

Abstract
The past decade has seen rapid growth in the use of diverse compound libraries in classical phenotypic screens to identify modulators of a given process. The subsequent process of identifying the molecular targets of active hits, also called ‘target deconvolution’, is an essential step for understanding compound mechanism of action and for using the identified hits as tools for further dissection of a given biological process. Recent advances in ‘omics’ technologies, coupled with in silico approaches and the reduced cost of whole genome sequencing, have greatly improved the workflow of target deconvolution and have contributed to a renaissance of ‘modern’ phenotypic profiling. In this review, we will outline how both new and old techniques are being used in the difficult process of target identification and validation as well as discuss some of the ongoing challenges remaining for phenotypic screening.
Introduction
When searching for biologically active molecules, phenotypic screening is often the most straightforward and intuitive way to discover relevant hits. The alternative is target-based screening in which a large number of compounds are screened against a single target protein, and subsequently, the active hits can be further optimized through medicinal chemistry efforts [1]. However, according to a recent analysis of new molecular entities, target-based approaches are not as efficient as traditional phenotype-based methods in terms of generating first-in-class small-molecule drugs [2]. One of the major limitations of target-based strategies is the fact that many compounds are found to interact with multiple targets, with most drug molecules interacting with six known molecular targets on average [3]. Therefore the one drug, ‘one target’ paradigm, thought to be the cornerstone of target-based methods, often does not hold true for compounds identified using target-based methods. This deficiency has lead to a paradigm shift, that, when coupled with recent technological advances in proteomics and genomics methods, has resulted in a renaissance for phenotype-based screening methods.
One of the major advantages of phenotype-based approaches is that they provide an unbiased way to find active compounds in the context of complex biological systems. Because phenotypic screening takes place in a physiologically relevant environment of cells or whole organism, the results from such screens provide a more direct view of the desired responses as well as highlight potential side effects. More importantly, phenotypic screens can lead to the identification of multiple proteins or pathways that may not have been previously linked to a given biological output. Therefore, identifying the molecular targets of active hits from phenotypic screens is a crucial process that is required to understand underlying mechanisms and to further optimize active compounds. Because target identification from phenotypic screens is expected to generate a spectrum of possible targets, the term ‘target deconvolution’ was coined to more accurately define the process.
Over the last decade, a number of technologies from a wide range of fields have been explored to identify targets from phenotypic screens. In particular, proteomics and genomics-based approaches have become more powerful when combined with whole genome sequencing [4]. High-throughput imaging platforms and computational analysis also have helped to find relevant pathways and proteins based on phenotype changes [5]. Recent advances in quantitative mass spectrometry techniques have facilitated quantitative analysis of proteins, and greatly enhanced the sensitivity of target detection [6]. In this review, we will focus on the most recent examples of target deconvolution techniques in modern phenotypic profiling.
Chemical proteomic-based approaches
The term ‘chemical proteomics’ is often used to define a specific focus area within the broader field of proteomics in which a small molecule is used to directly reduce the complexity of an entire proteome to focus only on proteins that interact with that target molecule. There are multiple approaches that can be employed in chemical proteomic workflows. These include small molecule affinity-and activity-based probes that can be used to isolate targets and more recently, label-free techniques to directly identify small molecule binding proteins. Since, many reviews have covered the general principles of these approaches [6–9], we will focus only on the most recent examples of each technique.
Affinity chromatography
Affinity purification is the most widely used technique to isolate specific target proteins from a complex proteome (Figure 1A). Small molecules identified in phenotypic screens are immobilized onto a solid support that can be used to isolate bound protein targets. This approach relies on extensive washing steps to remove non-binders, followed by specific methods to elute the proteins of interest. The eluted proteins can then either be directly identified using “shotgun” type sequencing methods with multi-dimensional liquid chromatography or be further separated by gel electrophoresis and analyzed by mass spectrometry. The identified peptide sequences can then be used in database searches to identify the target protein [10].
Figure 1.

Affinity Chromatography. (A) An active hit is directly linked to a solid support or tagging group such as biotin, and its target protein isolated via affinity pull-down. The isolated proteins are further analyzed by mass spectrometry; (B) An active hit is modified with a small ‘clickable’ group for in situ labeling with minimal structural perturbation; (C) A photo-reactive group such as diazirine group is added to induce covalent cross-link between the hit compound and its target.
Although the idea is simple, immobilizing a small molecule onto a solid support is a challenging task. Any modification of the active molecules has the potential to affect binding affinity to the target, therefore the process requires substantial knowledge of structure-activity relationship and often requires significant chemistry efforts to identify a site for attachment of the affinity tag. Moreover, for some molecules, the addition of any kind of bulky tag leads to a dramatic loss of activity. In order to overcome these problems, a relatively small azide or an alkyne tag has been widely used to minimize structural perturbation and to conjugate an affinity tag via ‘click chemistry’ after the active hit is bound to its target (Figure 1B) [11]. Since the modified hits do not contain a bulky tag, which can interfere with membrane permeability, this method is particularly useful to search for intracellular targets and has been used, for example, for isolating targets of kinase inhibitors in mammalian cells [12,13].
In addition to an affinity tag, it is also possible to use a photoreactive group to induce covalent cross-linking and secure the interaction between a weakly bound small molecule and a protein target (Figure 1C). For optimal identification, the hit compound needs to be modified with a small photoreactive group such as a benzophenone, diazirine, or arylazide, and also requires a reporter group for rapid isolation. One interesting example is the case of imatinib, also known as Gleevec [14]. This drug was rationally designed to target the Bcr-Abl oncogenic receptor tyrosine kinase, and marketed as an anti-cancer drug. However, imatinib has been reported to reduce hypertension [15] and also reduce β-amyloid in the brain [16]. Recently, imatinib was labeled with an aryl azide and used to identify γ-secretase activating protein (gSAP) as an additional molecular target [14].
Since photo-affinity labeling requires a photo-reactive group and a reporter tag, ‘all-in-one’ functional groups containing both components have been introduced to minimize structural modification without using radioisotopes [17]. Alternatively, a multifunctional benzophenone-based small molecule library was also developed for integrated screening and target isolation [18]. This multifunctional scaffold can serve as a photoreactive group, a CLICK tag and a protein-interacting functionality simultaneously. By embedding all three elements into one core scaffold, the process of phenotypic screening to target identification can be greatly accelerated.
In addition to effects from adding a tag for affinity purification, changing the composition of affinity beads can improve efficiency of purification by reducing false-positives and boosting interaction between small molecules and protein targets [19,20]. For example, using high-performance magnetic beads can reduce multiple washing and separation steps to one procedure. These magnetic beads have been applied to identify the molecular target of thalidomide, a sedative used in the early 60s that ended up having significant teratogenicity [19]. The drug is still used for leprosy and multiple myeloma, however, the reasons for its link to birth defects was never understood due to the lack of suitable affinity probe. By using high-performance beads decorated with thalidomide, cereblon was identified as the molecular target.
Activity-based protein profiling
Activity-based probes (ABPs) are small molecule tools that can be used to monitor the activity of specific classes of enzymes. Over the past decade and a half, various ABPs have been designed to study proteases, hydrolases, phosphatases, histone deacetylases, and glycosidases [21–23], and these probes have proven to be valuable in investigating enzyme-related disease mechanisms including cancer [24], microbial and parasitic pathogenesis [25,26], and metabolic disorders [27]. Typical activity-based protein profiling follows a similar overall workflow as affinity chromatography, including probe binding, protein separation, sequence analysis, and database searching (Figure 2). However, because most ABPs are designed to target a specific enzyme class, ABPs are particularly useful for phenotypic screening and lead optimization where a specific enzyme or enzyme family is suspected to be involved in a certain disease state or pathway.
Figure 2.

Activity-Based Protein Profiling. (A) Three main features of activity-based probes: 1) A reactive electrophile that allows covalent attachment of the ABP in the active site of target enzyme, and enables rapid isolation and analysis of the target protein, 2) A specificity region that directs the probe to the specific class of enzymes, and 3) A tagging group for detection; (B) General workflow for using an ABP. When an active hit is identified from a screen it can be used to directly affinity isolate the target (top). Similarly, the direct screening of ABPs and covalent inhibitors in various systems such as cells and animals greatly facilitates the overall process of target deconvolution. Furthermore, broad-spectrum ABPs provide a powerful tool to study disease related pathways and class-wide enzyme assay platform.
ABPs have three components; a reactive electrophile for covalent modification of enzyme active site, a linker or a specificity group for directing probes to specific enzymes, and a reporter or a tag for separating labeled enzymes (Figure 2A). Therefore, a library of ABPs can be directly used for phenotypic screening and target identification simultaneously [28,29]. Additionally, covalent enzyme inhibitors can be readily converted to ABPs by attaching a tagging group, and the combined use of covalent inhibitors and ABPs in phenotypic screening greatly facilitates target identification and also offers a powerful tool to study function and mechanism of identified proteins [30,31]. For example, Hall et al. successfully identified a small molecule that blocks the process of host cell invasion by the Toxoplasma gondii. The selected inhibitor, WRR-086 was then converted to an ABP by attaching an alkyne group for click chemistry, and used to identify TgDJ-1, a poorly characterized protein involved in oxidative stress response as a key player in host cell invasion [31].
In order to covalently attach ABPs to target proteins, an active site nucleophile such as cysteine or serine is required. However, not all enzymes have a nucleophile in their active site. One way to overcome this problem is to incorporate a photo-reactive group, as has been demonstrated for probes of γ-secretase [32] metalloproteases [33], and the proteasome [34]. Alternatively, an electrophile can be introduced to react with any cysteine residue in close proximity to the site of probe binding [35,36]. Incorporating a clickable group such as an alkyne or an azide on an ABP can also facilitate a direct link from phenotypic screening to target identification [31,37,38]. Additionally, to improve efficiency of two-step labeling, other tagging methods employing copper-free click chemistry [39], sulfo-click chemistry [40], Staudinger ligation [41], or Diels-Alder reaction [42] were developed as an alternative to conventional click chemistry.
ABPs are not only useful for target identification, but also are powerful tools for the discovery of disease related proteins. For example, a broad-spectrum cathepsin-C specific probe was used to show that dipeptidylpetidase1 (DPAP1)[29] plays a significant role in malarial infection and is potentially valuable drug target. In other examples, a broad-spectrum probe was used to link several serine hydrolases including retinoblastoma-binding protein 9 (RBBP9)[43], KIAA1363 [44], and monoacylglycerol lipase (MAGL) [45] to cancer progression. Furthermore, the broad-spectrum ABPs used for target validation are also useful to set up class-wide enzyme assays to identify new inhibitors or to test existing library of small molecules in phenotypic screening [28,46–48].
Label-free techniques
Label-free techniques have the advantage in that they do not require any chemical modification of an active compound, which can greatly facilitate the target identification process. This relatively new type of target identification strategy relies on changes in thermodynamic stability as the result of a protein-drug interaction. These methods are based on the concept that a protein has conformational flexibility in solution, making it more susceptible to proteolysis, however, once it binds to a small molecule, the overall complex will be more resistant to proteolysis [49]. One such label-free technique called DARTS (Drug Affinity Responsive Target Stability) was used to successfully identify cellular targets of Rapamycin, FK506, didemnin B, and resveratrol [50]. Similarly, a ‘pulse proteolysis’ technique demonstrated that ligand bound proteins are more stable upon protein denaturation and proteolysis compared to the samples without a ligand (Figure 3A) [51].
Figure 3.
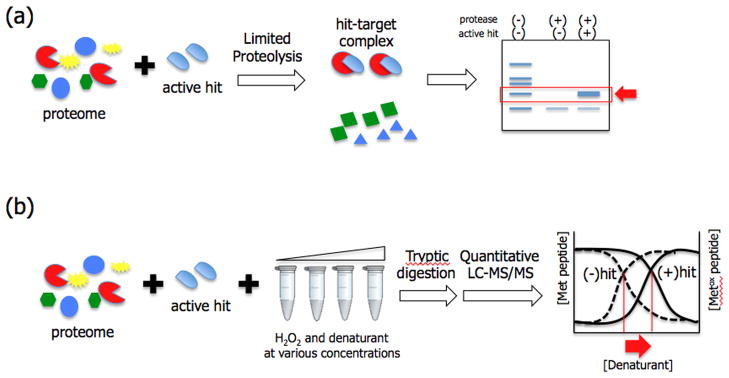
Label-free techniques for target deconvolution. (A) Limited proteolysis techniques such as DARTS and pulse proteolysis utilize stability of protein-ligand complex under proteolytic condition. Ligand bound proteins are more resistant to proteolysis in the presence of denaturant (pulse-proteolysis) or without denaturant (DARTS), and non-binding proteins are hydrolyzed to small peptides and amino acids. All proteolysis resistant proteins can be analyzed by SDS-PAGE and identified by mass spectrometry; (B) SPROX technique is based on a similar principle, however, it exploits protein-ligand interaction under oxidative conditions in various concentrations of denaturant. Ligand bound proteins are more resistant to oxidant, thus require higher amounts of denaturant to generate the same degree of oxidation compared to non-binders. These results can be plotted and protein-ligand interactions result in a right shift of the plot.
Another technique termed SPROX (Stability of Proteins from Rates of Oxidation) is a quantitative mass spectrometry-based approach that, like DARTS, utilizes thermodynamic stability of ligand-protein complexes but focuses on changes in stability under oxidative conditions (Figure 3B)[52]. This method utilizes an oxidizing agent (H2O2) in the presence of increasing concentration of a chemical denaturant to oxidize methionine residues in target proteins. After quenching the oxidation reaction, the amount of non-oxidized and oxidized methionine-containing peptides in each sample are quantified and plotted against concentrations of denaturant. Ligand bound proteins will show bigger shifts toward high-concentrations of denaturant compared to non-binders. Two cyclosporine A binding proteins were identified from a yeast proteome as a proof-of-principle study [53], and previously unknown target proteins of resveratrol were later identified [54]. Unlike DARTS and pulse proteolysis, which requires gel electrophoresis to separate proteolysis-resistant complexes, SPROX measures concentrations of trypsinized peptides from a complex mixture by using a tandem LC-MS/MS technique such as MudPIT. Hence, SPROX has the potential to be used for more direct global analysis of drug-protein interactions.
Expression cloning techniques
Expression cloning techniques utilize a library of cDNAs inserted into cloning vectors to express a library of proteins. A small molecule-protein interaction can be detected by adding a tagged small molecule followed by affinity purification. In a sense, expression cloning techniques are similar to typical affinity purification because they also require chemical modification to attach a tag. However, when the target of interest is low abundance or is unstable, expression cloning can be an excellent alternative.
One method to express a large-scale library of proteins is phage display (Figure 4A). Phage display is an affinity selection technique initially developed to identify antigen-antibody interactions and protein-protein interactions. A library of DNA sequences can be fused to a gene encoding a phage coat protein. Hence, the phage will display one unique protein on its surface per phage particle. Phage particles that bind to a small molecule target with high affinity can be isolated. The isolated phage can be used for subsequent rounds of selection that can lead to further enrichment. Although, traditional phage display techniques have identified molecular targets of many natural and synthetic ligands [55–57], more improved techniques have facilitated the process. In a recent example, Van Dorst et al. demonstrated that lytic (T7) cDNA phage display can be used as a fast, cost effective alternative to conventional filamentous phage (M13) display, and identified molecular targets of 17β estradiol [58].
Figure 4.

Expression cloning techniques. Proteins can be expressed using cloning vectors containing cDNA library, and these proteins exposed to small molecules for affinity selection. (A) Phage display. Small molecule captured phage particles can be selectively eluted and transfected into bacterial cells for further amplification and enrichment; (B) mRNA display. mRNA display utilizes an in vitro translation system to generate a library of mRNA-protein fusions, and this newly generated library can be exposed to an immobilized small molecule. After affinity selection, the cDNA of the captured proteins can be amplified by PCR and used to identify the target or for a next round of selection for further enrichment; (C) Yeast three hybrid screen. Screen constructs consists of a bait domain containing a DNA binding domain fused to a protein of interest, and a prey domain containing a transcriptional activator for a reporter gene linked to a library of proteins encoded by a cDNA library. When the bait and prey domains interact through the small molecule dimerizer, transcription of a reporter gene is activated.
As an alternative to phage, mRNA display was introduced as a method in which proteins could be expressed as a fusion to their corresponding mRNAs (Figure 4B) [59]. This allows direct affinity screening and rapid identification of the target protein by sequencing of the corresponding cDNA tag. However, since the initial proof-of-concept experiment a decade ago [60], few actual examples of target identification have been reported. Most examples of mRNA display applications have been focused on protein-protein interaction and lead discovery.
Another alternative display method is the yeast three-hybrid screen (Figure 4C). This is an adaptation of the commonly used two-hybrid system for identification of protein-protein interactions. The three-hybrid system makes use of the same elements but includes a ‘chemical dimerizer’ that is used to link the small molecule of interest to the bait protein so that interaction with the prey domain can be measured. Although the idea was introduced nearly two decades ago [61], there have been only a few reports using three-hybrid system for target identification of small molecules [62]. Recently, Chidley et al. incorporated a SNAP-tag to covalently label drug molecules inside yeast and were able to identify previously unknown targets for clinically approved drugs [63].
In Silico approach
Computer aided drug design is a major workhorse of target-based drug discovery. Based on docking studies and virtual screening, drug candidates with optimal potency and selectivity can be predicted. With the recent renaissance of phenotype-based drug discovery, these in silico technologies have found an important new role in the process of target prediction. Over the last two decades, extensive information regarding activity, structures and targets of small molecule libraries has been deposited into public databases such as ChEMBL [64], DrugBank [65] and ChemBank [66]. There are also public web services such as TarFisDock [67] and SEA (Similarity Ensemble Approach) for target prediction of small molecules [68,69]. Using these tools, targets of active compounds can be predicted based on similarities in structure between an active hit and well-characterized drugs in these databases. This computer aided target prediction has been widely used to identify new targets of known drugs [69,70], to predict the targets of active hits from a library screening [71–73], and to investigate the mechanism of action of hits discovered from phenotypic screens [74,75]. For example, Lounkine et al. used a public database ChEMBL, and performed a large-scale ‘ligand-based similarity search’ to predict target proteins, which lead to the identification of 73 unintended off targets of 656 marketed drugs [69]. Additionally, recent advances in high-content screening platforms using automated imaging systems enable the establishment of phenotypic-SAR thus improving confidence levels in target prediction and providing important information regarding drug mode of action [76,77].
Conclusion and perspective
Small molecules have long been used as tools to manipulate biological systems. In addition to acting as therapeutic agents where the primary focus is on ultimate effects rendered by treatment with the compounds, small molecules also have the potential to function as reagents that allow detailed studies of the functional roles of diverse target proteins. History has proven that it is relatively simple to find small molecules that have a given biological effect on a cell or organism, however, the process by which the mechanism of action of the compound can be identified on a molecular level remains a major challenge. This is largely due to the fact that most small molecules do not simply bind to one target. Therefore finding ways to deconvolute the list of possible players is of utmost importance. In this review, we have outlined some of the more recent advances in methods that can be used to link specific small molecules identified in a phenotypic screen to a valid target. You may have noticed that the list of recent examples where a given technique has been used to identify novel targets is somewhat short, especially given the rapid growth in the use of phenotypic screening methods over the past decade. We believe this is due to significant challenges that still exist in globally mapping out small molecule-target interactions. However, we also believe that rapid advances in analytical methods such as mass spectrometry coupled with advances in genetic methods and genome-wide sequencing are likely to have a big impact on our ability to more rapidly and efficiently identify targets and furthermore to provide direct causal links between a hit binding to its target and a phenotypic outcome. We therefore feel the future is bright for phenotypic screening and future reviews on this topic are likely to have increasingly more concrete examples of how the techniques presented here have been applied in basic biology and drug discovery research.
HIGHLIGHTS.
Discussion of the concept of target deconvolution
Discussion of why new technology is helping to advance target deconvolution
Recent examples of various methods used to find targets of small molecule hits
Discussion of the future for phenotypic screening methods
Acknowledgments
This work was funding by NIH grants R01 EB005011 and R01 AI078947 (to MB). J.L. was supported by the Sungshin Women’s University Research Grant of 2012-2-21-003/1.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 2.Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- 3.Mestres J, Gregori-Puigjane E, Valverde S, Sole RV. The topology of drug-target interaction networks: implicit dependence on drug properties and target families. Mol Biosyst. 2009;5:1051–1057. doi: 10.1039/b905821b. [DOI] [PubMed] [Google Scholar]
- 4.Roemer T, Davies J, Giaever G, Nislow C. Bugs, drugs and chemical genomics. Nat Chem Biol. 2012;8:46–56. doi: 10.1038/nchembio.744. [DOI] [PubMed] [Google Scholar]
- 5.Feng Y, Mitchison TJ, Bender A, Young DW, Tallarico JA. Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat Rev Drug Discov. 2009;8:567–578. doi: 10.1038/nrd2876. [DOI] [PubMed] [Google Scholar]
- 6.Schirle M, Bantscheff M, Kuster B. Mass spectrometry-based proteomics in preclinical drug discovery. Chem Biol. 2012;19:72–84. doi: 10.1016/j.chembiol.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 8.Bantscheff M, Scholten A, Heck AJ. Revealing promiscuous drug-target interactions by chemical proteomics. Drug Discov Today. 2009;14:1021–1029. doi: 10.1016/j.drudis.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Raida M. Drug target deconvolution by chemical proteomics. Curr Opin Chem Biol. 2011;15:570–575. doi: 10.1016/j.cbpa.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 10.Sato S, Murata A, Shirakawa T, Uesugi M. Biochemical target isolation for novices: affinity-based strategies. Chem Biol. 2010;17:616–623. doi: 10.1016/j.chembiol.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 11.Mamidyala SK, Finn MG. In situ click chemistry: probing the binding landscapes of biological molecules. Chem Soc Rev. 2010;39:1252–1261. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- 12.Shi H, Cheng X, Sze SK, Yao SQ. Proteome profiling reveals potential cellular targets of staurosporine using a clickable cell-permeable probe. Chem Commun (Camb) 2011;47:11306–11308. doi: 10.1039/c1cc14824a. [DOI] [PubMed] [Google Scholar]
- 13.Shi H, Zhang CJ, Chen GY, Yao SQ. Cell-based proteome profiling of potential dasatinib targets by use of affinity-based probes. J Am Chem Soc. 2012;134:3001–3014. doi: 10.1021/ja208518u. [DOI] [PubMed] [Google Scholar]
- •14.He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, et al. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature. 2010;467:95–98. doi: 10.1038/nature09325. This paper demonstrates how a chemical affinity probe can be used to identify off-target proteins of a known drug and to investigate its molecular mechanism. In this paper, a kinase inhibitor, imatinib, was modified with a photoreactive group and I125, and gamma-secretase activating protein was identified as an additional target protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tapper EB, Knowles D, Heffron T, Lawrence EC, Csete M. Portopulmonary hypertension: imatinib as a novel treatment and the Emory experience with this condition. Transplant Proc. 2009;41:1969–1971. doi: 10.1016/j.transproceed.2009.02.100. [DOI] [PubMed] [Google Scholar]
- 16.Netzer WJ, Dou F, Cai D, Veach D, Jean S, Li Y, Bornmann WG, Clarkson B, Xu H, Greengard P. Gleevec inhibits beta-amyloid production but not Notch cleavage. Proc Natl Acad Sci U S A. 2003;100:12444–12449. doi: 10.1073/pnas.1534745100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar NS, Young RN. Design and synthesis of an all-in-one 3-(1,1-difluoroprop-2-ynyl)-3H-diazirin-3-yl functional group for photo-affinity labeling. Bioorg Med Chem. 2009;17:5388–5395. doi: 10.1016/j.bmc.2009.06.048. [DOI] [PubMed] [Google Scholar]
- ••18.Cisar JS, Cravatt BF. Fully functionalized small-molecule probes for integrated phenotypic screening and target identification. J Am Chem Soc. 2012;134:10385–10388. doi: 10.1021/ja304213w. This paper demonstrated an integrated approach for phenotypic screening and target identification by using a multifunctional chemical library. The most interesting aspect in this work is that each molecule labeled markedly different proteins in intact cells and cell lysates indicating that these small subsets of diversity elements were sufficient to investigate diverse protein-small molecule interactions. Moreover, introducing various functional groups did not significantly affect reactivity of photo-cross linking and click chemistry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •19.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–1350. doi: 10.1126/science.1177319. This work described the molecular target of thalidomide and its mechanism of inducing birth-defects. By using high-performance affinity beads, the authors were able to reduce false-positives and isolate promising target proteins. Further competative experiments with thalidomide confirmed cereblon as its taret protein. [DOI] [PubMed] [Google Scholar]
- 20.Hu L, Iliuk A, Galan J, Hans M, Tao WA. Identification of drug targets in vitro and in living cells by soluble-nanopolymer-based proteomics. Angew Chem Int Ed Engl. 2011;50:4133–4136. doi: 10.1002/anie.201006459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 22.Sadaghiani AM, Verhelst SH, Bogyo M. Tagging and detection strategies for activity-based proteomics. Curr Opin Chem Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 23.Deu E, Verdoes M, Bogyo M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nat Struct Mol Biol. 2012;19:9–16. doi: 10.1038/nsmb.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nomura DK, Dix MM, Cravatt BF. Activity-based protein profiling for biochemical pathway discovery in cancer. Nat Rev Cancer. 2010;10:630–638. doi: 10.1038/nrc2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puri AW, Bogyo M. Using small molecules to dissect mechanisms of microbial pathogenesis. ACS Chem Biol. 2009;4:603–616. doi: 10.1021/cb9001409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Child MA, Bogyo M. Proteases as regulators of pathogenesis: examples from the Apicomplexa. Biochim Biophys Acta. 2012;1824:177–185. doi: 10.1016/j.bbapap.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long JZ, Cravatt BF. The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem Rev. 2011;111:6022–6063. doi: 10.1021/cr200075y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puri AW, Lupardus PJ, Deu E, Albrow VE, Garcia KC, Bogyo M, Shen A. Rational design of inhibitors and activity-based probes targeting Clostridium difficile virulence factor TcdB. Chem Biol. 2010;17:1201–1211. doi: 10.1016/j.chembiol.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deu E, Leyva MJ, Albrow VE, Rice MJ, Ellman JA, Bogyo M. Functional studies of Plasmodium falciparum dipeptidyl aminopeptidase I using small molecule inhibitors and active site probes. Chem Biol. 2010;17:808–819. doi: 10.1016/j.chembiol.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arastu-Kapur S, Ponder EL, Fonovic UP, Yeoh S, Yuan F, Fonovic M, Grainger M, Phillips CI, Powers JC, Bogyo M. Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum. Nat Chem Biol. 2008;4:203–213. doi: 10.1038/nchembio.70. [DOI] [PubMed] [Google Scholar]
- ••31.Hall CI, Reese ML, Weerapana E, Child MA, Bowyer PW, Albrow VE, Haraldsen JD, Phillips MR, Sandoval ED, Ward GE, et al. Chemical genetic screen identifies Toxoplasma DJ-1 as a regulator of parasite secretion, attachment, and invasion. Proc Natl Acad Sci U S A. 2011;108:10568–10573. doi: 10.1073/pnas.1105622108. This is a recent example of how activity-based probes can streamline the whole processes of phenotypic screening, target identification and validation in a disease model. The authors used a library of ABPs and covalent inhibitors of cysteine proteases and found active compounds regulating T. gondii invasion. Active hits were converted to ABPs to further investigate molecular mechanism of host invasion process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crump CJ, am Ende CW, Ballard TE, Pozdnyakov N, Pettersson M, Chau DM, Bales KR, Li YM, Johnson DS. Development of clickable active site-directed photoaffinity probes for gamma-secretase. Bioorg Med Chem Lett. 2012;22:2997–3000. doi: 10.1016/j.bmcl.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sieber SA, Niessen S, Hoover HS, Cravatt BF. Proteomic profiling of metalloprotease activities with cocktails of active-site probes. Nat Chem Biol. 2006;2:274–281. doi: 10.1038/nchembio781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geurink PP, Florea BI, Van der Marel GA, Kessler BM, Overkleeft HS. Probing the proteasome cavity in three steps: bio-orthogonal photo-reactive suicide substrates. Chem Commun (Camb) 2010;46:9052–9054. doi: 10.1039/c0cc03322g. [DOI] [PubMed] [Google Scholar]
- 35.Barglow KT, Saikatendu KS, Bracey MH, Huey R, Morris GM, Olson AJ, Stevens RC, Cravatt BF. Functional proteomic and structural insights into molecular recognition in the nitrilase family enzymes. Biochemistry. 2008;47:13514–13523. doi: 10.1021/bi801786y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pace NJ, Pimental DR, Weerapana E. An Inhibitor of Glutathione S-Transferase Omega 1 that Selectively Targets Apoptotic Cells. Angew Chem Int Ed Engl. 2012;51:8365–8368. doi: 10.1002/anie.201203730. [DOI] [PubMed] [Google Scholar]
- 37.Wright AT, Song JD, Cravatt BF. A suite of activity-based probes for human cytochrome P450 enzymes. J Am Chem Soc. 2009;131:10692–10700. doi: 10.1021/ja9037609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saario SM, McKinney MK, Speers AE, Wang C, Cravatt BF. Clickable, photoreactive inhibitors to probe the active site microenvironment of fatty acid amide hydrolase. Chem Sci. 2012;3:77–83. doi: 10.1039/C1SC00336D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van der Linden WA, Li N, Hoogendoorn S, Ruben M, Verdoes M, Guo J, Boons GJ, van der Marel GA, Florea BI, Overkleeft HS. Two-step bioorthogonal activity-based proteasome profiling using copper-free click reagents: a comparative study. Bioorg Med Chem. 2012;20:662–666. doi: 10.1016/j.bmc.2011.06.037. [DOI] [PubMed] [Google Scholar]
- 40.Kulkarni SS, Hu X, Doi K, Wang HG, Manetsch R. Screening of protein-protein interaction modulators via sulfo-click kinetic target-guided synthesis. ACS Chem Biol. 2011;6:724–732. doi: 10.1021/cb200085q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berry AF, Heal WP, Tarafder AK, Tolmachova T, Baron RA, Seabra MC, Tate EW. Rapid multilabel detection of geranylgeranylated proteins by using bioorthogonal ligation chemistry. Chembiochem. 2010;11:771–773. doi: 10.1002/cbic.201000087. [DOI] [PubMed] [Google Scholar]
- 42.Willems LI, Verdoes M, Florea BI, van der Marel GA, Overkleeft HS. Two-step labeling of endogenous enzymatic activities by Diels-Alder ligation. Chembiochem. 2010;11:1769–1781. doi: 10.1002/cbic.201000280. [DOI] [PubMed] [Google Scholar]
- ••43.Shields DJ, Niessen S, Murphy EA, Mielgo A, Desgrosellier JS, Lau SK, Barnes LA, Lesperance J, Bouvet M, Tarin D, et al. RBBP9: a tumor-associated serine hydrolase activity required for pancreatic neoplasia. Proc Natl Acad Sci U S A. 2010;107:2189–2194. doi: 10.1073/pnas.0911646107. This work shows that a broad spectrum ABP can be used to identify disease related proteins. In this work, the authors used a broad spectrum serine hydrolase inhibitor to study activity levels of tumor-associated proteases and discovered that RBB9 is particulary elevated in pancreatic neoplasia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiang KP, Niessen S, Saghatelian A, Cravatt BF. An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem Biol. 2006;13:1041–1050. doi: 10.1016/j.chembiol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 45.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140:49–61. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 2009;27:387–394. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deu E, Yang Z, Wang F, Klemba M, Bogyo M. Use of activity-based probes to develop high throughput screening assays that can be performed in complex cell extracts. PLoS One. 2010;5:e11985. doi: 10.1371/journal.pone.0011985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •48.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, et al. Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc Natl Acad Sci U S A. 2011;108:6811–6816. doi: 10.1073/pnas.1015248108. This paper demonstrated that ABPs can be used for high-throughput screening in the lead optimization process. In this work, the authors screened the 300,000+ member NIH library via fluorescence polarization method using a broad spectrum ABP, and identified the aza-β-lactams as covalent inhibitors of PME-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontana A, de Laureto PP, Spolaore B, Frare E, Picotti P, Zambonin M. Probing protein structure by limited proteolysis. Acta Biochim Pol. 2004;51:299–321. [PubMed] [Google Scholar]
- 50.Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, Wang J, Wu RP, Gomez F, Loo JA, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A. 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chang Y, Schlebach JP, Verheul RA, Park C. Simplified proteomics approach to discover protein-ligand interactions. Protein Sci. 2012;21:1280–1287. doi: 10.1002/pro.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.West GM, Tang L, Fitzgerald MC. Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Anal Chem. 2008;80:4175–4185. doi: 10.1021/ac702610a. [DOI] [PubMed] [Google Scholar]
- 53.West GM, Tucker CL, Xu T, Park SK, Han X, Yates JR, 3rd, Fitzgerald MC. Quantitative proteomics approach for identifying protein-drug interactions in complex mixtures using protein stability measurements. Proc Natl Acad Sci U S A. 2010;107:9078–9082. doi: 10.1073/pnas.1000148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •54.Dearmond PD, Xu Y, Strickland EC, Daniels KG, Fitzgerald MC. Thermodynamic analysis of protein-ligand interactions in complex biological mixtures using a shotgun proteomics approach. J Proteome Res. 2011;10:4948–4958. doi: 10.1021/pr200403c. This is a proof-of-principle study to demonstrate a label-free strategy based on shotgun proteomics protocols and thermodynamic stability of proteins under oxidation conditions. Modified protocols with enrichment steps and isobaric mass tags improved peptides and protein coverage up to 2-fold compared to previously reported methods [53] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Dorst B, Mehta J, Rouah-Martin E, Somers V, De Coen W, Blust R, Robbens J. cDNA phage display as a novel tool to screen for cellular targets of chemical compounds. Toxicol In Vitro. 2010;24:1435–1440. doi: 10.1016/j.tiv.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 56.Jung HJ, Shim JS, Lee J, Song YM, Park KC, Choi SH, Kim ND, Yoon JH, Mungai PT, Schumacker PT, et al. Terpestacin inhibits tumor angiogenesis by targeting UQCRB of mitochondrial complex III and suppressing hypoxia-induced reactive oxygen species production and cellular oxygen sensing. J Biol Chem. 2010;285:11584–11595. doi: 10.1074/jbc.M109.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takakusagi Y, Takakusagi K, Sugawara F, Sakaguchi K. Use of phage display technology for the determination of the targets for small-molecule therapeutics. Expert Opin Drug Discov. 2010;5:361–389. doi: 10.1517/17460441003653155. [DOI] [PubMed] [Google Scholar]
- 58.Van Dorst B, Mehta J, Rouah-Martin E, De Coen W, Blust R, Robbens J. The identification of cellular targets of 17beta estradiol using a lytic (T7) cDNA phage display approach. Toxicol In Vitro. 2011;25:388–393. doi: 10.1016/j.tiv.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 59.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McPherson M, Yang Y, Hammond PW, Kreider BL. Drug receptor identification from multiple tissues using cellular-derived mRNA display libraries. Chem Biol. 2002;9:691–698. doi: 10.1016/s1074-5521(02)00148-5. [DOI] [PubMed] [Google Scholar]
- 61.Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc Natl Acad Sci U S A. 1996;93:12817–12821. doi: 10.1073/pnas.93.23.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becker F, Murthi K, Smith C, Come J, Costa-Roldan N, Kaufmann C, Hanke U, Degenhart C, Baumann S, Wallner W, et al. A three-hybrid approach to scanning the proteome for targets of small molecule kinase inhibitors. Chem Biol. 2004;11:211–223. doi: 10.1016/j.chembiol.2004.02.001. [DOI] [PubMed] [Google Scholar]
- ••63.Chidley C, Haruki H, Pedersen MG, Muller E, Johnsson K. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat Chem Biol. 2011;7:375–383. doi: 10.1038/nchembio.557. A new exciting improvement in yeast three hybrid method for target identification. In this work, the authors incorporated a snap tag to solve two major limidations - binding affinity and membrane permeability of drug molecules. Derivatives of five clinically available drugs were screened against a human cDNA library to identify a new protein target of the drug sulfasalazine. [DOI] [PubMed] [Google Scholar]
- 64.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668–672. doi: 10.1093/nar/gkj067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Seiler KP, George GA, Happ MP, Bodycombe NE, Carrinski HA, Norton S, Brudz S, Sullivan JP, Muhlich J, Serrano M, et al. ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008;36:D351–359. doi: 10.1093/nar/gkm843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koutsoukas A, Simms B, Kirchmair J, Bond PJ, Whitmore AV, Zimmer S, Young MP, Jenkins JL, Glick M, Glen RC, et al. From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics. 2011;74:2554–2574. doi: 10.1016/j.jprot.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 68.Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, Jensen NH, Kuijer MB, Matos RC, Tran TB, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••69.Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Cote S, et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature. 2012;486:361–367. doi: 10.1038/nature11159. A large-scale analysis of chemical databases and in silico prediction of binding affinity yielded 73 new protein targets that have been associated with side effects of 656 known drugs. The authors also predicted new drug-target associations that can help understand previously unknown mechanisms of off-target effects. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •70.Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, Uren A, Brown ML, Byers SW. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012;55:6832–6848. doi: 10.1021/jm300576q. A reverse docking study of 3,671 U.S. approved drugs against human proteins showed that mebendazole, an anti-hook worm medication, inhibited VEGFR2 activity, and celecoxib, an anti-inflammatory drug, bound to a putative target involved in arthritis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Areias FM, Brea J, Gregori-Puigjane E, Zaki ME, Carvalho MA, Dominguez E, Gutierrez-de-Teran H, Proenca MF, Loza MI, Mestres J. In silico directed chemical probing of the adenosine receptor family. Bioorg Med Chem. 2010;18:3043–3052. doi: 10.1016/j.bmc.2010.03.048. [DOI] [PubMed] [Google Scholar]
- 72.Flachner B, Lorincz Z, Carotti A, Nicolotti O, Kuchipudi P, Remez N, Sanz F, Tovari J, Szabo MJ, Bertok B, et al. A chemocentric approach to the identification of cancer targets. PLoS One. 2012;7:e35582. doi: 10.1371/journal.pone.0035582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Laggner C, Kokel D, Setola V, Tolia A, Lin H, Irwin JJ, Keiser MJ, Cheung CY, Minor DL, Jr, Roth BL, et al. Chemical informatics and target identification in a zebrafish phenotypic screen. Nat Chem Biol. 2012;8:144–146. doi: 10.1038/nchembio.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Plouffe D, Brinker A, McNamara C, Henson K, Kato N, Kuhen K, Nagle A, Adrian F, Matzen JT, Anderson P, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci U S A. 2008;105:9059–9064. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Young DW, Bender A, Hoyt J, McWhinnie E, Chirn GW, Tao CY, Tallarico JA, Labow M, Jenkins JL, Mitchison TJ, et al. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat Chem Biol. 2008;4:59–68. doi: 10.1038/nchembio.2007.53. [DOI] [PubMed] [Google Scholar]
- 76.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lounkine E, Kutchukian P, Petrone P, Davies JW, Glick M. Chemotography for multi-target SAR analysis in the context of biological pathways. Bioorg Med Chem. 2012;20:5416–5427. doi: 10.1016/j.bmc.2012.02.034. [DOI] [PubMed] [Google Scholar]