Ginsberg AM. Drugs in development for tuberculosis. Drugs. 2010;70:2201–14. doi: 10.2165/11538170-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Cole ST, Riccardi G. New tuberculosis drugs on the horizon. Curr Opin Microbiol. 2011;14:570–6. doi: 10.1016/j.mib.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 4.Koul A, Dendouga N, Vergauwen K, et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol. 2007;3:323–4. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 5.Koul A, Vranckx L, Dendouga N, et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J Biol Chem. 2008;283:25273–80. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- 6.Diacon AH, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N Engl J Med. 2009;360:2397–405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 7.Bald D, Koul A. Respiratory ATP synthesis: the new generation of mycobacterial drug targets? FEMS Microbiol Lett. 2010;308:1–7. doi: 10.1111/j.1574-6968.2010.01959.x. [DOI] [PubMed] [Google Scholar]
- 8.Weinstein EA, Yano T, Li LS, et al. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci USA. 2005;102:4548–53. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melo AMP, Bandeiras TM, Teixeira M. New insights into type II NAD(P)H:quinone oxidoreductases. Microbiol Mol Biol Rev. 2004;68:603–16. doi: 10.1128/MMBR.68.4.603-616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao SPS, Alonso S, Rand L, et al. The proton motive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2008;105:11945–50. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yano T, Kassovska-Bratinova S, Teh JS, et al. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase. A pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem. 2011;286:10276–87. doi: 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 13.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 2003;100:12989–94. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griffin JE, Gawronski JD, DeJesus MA, et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amaral L, Kristiansen JE, Abebe LS, et al. Inhibition of the respiration of multi-drug resistant clinical isolates of Mycobacterium tuberculosis by thioridazine: potential use for initial therapy of freshly diagnosed tuberculosis. J Antimicrob Chemother. 1996;38:1049–53. doi: 10.1093/jac/38.6.1049. [DOI] [PubMed] [Google Scholar]
- 16.Crowle AJ, Douvas GS, May MH. Chlorpromazine—a drug potentially useful for treating mycobacterial infections. Chemotherapy. 1992;38:410–9. doi: 10.1159/000239036. [DOI] [PubMed] [Google Scholar]
- 17.Bate AB, Kalin JH, Fooksman EM, et al. Synthesis and antitubercular activity of quaternized promazine and promethazine derivatives. Bioorg Med Chem Lett. 2007;17:1346–8. doi: 10.1016/j.bmcl.2006.11.091. [DOI] [PubMed] [Google Scholar]
- 18.Martins M, Schelz Z, Martins A, et al. In vitro and ex vivo activity of thioridazine derivatives against Mycobacterium tuberculosis. Int J Antimicrob Agents. 2007;29:338–40. doi: 10.1016/j.ijantimicag.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Abbate E, Vescovo M, Natiello M, et al. Successful alternative treatment of extensively drug-resistant tuberculosis in Argentina with a combination of linezolid, moxifloxacin and thioridazine. J Antimicrob Chemother. 2012;67:473–7. doi: 10.1093/jac/dkr500. [DOI] [PubMed] [Google Scholar]
- 20.van Soolingen D, Hernandez-Pando R, Orozco H, et al. The antipsychotic thioridazine shows promising therapeutic activity in a mouse model of multidrug-resistant tuberculosis. PLoS ONE. 2010;5:e12640. doi: 10.1371/journal.pone.0012640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yano T, Li LS, Weinstein E, et al. Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2) J Biol Chem. 2006;281:11456–63. doi: 10.1074/jbc.M508844200. [DOI] [PubMed] [Google Scholar]
- 22.Boshoff HIM, Myers TG, Copp BR, et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004;279:40174–84. doi: 10.1074/jbc.M406796200. [DOI] [PubMed] [Google Scholar]
- 23.Glassman AH, Bigger JT. Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am J Psychiatry. 2001;158:1774–82. doi: 10.1176/appi.ajp.158.11.1774. [DOI] [PubMed] [Google Scholar]
- 24.Maurer I, Möller H-J. Inhibition of complex I by neuroleptics in normal human brain cortex parallels the extrapyramidal toxicity of neuroleptics. Mol Cell Biochem. 1997;174:255–9. [PubMed] [Google Scholar]
- 25.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. 3rd edn. Cold Spring Harbor: Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- 26.Fisher N, Warman AJ, Ward SA, et al. Type II NADH:quinone oxidoreductases of Plasmodium falciparum and Mycobacterium tuberculosis: kinetic and high-throughput assays. Meth Enzymol. 2009;456:303–20. doi: 10.1016/S0076-6879(08)04417-0. [DOI] [PubMed] [Google Scholar]
- 27.Fisher N, Majid RA, Antoine T, et al. Cytochrome b mutation Y268S conferring atovaquone resistance phenotype in malaria parasite results in reduced parasite bc1 catalytic turnover and protein expression. J Biol Chem. 2012;287:9731–41. doi: 10.1074/jbc.M111.324319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuboyama M, Yong FC, King TE. Studies on cytochrome oxidase. J Biol Chem. 1972;247:6375–83. [PubMed] [Google Scholar]
- 29.Wayne LG, Hayes LG. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun. 1996;64:2062–9. doi: 10.1128/iai.64.6.2062-2069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartkoorn RC, Chandler B, Owen A, et al. Differential drug susceptibility of intracellular and extracellular tuberculosis, and the impact of P-glycoprotein. Tuberculosis. 2007;87:248–55. doi: 10.1016/j.tube.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Berenbaum MC. Method for testing for synergy with any number of agents. J Infect Dis. 1978;137:122–30. doi: 10.1093/infdis/137.2.122. [DOI] [PubMed] [Google Scholar]
- 32.de Steenwinkel JEM, de Knegt GJ, ten Kate MT, et al. Time–kill kinetics of antituberculosis drugs, and emergence of resistance, in relation to metabolic activity of Mycobacterium tuberculosis. J Antimicrob Chemother. 2010;65:2582–9. doi: 10.1093/jac/dkq374. [DOI] [PubMed] [Google Scholar]
- 33.Murugasu-Oei B, Dick T. Bactericidal activity of nitrofurans against growing and dormant Mycobacterium bovis BCG. J Antimicrob Chemother. 2000;46:917–9. doi: 10.1093/jac/46.6.917. [DOI] [PubMed] [Google Scholar]
- 34.Marroquin LD, Hynes J, Dykens JA, et al. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–47. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 35.Wayne LG, Sramek HA. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1994;38:2054–8. doi: 10.1128/aac.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barton V, Fisher N, Biagini GA, et al. Inhibiting Plasmodium cytochrome bc1: a complex issue. Curr Opin Chem Biol. 2010;14:440–6. doi: 10.1016/j.cbpa.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 37.Nantapong N, Otofuji A, Migita CT, et al. Electron transfer ability from NADH to menaquinone and from NADPH to oxygen of type II NADH dehydrogenase of Corynebacterium glutamicum. Biosci Biotechnol Biochem. 2005;69:149–59. doi: 10.1271/bbb.69.149. [DOI] [PubMed] [Google Scholar]
- 38.Yang Y, Yamashita T, Nakamaru-Ogiso E, et al. Reaction mechanism of single subunit NADH-ubiquinone oxidoreductase (Ndi1) from Saccharomyces cerevisiae. Evidence for a ternary complex mechanism. J Biol Chem. 2011;286:9287–97. doi: 10.1074/jbc.M110.175547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoll VS, Blanchard JS. Kinetic mechanism and nucleotide specificity of NADH peroxidase. Arch Biochem Biophys. 1988;260:752–62. doi: 10.1016/0003-9861(88)90505-x. [DOI] [PubMed] [Google Scholar]
- 40.Ohnishi T, Moser CC, Page CC, et al. Simple redox-linked proton-transfer design: new insights from structures of quinol-fumarate reductase. Struct Fold Design. 2000;8:R23–32. doi: 10.1016/s0969-2126(00)00098-8. [DOI] [PubMed] [Google Scholar]
- 41.Thauer RK, Jungermann K, Decker K. Energy conservation in chemotropic anaerobic bacteria. Bacteriol Rev. 1977;41:100–80. doi: 10.1128/br.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kotlyar AB, Gutman M, Ackrell BAC. Interaction of ubiquinone and vitamin K3 with mitochondrial succinate-ubiquinone oxidoreductase. Biochem Biophys Res Commun. 1992;186:1656–62. doi: 10.1016/s0006-291x(05)81598-0. [DOI] [PubMed] [Google Scholar]
- 43.Amaral L, Martins M, Viveiros M, et al. Promising therapy of XDR-TB/MDR-TB with thioridazine an inhibitor of bacterial efflux pumps. Curr Drug Targ. 2008;9:816–9. doi: 10.2174/138945008785747798. [DOI] [PubMed] [Google Scholar]
- 44.Amaral L, Viveiros M. Why thioridazine in combination with antibiotics cures extensively drug-resistant Mycobacterium tuberculosis infections. Int J Antimicrob Agents. 2012;39:376–80. doi: 10.1016/j.ijantimicag.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 45.Boshoff HI, Mizrahi V, Barry CE. Effects of pyrazinamide on fatty acid synthesis by whole mycobacterial cells and purified fatty acid synthase I. J Bacteriol. 2002;184:2167–72. doi: 10.1128/JB.184.8.2167-2172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie ZF, Siddiqi N, Rubin EJ. Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2005;49:4778–80. doi: 10.1128/AAC.49.11.4778-4780.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Argyrou A, Vetting MW, Blanchard JS. New insight into the mechanism of action of and resistance to isoniazid: interaction of Mycobacterium tuberculosis enoyl-ACP reductase with INH-NADP. J Am Chem Soc. 2007;129:9582–3. doi: 10.1021/ja073160k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100:13881–6. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee ASG, Teo ASM, Wong S-Y. Novel mutations in ndh in isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob Agents Chemother. 2001;45:2157–9. doi: 10.1128/AAC.45.7.2157-2159.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miesel L, Weisbrod TR, Marcinkeviciene JA, et al. NADH dehydrogenase defects confer isoniazid resistance and conditional lethality in Mycobacterium smegmatis. J Bacteriol. 1998;180:2459–67. doi: 10.1128/jb.180.9.2459-2467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schlitzer M. Antimalarial drugs—what is in use and what is in the pipeline. Arch Pharm (Weinheim) 2008;341:149–63. doi: 10.1002/ardp.200700184. [DOI] [PubMed] [Google Scholar]
- 52.Thanacoody HKR. Thioridazine: resurrection as an antimicrobial agent? Br J Clin Pharmacol. 2007;64:566–74. doi: 10.1111/j.1365-2125.2007.03021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ordway D, Viveiros M, Leandro C, et al. Clinical concentrations of thioridazine kill intracellular multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2003;47:917–22. doi: 10.1128/AAC.47.3.917-922.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodvold KA, George JM, Yoo L. Penetration of anti-infective agents into pulmonary epithelial lining fluid: focus on antibacterial agents. Clin Pharmacokinet. 2011;50:637–64. doi: 10.2165/11594090-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 55.Rodvold KA, Yoo L, George JM. Penetration of anti-infective agents into pulmonary epithelial lining fluid: focus on antifungal, antitubercular and miscellaneous anti-infective agents. Clin Pharmacokinet. 2011;50:689–704. doi: 10.2165/11592900-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 56.Lee M, Lee J, Carroll MW, et al. Linezolid for treatment of chronic extensively drug-resistant tuberculosis. New Engl J Med. 2012;367:1508–18. doi: 10.1056/NEJMoa1201964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Odds FC. Synergy, antagonism and what the chequerboard puts between them. J Antimicrob Chemother. 2003;52:1. doi: 10.1093/jac/dkg301. [DOI] [PubMed] [Google Scholar]