

Abstract
In this report, we describe a male subject who presents with a complex phenotype of myopia associated with cone dysfunction and a protan vision deficiency. Retinal imaging demonstrates extensive cone disruption, including the presence of non-waveguiding cones, an overall thinning of the retina, and an irregular mottled appearance of the hyper reflective band associated with the inner segment ellipsoid portion of the photoreceptor. Mutation screening revealed a novel p.Glu41Lys missense mutation in a hybrid L/M opsin gene. Spectral analysis shows that the mutant opsin fails to form a pigment in vitro and fails to be trafficked to the cell membrane in transfected Neuro2a cells. Extensive sequence and quantitative PCR analysis identifies this mutant gene as the only gene present in the affected subject’s L/M opsin gene array, yet the presence of protanopia indicates that the mutant opsin must retain some activity in vivo. To account for this apparent contradiction, we propose that a limited amount of functional pigment is formed within the normal cellular environment of the intact photoreceptor, and that this requires the presence of chaperone proteins that promote stability and normal folding of the mutant protein.
Keywords: Colour vision, visual pigments, dichromacy, opsin mutation, cone dysfunction, retinal imaging
1. Introduction
Normal colour vision in humans is described as trichromatic as it arises from the presence of three classes of cone photoreceptors that are defined as short-, middle-, and long-wavelength-sensitive (S, M, and L), with overlapping sensitivities that peak in the violet, green and yellow regions of the spectrum, respectively. Inherited defects in red-green colour perception are common, affecting around one in 12 white males, and arise most often either from the absence of the L or M gene and associated pigment and cone class, or from the presence of hybrid L/M genes that specify pigments with altered spectral properties (Deeb et al., 1992, Jagla et al., 2002, Nathans et al., 1986, Neitz et al., 2004, Ueyama et al., 2003). In both cases, the underlying cause is an alteration in the genomic structure of the L and M genes, which lie in a head-to-tail array on the X-chromosome, at Xq28 (Nathans, Thomas & Hogness, 1986). These genes share 98% nucleotide identity and show a high incidence of non-homologous pairing and recombination. Depending on the precise site of the exchange, this can generate gene loss (or gene gain) and the production of L/M gene hybrids, leading either to the complete absence of the L or M pigment (dichromats), or the production of hybrid pigments with altered spectral properties (anomalous trichromats). Normal visual acuity is preserved, or even enhanced (Jägle et al., 2006), in such individuals, as each cone will express a functional pigment. A second form of dichromacy arises from missense mutations in the L and M opsins (Neitz et al., 2004, Ueyama et al., 2004, Ueyama et al., 2003, Winderickx et al., 1992). In such cases, visual acuity may also be affected and the disorder has been associated with myopia (Michaelides et al., 2005).
One such example of dichromacy associated with myopia and reduced visual acuity was reported by Michaelides et al. (2005). In this case, the affected subject carried a mutation in both the L and M opsin genes that encoded a p.Cys203Arg substitution which is known to result in the loss of an important disulfide bond required for the correct folding of the opsin protein in the production of a functional pigment (Karnik et al., 1988, Kazmi, Sakmar & Ostrer, 1997). A more recent study by Carroll et al. (2009) showed a significant reduction in cone density, disruption in the cone mosaic, and thinning of the outer nuclear layer of the retina in two protanope subjects with the p.Cys203Arg substitution. These data are consistent with the degeneration of those cones that expressed the Cys203Arg pigment.
In this report, we describe a male subject who presents with a complex phenotype of myopia associated with cone dysfunction and a colour vision deficiency. Molecular evidence has identified a novel missense opsin gene mutation that would appear to be directly responsible for the cone and colour vision aspects of this disorder. We sought to assess the consequence of this mutation on the structure of the cone photoreceptors using high-resolution retinal imaging tools.
2. Materials and Methods
2.1 Origin and isolation of DNA
Following informed consent, blood samples were collected for the isolation of genomic DNA for subsequent genetic analysis, using an extraction kit (Nucleon Biosciences, Coatbridge, UK). The study was approved by the local research ethics committee and all investigations were conducted in accordance with the principles of the Declaration of Helsinki (World Medical Association, 1997).
2.2 Clinical assessment
Full medical and ophthalmic histories were taken prior to performing a complete ophthalmological examination. Electroretinography (ERG) was performed on the affected individual that conformed to the International Society for Clinical Electrophysiology of Vision (ISCEV) standard (Marmor & Zrenner, 1993, Marmor & Zrenner, 1995), but with a modified protocol using skin electrodes when tested in infancy, and corneal electrodes when re-tested in childhood. Colour vision testing included the use of the American Optical Company, Hardy, Rand and Rittler (HRR) plates, SPP2 plates for acquired colour deficiency, Farnsworth–Munsell (FM) 100-hue test, Farnsworth D-15, the Mollon-Reffin (MR) minimal test (Mollon, Astell & Reffin, 1991), the Cambridge Colour Test (Regan, Reffin & Mollon, 1994), and anomaloscopy. The FM 100-hue, Farnsworth D-15, and the MR test were all performed under CIE Standard Illuminant C from a MacBeth Easel lamp. The MR minimal test is a saturation discrimination-type test (Mollon, Astell & Reffin, 1991).
2.3 Spectral Domain Optical Coherence Tomography (OCT)
Volumetric images of the macula were obtained using a Zeiss Cirrus™ HD-OCT (Carl Zeiss Meditec, Dublin, California, USA). Volumes were nominally 6 mm × 6 mm and consisted of 128 B-scans (512 A-scans/B-scan). Retinal thickness was calculated using the built in Macular Analysis software on the Cirrus (software version 6.0), which automatically determines the difference between the inner limiting membrane and retinal pigment epithelium boundaries. High-density line scans (1000 A-scans/B-scan, 100 repeated B scans) were acquired through the foveal centre using the Bioptigen SD-OCT system (Research Triangle Park, North Carolina, USA). Line scans were registered and averaged to reduce speckle noise in the image as previously described (Tanna et al., 2010).
2.4 Adaptive Optics Retinal Imaging
Images of the photoreceptor mosaic were obtained with an adaptive optics (AO) fundus camera using a previously described protocol (Carroll et al., 2009, Rha et al., 2009). In brief, in a continuous closed-loop fashion, the monochromatic aberrations of the eye were measured over a 6.4mm pupil with a Shack-Hartmann wavefront sensor and corrected for with a 52-channel deformable mirror (Imagine Eyes, Orsay, France). Once a wavefront correction was obtained, a retinal image was acquired by illuminating the retina with a 1.8-degree diameter, 500ms flash of light controlled by a mechanical shutter. A back-illuminated, scientific-grade, 12-bit charge-coupled device (Cam1M100-SFT, Sarnoff Imaging) captured images of the retina. During each 500ms shutter controlled exposure, continuously triggered images of the retina were collected at a frame rate of 167 Hz, giving a duration of 6ms for individual exposures. Images were collected at different locations across the central fovea. After background correction, all images were registered and averaged using a custom Matlab (The Mathworks, Inc., Natick, Massachusetts, USA) program.
2.5 Amplification of the first and downstream genes of the L/M array
In order to amplify the L and M genes separately, the protocol from Oda et al. (2003) was followed using PCR primers (Table 1) specific to either the promoter region of the upstream L gene (primer FG) or the promoter region of the downstream M gene (primer DG) (Figure 1). In both cases, the same reverse primer (E6) was used. Amplifications were visualised on a 0.5% gel and the isolated products used as templates for subsequent PCR amplifications (using BIOTaq, Bioline, London, UK) of exons 2 to 5 using the primers listed in Table 1. The promoter was screened with primers PF2 and PR that hybridise to a region of both promoters that have an identical sequence. All PCR products were sequenced.
Table 1.
Primers used for screening the first and downstream genes and for qPCR analysis of the L and M genes.
| Primer | Sequence (5′ – 3′) |
|---|---|
| FG | GAGGCGAGGCTACGGAGT |
| DG | TTAGTCAGGCTGGTCGGGAACT |
| PF2 | GAGGAGGAGGTCTAAGTCCC |
| PR | GGCTATGGAAAGCCCTGTCCC |
| E6 | GCAGTGAAAGCCTCTGTGACT |
| L/M5F | TTTGCTGCTGCCAACCCT |
| L/M5R | TTGCTTACCTGCCGGTTCATAA |
| L probe | 6FAM-CCTGCCGGCCTACTTTGCCAAA |
| M probe | JOE-CCTGCCGGCCTTCTTTGCCAAA |
| 2F | GCACTGGTATCGACAGGCG |
| 2R | CAGTATATGGATGTGAGGC |
| 3F | CTCAGTCCGTGGAGCCCTGAATTC |
| 3R | ACATTGATAGACATTGCACGCTCA |
| 4F | GGTGACTCCACAGAATTGAT |
| 4R | CTGATTCTCATCGCTGGATCT |
| 5F | CTATGCCTGGGTCACCTGCCTC |
| 5R | CTTATCAGAGACATGATTCCAGGTCC |
Figure 1.
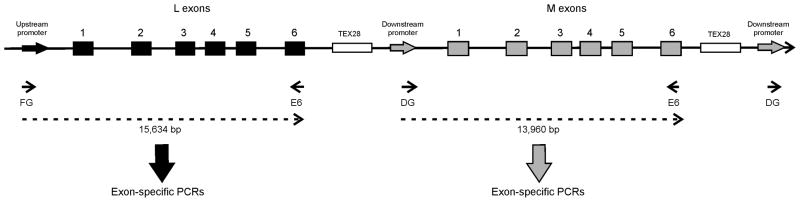
The L and M opsin genes are present in a tandem array on Xq28. The L opsin generally sits in the proximal position with multiple M genes downstream. The upstream promoter differs in sequence to the downstream promoters. Primer FG binds specifically to the upstream promoter whilst DG binds specifically to the downstream promoter. The reverse primer E6 binds to a common sequence in exon 6 of both L and M genes. The complete first gene in the array was amplified by use of primers FG and E6 and the amplicon generated used as template in subsequent exon-specific PCRs to determine the coding sequence. Any downstream genes present in the array were amplified together using primers DG and E6 and subsequent exon-specific PCRs identified whether the downstream genes were WT or L/M hybrids.
2.6 Ratio of L to M genes
The protocol described in Neitz & Neitz (2000) using a single primer set that hybridised to exon 5 of both the L and M opsin genes (L/M5F and L/M5R in Table 1) was followed. Probes specific for either the L or M exon 5 sequence were subsequently used to detect specific amplicons. The L probe was tagged at the 5′ end with 6FAM and the M probe with JOE fluorochrome dyes, respectively. Reactions were run in triplicate and repeated on three separate occasions. Each reaction contained 10ng of purified DNA, 140nM L probe, 250nM M probe, 900nM of both the forward and reverse primers, 12.5μl TaqMan Gene Expression Master Mix (Applied Biosystems, Warrington, UK). Reactions were made up to a final volume of 25μl with molecular biology grade water. The cycling schedule involved an initial incubation at 50°C for 2 minutes followed by denaturation at 95°C for 3 minutes. 3 cycles of 95°C for 30 s and 67°C for 1 minutes were then followed by 27 cycles of 95°C for 15 s and 67°C for 1 minute. The data were analysed using the comparative-Ct method.
2.7 Production and expression of variant opsins
Exons 1 to 6 of wild-type L and M opsin genes were amplified with primers designed specifically for use in SPLICE reactions (Davies, Carvalho & Hunt, 2007, 2009). The coding L and M exons were amplified separately from genomic DNA using primers listed in Table 2, and their sequences confirmed. Different combinations of exons were then mixed together as template in SPLICE reactions to create full-length L, M and hybrid coding sequences with and without the p.Glu41Lys mutation. The coding sequences designated as mutant contained the mutant exon 2 identified in the family presented here. Coding sequences were then ligated into a pMT4 mammalian expression vector, which introduced the C-terminal bovine rod opsin (1D4) tag at the 3′-end of the coding sequence. The resulting constructs were transfected into HEK293T cells using the GeneJuice Transfection Reagent (Merck, Nottingham, UK). Twelve 140 mm petri dishes were used for each opsin transfection. Expressed opsins were isolated by affinity immunochromatography and regenerated with 11-cis-retinal as described previously (Davies et al., 2009). Absorbance spectra were recorded in triplicate in the dark using a Shimadzu UV-visible spectrophotometer (UV-2550) (Shimadzu, Milton Keynes, UK) and bleached by exposure to broad-spectrum white fluorescent light for 1 h before a second spectrum was recorded. The peak sensitivity, λmax, for each pigment was determined by subtracting the bleached spectrum from the dark spectrum of each pigment to produce a difference spectrum. This was then fitted to a modified Govardovskii rhodopsin/vitamin-A1 template (Govardovskii et al., 2000) with a subtracted bleached retinal curve. Best-fit curves were obtained using the Solver add-in function in Microsoft Excel to determine the λmax by the method of least squares as previously described (Davies et al., 2007).
Table 2.
Primers designed for the generation of coding sequences for L and M opsin using the SPLICE method*.
| Primer | Sequence |
|---|---|
| HSLM1F | GCGCGAATTCCACCATGGCCCAGCAGTGGAGCCTC |
| HSLM1R | GCCTTCGTTGGGGCCTCTGGTGGAGTTGCT |
| HSML2F | CAACTCCACCAGAGGCCCCTTCGAAGGCCC |
| HSLM2R | AGAGACCTGTGATCCCACACAGGGAGACGG |
| HSLM3F | CGTCTCCCTGTGTGGGATCACAGGTCTCTG |
| HSLM3R | CCGTGGGGCCAGTACCTGCTCCAACCAAAG |
| HSLM4F | CTTTGGTTGGAGCAGGTACTGGCCCCACGG |
| HSLM4R | CTGCTGCTTTGCCACCGCTCGGATGGCCAG |
| HSLM5F | CTGGCCATCCGAGCGGTGGCAAAGCAGCAG |
| HSLM5R | ATGCAGTTTCGAAACTGCCGGTTCATAAAG |
| HSLMF6 | TTTATGAACCGGCAGTTTCGAAACTGCATC |
| HSLMF2mut | ACTCCACCAGAGGCCCCTTCAAAGGCCCGA |
| HSLMR1EX2mut | TCGGGCCTTTGAAGGGGCCTCTGGTGGAGT |
2.8 Immunocytochemistry
Full-length expression constructs (3 μg) containing both wild-type and mutant L and M opsins were used to transfect a neuroblastoma (Neuro2a) cell-line grown over glass slips in 5 cm wells, using using GeneJuice Transfection Reagent (Merck, Nottingham, UK). After 48 h, the medium was removed and the cells were washed in PBS. The cells were then fixed with 4% PFA for 10 min at 4°C. The PFA was removed and the cells were washed twice in PBS, before being permeabilised in PBS containing 0.05% Tween-20 for 10 min. The cells were washed a further three times in PBS, before blocking with 10% donkey serum in PBS for 1 h. The blocking solution was removed and the primary mouse anti-1D4 antibody, diluted 1 in 2,000 in 10% donkey serum in PBS, was added and left for 1 h. This was removed and the cells were washed five times in PBS, before adding the secondary Alexa Fluor® 488 donkey anti-mouse antibody (Life Technologies, Paisley, UK), diluted 1 in 500 in 10% donkey serum in PBS. This was incubated for 45 min, before washing the cells five times in PBS followed by a single wash with distilled water. The cells were immediately mounted with DAPI ProLong Gold anti-fade mounting reagent (Life Technologies, Paisley, UK). Images were taken on a LSM710 confocal microscope (Zeiss, Cambridge, UK).
3. Results
3.1 Colour vision phenotype
The patient was first seen at his local eye department at the age of 3 for assessment because of a family history of strabismus. During a follow-up examination, his vision remained reduced despite correction of his myopic refractive error. He was also noted to be mildly photophobic. He was first seen in our clinic at the age of 7 with a visual acuity of 6/24 in the right eye, 6/18 in the left eye, and was found to have a colour vision defect, myopia and astigmatism (OD: −4.75/−1.00 × 80, OS: −4.75/−1.00 × 120). Fundoscopy revealed myopic fundi with tilted optic discs, whilst ERG showed evidence of generalised cone dysfunction with normal rod responses. The patient’s maternal grandfather and a male cousin of his mother were both colour blind and suffered from reduced visual acuity from a young age, that was not fully corrected by spectacles. Neither was willing to undergo psychophysical testing.
At the age of 9, detailed psychophysical testing was undertaken. On the City University colour vision test and HRR plates, a red-green deficiency was detected with mainly a protan defect, and no tritan errors. On the MR test he showed excellent tritan discrimination. On the D-15, the subject showed a predominantly protan sequence of errors with each eye (Figure 2A). At the anomaloscope, he showed a protan spectral sensitivity (Figure 2B) and on Cambridge computerised colour testing (Regan, Reffin & Mollon, 1994), his discrimination ellipse was oriented in a protan direction (Figure 2C). His mother was found to have entirely normal colour vision. These behavioral data are consistent with the presence of functional S and M cone pigments of normal spectral sensitivity. His brightness settings on the anomaloscope were classically protan, and were not consistent with a diagnosis of achromatopsia or blue cone monochromatism. His mother was found to have normal trichromatic colour vision (data not shown).
Figure 2.
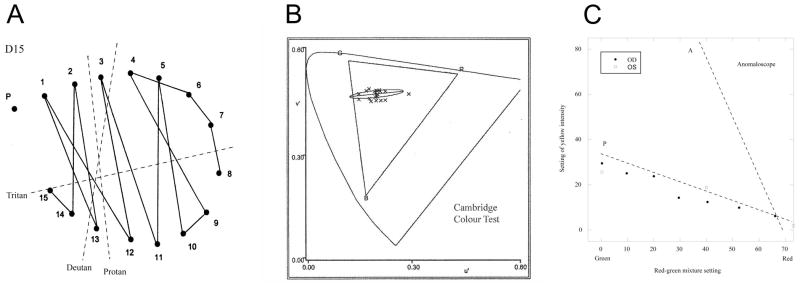
Psychophysical results for the propositus at age 9. (A) Arrangement of D15 tokens using right eye. The dotted lines indicate the direction of confusion expected for each type of dichromat. The propositus shows a predominantly protan pattern of confusions. (B) Discrimination ellipse on the Cambridge Colour Test. The data are plotted in the CIE u′v′ diagram and show how far the target has to differ in chromaticity from the grey background in order to be discriminated. The thresholds are extended in a classically protan direction, but discrimination in the orthogonal direction is good. The triangle marked RGB shows the gamut of chromaticities available on the monitor. (C) Brightness settings of the yellow field on the Nagel anomaloscope for right and left eyes. The propositus was able to make complete matches at all values of the red-green mixture. The dotted line marked P indicates typical settings of a protanope (from Pokorny, J. et al. 1979) and the dotted line marked A indicates typical settings for a rod achromat. The results show that the propositus has a photoreceptor with a spectral sensitivity similar to that of normal M cones.
The subject was re-examined at age 18 and had a visual acuity of 6/24 in each eye. FM-100 and HRR plate testing again showed a protan colour vision defect. Repeat electrophysiological assessment revealed no evidence of progression and there was again generalised cone dysfunction and normal rod responses.
3.2 Imaging of retina
To examine the structural basis for the observed cone dysfunction, high-resolution imaging of the retina in the affected subject was undertaken at age 18. Volumetric SD-OCT imaging of the macula revealed significant retinal thinning (Figure 3A). The high-resolution line scan revealed the presence of all major retinal layers, although the hyper-reflective band associated with the inner segment ellipsoid portion of the photoreceptor (ISe) had an irregular mottled appearance (Figure 3B).
Figure 3.

Glu41Lys disrupts cone photoreceptor structure. Images from 0.5deg (A,B) and 2deg (C,D) reveal a disrupted mosaic in the patient with the p.Glu41Lys mutation (B,D) compared to that of a normal control (A,C). Near the fovea in the p.Glu41Lys retina (B), there was a sparse population of strongly waveguiding cones. In the parafoveal images of the same patient (D), there were a reduced number of cones and those present had severely diminished waveguiding. Normal-appearing rods are visible in-between the dark cone structures. Scale bar is 50 μm.
Images of the photoreceptor mosaic at 0.5 deg and 2 deg eccentricity were obtained using an adaptive optics fundus camera. Near the fovea, the cone mosaic was disrupted compared to normal, with only a sparse population of strongly waveguiding cones remaining (Figure 4A,B). In the parafoveal image (2 deg), normal-appearing rods were observed dispersed amongst a reduced number of cones with severely diminished wave guiding compared to normal (Figure 4C,D).
Figure 4.

Altered retinal morphology in the affected subject assessed with OCT. (A) Topographical retinal thickness maps with early treatment diabetic retinopathy study (ETDRS) grids plotted below each thickness map showing moderate retinal thinning. (B) High resolution line scan through the fovea of the right eye, showing presence of all retinal layers, with a mottled appearance of the ISe layer (yellow arrow).
Imaging of the mother’s retina revealed a largely contiguous mosaic of photoreceptors with reduced density at all parafoveal locations, as has been reported for carriers of blue cone monochromacy (BCM) (Carroll et al., 2010). This is consistent with at least some of the cones expressing the mutant p.Glu41Lys pigment undergoing early degeneration.
3.3 Molecular genetics
The PCR amplification and sequencing of exons 3 and 4 of the L and M opsin genes did not reveal either a p.Cys203Arg mutation (Karnik et al., 1988, Kazmi, Sakmar & Ostrer, 1997) or a variant exon 3 sequence (Carroll et al., 2004, McClements et al., 2010) as found in families displaying a similar phenotype of cone dysfunction, dichromacy and myopia. However, the amplification and sequencing of exon 2 revealed a new mutation, c.121G>A, that encodes for a glutamate to lysine substitution at site 41 (p.Glu41Lys) in both the carrier mother and the affected male offspring (Figure 5A). Note that the direct sequencing of exon 2 in the affected male gave a non-ambiguous adenosine (A) peak at site 121, whereas the carrier mother had a double peak of guanine (G) and A, indicating the presence of both mutant and wild type versions of L exon 2 in the maternal DNA but only a single mutant version in the affected son.
Figure 5.

Mutation and opsin gene arrays in mother and son. (A) Sequence data revealing the exon 2 mutation c.121G>A (black line)) encoding a p.Glu41Lys substitution. (B) Exon 3 sequences. Note the variation in the maternal sequence at three sites that distinguish L and M exon 3. (C) Promoter sequences. Note the variation in the maternal sequence at five sites that distinguish L and M promoters. (D) Hybrid gene showing the position of the c.121G>A change in exon 2 found in affected male subject and in his mother. (E) Wild type L and M genes also present in the mother on her second X chromosome. (n) indicates multiple M cone opsin sequences in maternal L/M array.
The Glu residue present at this site is completely conserved in the L (LWS) pigments of species drawn from across the animal kingdom (Supplementary Figure 1). In three of the other four classes of vertebrate visual pigments, the residue at site 41 is either conserved as Glu or replaced by Asp, the latter change representing a conservative substitution that retains the negative charge. The exception is the cone SWS2 pigments where an uncharged Leu residue is present. Note also that the p.Glu41Lys substitution is within two residues of site responsible for the most common form of dominant retinitis pigmentosa (Dryja et al., 1990), the Pro to His change at site 23/39 in rod/L cone opsin numbering.
Long-range PCR across exons 1–6 followed by exon-specific PCR amplifications and sequencing of each exon showed that the mutation was in a complex hybrid gene comprising L exon 2, M exon 3, L exon 4 and M exon 5. Since exons 1 and 6 of the L and M genes are identical, the hybrid sequence is abbreviated to L1-2M3L4M5-6. This exon 2 mutant hybrid L1-2M3L4M5-6 gene is also present in the maternal carrier along with a normal L opsin gene and normal downstream M opsin genes. Amplification and direct sequencing of exon 3 again revealed only a single M sequence in the affected male but mixed L and M sequences in the mother (Figure 5B). Further confirmation of the single gene array in the affected male was obtained from three independent repeats of exon 2 and exon 3 amplification from genomic DNA, followed by sequencing that revealed the presence of only a single gene sequence in the affected male, whereas both L and M sequences were identified in the maternal genome.
In order to determine the genomic structure of the opsin array, the L and M genes were separately amplified using promoter specific primers (Oda et al., 2003). In the affected male, the mutant hybrid gene is adjacent to the L promoter in the proximal position in the opsin gene array but all attempts to amplify downstream genes failed, again indicating that his opsin gene array is limited to just this single gene. In contrast, the mutant hybrid gene array was present in the carrier mother along with a second array that comprised a normal L opsin gene with normal downstream M opsin genes.
Finally, the L and M promoters were screened in both the mother and the affected male using a primer pair that simultaneously amplifies both promoter regions. As shown in Figure 5C, the sequence obtained from maternal DNA showed nucleotide variation at sites known to differ between the L and M promoters (Nathans, Thomas & Hogness, 1986). Moreover, the peak height in the electropherogram of the nucleotide present in the M promoter was substantially higher than that of the nucleotide present in the L promoter, indicating the presence of additional copies of the M gene in the mother’s opsin array. In contrast, the sequence of the promoter in the affected son did not show any nucleotide variation, with the sequence following that for the upstream promoter, entirely consistent therefore with the presence of just a single gene in the affected subject’s array. The data are summarised in Figure 5D.
Quantitative PCR (qPCR) amplification of exon 5 of the L and M opsin genes using primers to exon sequences common to both genes was used to assess the ratio of L to M gene sequences. The ratios obtained are consistent with the sequencing data; the affected subject lacks any L exon 5 and has just a single M exon 5 (Figure 6). In contrast, his mother possesses a single L exon 5 and seven copies of M exon 5. This would imply that her non-mutant opsin array comprises a single L gene plus six M genes. Multiple M genes are not uncommon within the L/M opsin gene array (Neitz & Neitz, 1995).
Figure 6.
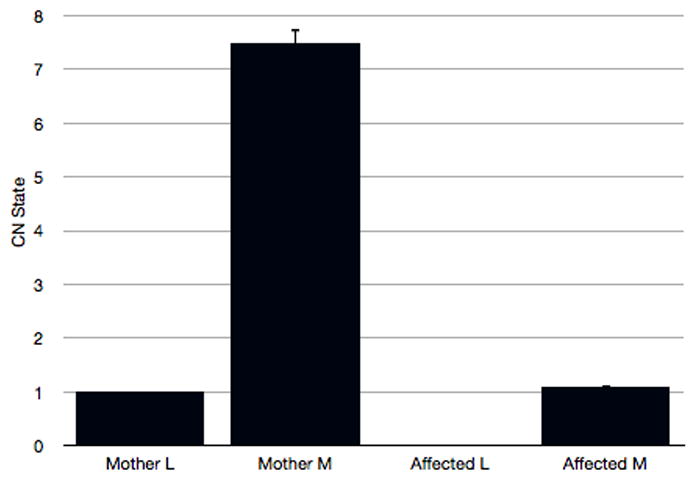
Quantitative PCR analysis of L and M exon 5 sequences in the L/M opsin gene array of the affected male subject and his mother. Note the complete absence of L opsin exon 5 in the affected son.
3.4 Pigment regeneration
The p.Glu41Lys mutation was introduced into wild type L, M and hybrid L1-2M3L4M5-6 coding sequences; these were generated by the SPLICE method of gene assembly (Davies, Carvalho & Hunt, 2007). The resulting sequences were expressed in vitro and the isolated opsins reconstituted with 11-cis-retinal alongside L, M and the hybrid sequences lacking the mutation. As shown in Figure 7, all three mutant opsins failed to form spectrally functional pigments. In contrast, all non-mutant opsins formed viable pigments with absorbance peaks at 536 nm for the hybrid, 562 nm for wild type L and 528 nm for wild type M. The peak absorbances for the L and M pigments are consistent with previous reports (Merbs & Nathans, 1992a, Merbs & Nathans, 1992b, Merbs & Nathans, 1993). Similarly, the peak absorbance for the hybrid is consistent with M exons 3 and 5 of the hybrid gene encoding the key tuning residues at sites 180, 277 and 285 in the opsin protein (Asenjo, Rim & Oprian, 1994, Merbs & Nathans, 1992b).
Figure 7.
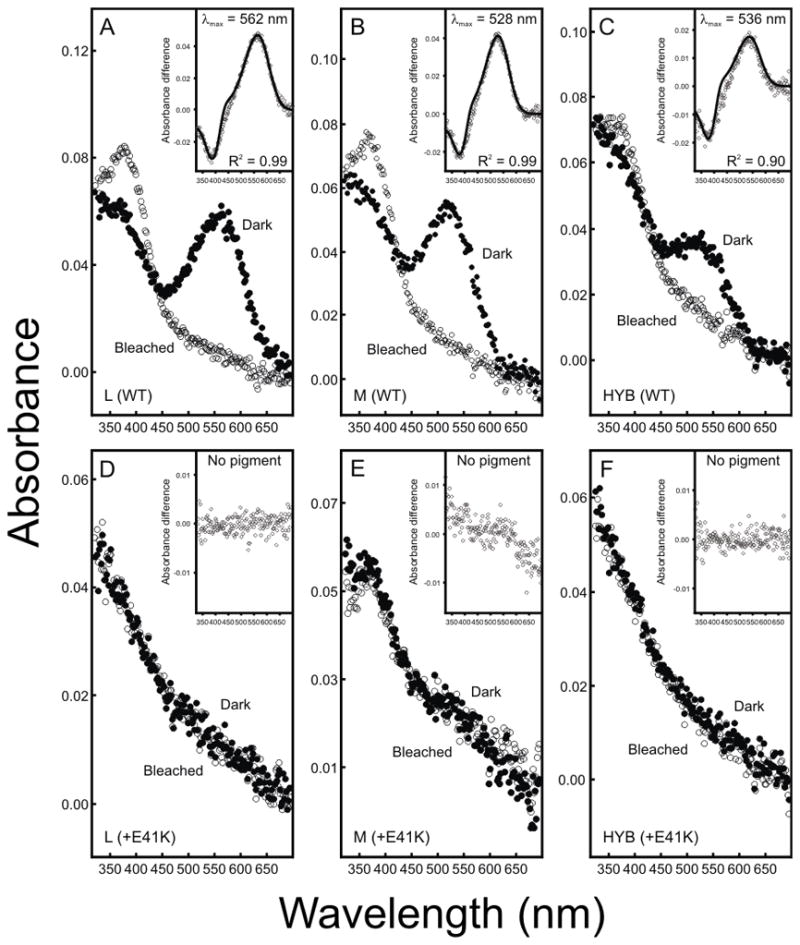
Dark, bleached and difference spectra for pigments regenerated in vitro. Difference spectra shown in insets were obtained by subtracting bleached from dark spectra data. (A) L (WT), (B) M (WT), (C) L1-2M3L4M5-6 hybrid (HYB), (D) Mutant (E41K) L, (E) Mutant (E41K) M, (F) Mutant (E41K) L1-2M3L4M5-6 hybrid.
3.5 Intracellular transport of mutant opsins
Wild-type and exon 2 mutant L, M and hybrid L1-2M3L4M5-6 opsin variants were expressed in the murine Neuro2a cell line and the cellular location of the expressed protein determined by immunocytochemistry. For cells transfected with the wild-type L, M, and hybrid sequences, opsin staining was located both intracellularly and at the cell surface, indicating that all three opsins were successfully transported to the plasma membrane (Figure 8A–C). In contrast, with the exon 2 mutant versions of these opsins, staining was only observed within the cytoplasm, indicating that although the opsin was produced, it was not trafficked successfully to the cell membrane (Figure 8B–F).
Figure 8.

Confocal microscopy images of transfected Neuro2a cells expressing wild-type and mutant E41K opsins. In all cases, opsin was visualized by the green fluorescence of the Alexa Fluor® 488 dye conjugated to the secondary antibody. (A) L (WT), (B) M (WT), (C) L12M3L4M56 hybrid (HYB), (D) Mutant (E41K) L, (E) Mutant (E41K) M, (F) Mutant (E41K) L1-2M3L4M5-6 hybrid. Note the presence of membrane-associated fluorescence only in cells transfected with the wild-type and hybrid constructs (i.e. without the E41K substitution) (white arrows). Cytoplasmic-associated fluorescence is also indicated (orange arrows).
4. Discussion
The molecular genetic data reveal that the affected male subject in the family described in this report has only a single opsin gene in the L/M gene array. This gene is a complex hybrid comprising L exon 2, M exon 3, L exon 4 and M exon 5 (L1-2M3L4M5-6) and contains a novel mutation in exon 2 that results in a p.Glu41Lys amino acid substitution. In vitro expression shows that this mutation prevents the production of a functional pigment irrespective of the nature of the opsin protein; mutant hybrid, M, and L opsins all failed to form functional pigments when expressed in vitro. In addition, cell trafficking experiments with neuronal cells transfected with wild-type and mutant gene constructs showed a failure in the transportation of the mutant pigment to the cell membrane. In summary, therefore, the p.Glu41Lys substitution would appear to cause a dysfunctional pigment in vitro. Glu41 is highly conserved across LWS pigments and is either retained or replaced by another negatively charged residue, Asp, in all other visual opsins except the SWS2 cone class. The presence of a negatively charged residue at this site would appear therefore to be important and it is not surprising that replacement with positively charged Lys is detrimental.
Since this mutant gene is the only L/M opsin gene present in the affected subject, this would imply that he lacks any functional L/M pigment, retaining only an S gene encoding a functional S pigment. If this were the case, his behavioral phenotype would be expected to be that of a blue cone monochromat. However, he clearly retains M-cone function and phenotypically, his disorder is similar to Bornholm Eye Disease (BED) where a generalised cone dysfunction and protanopia is found alongside myopia (Haim, Fledelius & Skarsholm, 1988, Michaelides et al., 2005, Young et al., 2004). Retinal imaging demonstrates extensive cone depletion, the presence of non-waveguiding cones and an overall thinning of the retina. The latter resembles that seen in BED patients (Neitz et al., 2011) whereas the presence of non-waveguiding cones that indicate the disruption of the cone outer segments is more like that seen in BCM (Carroll et al., 2012). The hyper reflective band associated with the ISe had an irregular mottled appearance, which contrasts with lowered intensity of the ISe seen in patients with diminished cone function (Hood et al., 2011). This may indicate an alteration of the inner segment of the photoreceptors that could be due to the retention of pigment. What is abundantly clear however from psychophysical testing and retinal imaging is that there is a robust cone population remaining in the affected subject’s retina. The presence of residual protanopic colour vision implies that at least some of these cones are M cones, and that they are functioning to some degree. Colour deficient individuals with reduced acuity and, in some cases, myopia have been previously reported by Mizrahi-Meissonnier et al. (2010). These individuals possess a single L/M opsin gene with a rare exon 3 haplotype that has been associated with the disruption of cone function (Carroll et al., 2004). However, as for the p.Glu41Lys mutant opsin, some functional pigment must be produced to account for the retention of some colour vision.
The presence of dichromacy in the affected subject implies the presence of at least one functional L/M opsin gene, and a protan defect indicates that the encoded pigment has an M cone-like spectral sensitivity. Significantly, the L1-2M3L4M5-6 hybrid opsin with M exons 3 and 5 generates a pigment with an M cone-like λmax at 536 nm. To account therefore for the protan colour vision defect, the mutant hybrid opsin must be able to produce a functional pigment. This is not however borne out by the in vitro experiments detailed in this report: mutant opsin generated in HEK293T cells failed to yield a functional pigment and trafficking experiments with Neuro2a cells showed that the mutant opsin failed to be transported to the plasma membrane. However, in both of these experiments, molecular chaperones that are known to be important for protein trafficking and folding, are missing. Within photoreceptors, a chaperone function has been proposed for the NinaA glycoprotein of Drosophila in the trafficking of Rh1 opsin (Stamnes et al., 1991) and the Ran-binding protein 2 (RanBP2) in mammals in the trafficking of the L cone opsin (Ferreira et al., 1996). More recently, Kosmaoglou et al. (2009) have shown that EDEM1 (ER degradation enhancing α-mannosidase-like 1) acts as a chaperone to promote the correct folding of rod opsin. Note also that 11-cis-retinal has been shown to promote the trafficking of mutant opsins (Insinna et al., 2012) and to protect against cone loss (Rohrer et al., 2005). It is possible therefore that either a chaperone protein or 11-cis-retinal is able to promote the trafficking and folding of the mutant p.Glu41Lys opsin in vivo to yield a functional pigment with an M cone-like spectral sensitivity. Since 11-cis-retinal is produced outside the photoreceptor cells, the absolute levels available may show local variations. If higher levels of 11-cis-retinal promote opsin trafficking and normal folding of the mutant opsin to give a functional pigment, then this may explain why cell loss and dysfunction does not affect all L/M cones in the subject’s retina. We propose therefore that a functional pigment is present in a sub-set of L/M cones that is perhaps retained in the inner segments. The presence of this pigment is sufficient to give a protan-like phenotype but insufficient to ensure the survival of all L/M cones. It remains to be established whether there is loss of further cone cells expressing the mutant opsin over time; repeat high-resolution retinal imaging may provide further insight into the issue of cone survival.
In summary, X-linked cone dysfunction, dichromacy and myopia have been shown to arise from a novel mutation of p.Glu41Lys in a hybrid L/M opsin gene. In vitro reconstitution of the mutant hybrid pigment has revealed that this mutation inhibits the ability of the opsin to reach the cell membrane and to bind chromophore in the production of a viable visual pigment. The L/M gene complement of the affected male subject is limited to a single copy of this mutant gene; the presence of protanopia rather than isolated tritan discrimination as found in BCM indicates that in vivo the encoded pigment must be functional, albeit in a sub-set of cone photoreceptors. In others, the accumulation of mutant opsin may be responsible for cone dysfunction and degeneration.
Supplementary Material
Amino acid alignment of the N-terminal region of vertebrate visual opsins representing the four cone (LWS, RH2, SWS2 and SWS1) and rod (RH1) classes. Human LWS, NM_020061; Human MWS, NG_011606; Macaque LWS, AF158968; Marmoset LWS, XM_002763410; Cow LWS, NM_174566; Mouse LWS, NM_008106; Wallaby LWS, AY286018; Dunnart LWS, EU232013; Honey possum LWS, AY772470; Platypus LWS, EU624413; Pigeon LWS, AF149243; Anolis LWS, ACU08131; Zebrafish LWS, NM_131175; Lamprey LWS, LWSAY366491; Pigeon RH2, AF149232; Anolis RH2, S79167; Zebrafish RH2, NM_131253; Lamprey RHA, AY366493; Lamprey RHB, AY366494; Platypus SWS2, EU624412; Pigeon SWS2, AF149238; Anolis SWS2, AF133907; Xenopus SWS2, NM_001089142; Zebrafish, SWS2, NM_131192; Lamprey SWS2, AY366492; Human SWS1, NM_001708; Macaque SWS1, AF158976; Cow SWS1, NM_174567; Mouse SWS1, NM_007538; Wallaby SWS1, AY286017; Honey Possum SWS1, AY772472; Dunnart SWS1, AY442173; Pigeon SWS1, AF149234; Anolis SWS1, AF134192; Zebrafish SWS1, NM_131319; Lamprey SWS1, AY366495; Human RH1, NM_000539; Macaque RH1, S76579; CowRH1NM_001014890; Mouse RH1, NM_145383; Dunnart RH1, AY159786; Platypus RH1, EF050076; Pigeon RH1, AF149230; Xenopus RH1, NM_001087048; Zebrafish RH1, BC045288.
Highlights.
Identification of a new missense mutation in L opsin gene.
Deleterious effect on pigment production confirmation by in vitro expression.
Single deleterious mutant gene in the subject’s L/M opsin gene array.
Protanopia and cone dysfunction associated with this mutant gene.
Retention of some in vivo activity of mutant opsin.
Acknowledgments
This work was supported by the following grants: UK Fight for Sight (DMH/ATM/MM and REM); Australian National Health and Medical Research Council (DMH); Foundation Fighting Blindness USA (ATM/MM and JC/JR); National Institute for Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology (ATM/MM and REM); NIHR Oxford Biomedical Research Centre (MMc/REM); Moorfields Eye Hospital Special Trustees (ATM/MM); Research to Prevent Blindness (JC/JR); the Gene and Ruth Posner Foundation (JC/JR); the Thomas M. Aaberg, Sr. Retina Research Fund (JC/JR); National Institute of Health USA grants R01EY017607 and P30EY001931 (JC/JR); Research to Prevent Blindness (MN); Core Grant for Vision Research P30EY01730 (MN); and National Eye Institute award R01EY09620 (MN). This investigation was conducted in part in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR016511 awarded to JC from the National Center for Research Resources, National Institutes of Health. MM is supported by a Foundation Fighting Blindness USA Career Development Award. The authors are grateful to Phyllis Summerfelt for assistance with imaging.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michelle McClements, Email: michelle.mcClements@stx.ox.ac.uk.
Wayne I L Davies, Email: w.davies13@gmail.con.
Michel Michaelides, Email: michel.michaelides@ucl.ac.uk.
Joseph Carroll, Email: jcarroll@mcw.edu.
Jungate Rha, Email: jungtae.rha@gmail.com.
John D Mollon, Email: jm123@cam.ac.uk.
Maureen Neitz, Email: mneitz@uw.edu.
Robert E MacLaren, Email: robert.maclaren@eye.ox.ac.uk.
Anthony T Moore, Email: tony.moore@ucl.ac.uk.
David M Hunt, Email: david.hunt@uwa.edu.au.
References
- Asenjo AB, Rim J, Oprian DD. Molecular determinants of human red/green color discrimination. Neuron. 1994;12(5):1131–1138. doi: 10.1016/0896-6273(94)90320-4. [DOI] [PubMed] [Google Scholar]
- Carroll J, Baraas RC, Wagner-Schuman M, Rha J, Siebe CA, Sloan C, Tait DM, Thompson S, Morgan JI, Neitz J, Williams DR, Foster DH, Neitz M. Cone photoreceptor mosaic disruption associated with Cys203Arg mutation in the M-cone opsin. Proc Natl Acad Sci U S A. 2009;106(49):20948–20953. doi: 10.1073/pnas.0910128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Dubra A, Gardner JC, Mizrahi-Meissonnier L, Cooper RF, Dubis AM, Nordgren R, Genead M, Connor TB, Jr, Stepien KE, Sharon D, Hunt DM, Banin E, Hardcastle AJ, Moore AT, Williams DR, Fishman G, Neitz J, Neitz M, Michaelides M. The effect of cone opsin mutations on retinal structure and the integrity of the photoreceptor mosaic. Invest Ophthalmol Vis Sci. 2012;53(13):8006–8015. doi: 10.1167/iovs.12-11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Neitz M, Hofer H, Neitz J, Williams DR. Functional photoreceptor loss revealed with adaptive optics: an alternate cause of color blindness. Proc Natl Acad Sci U S A. 2004;101(22):8461–8466. doi: 10.1073/pnas.0401440101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll J, Rossi EA, Porter J, Neitz J, Roorda A, Williams DR, Neitz M. Deletion of the X-linked opsin gene array locus control region (LCR) results in disruption of the cone mosaic. Vision Res. 2010;50(19):1989–1999. doi: 10.1016/j.visres.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies WL, Carvalho LS, Hunt DM. SPLICE: a technique for generating in vitro spliced coding sequences from genomic DNA. Biotechniques. 2007;43(6):785–789. doi: 10.2144/000112588. [DOI] [PubMed] [Google Scholar]
- Davies WL, Carvalho LS, Tay BH, Brenner S, Hunt DM, Venkatesh B. Into the blue: gene duplication and loss underlie color vision adaptations in a deep-sea chimaera, the elephant shark Callorhinchus milii. Genome Res. 2009;19(3):415–426. doi: 10.1101/gr.084509.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies WL, Cowing JA, Carvalho LS, Potter IC, Trezise AE, Hunt DM, Collin SP. Functional characterization, tuning, and regulation of visual pigment gene expression in an anadromous lamprey. FASEB J. 2007;21(11):2713–2724. doi: 10.1096/fj.06-8057com. [DOI] [PubMed] [Google Scholar]
- Deeb SS, Lindsey DT, Hibiya Y, Sanocki E, Winderickx J, Teller DY, Motulsky AG. Genotype-phenotype relationships in human red/green color-vision defects: molecular and psychophysical studies. Am J Hum Genet. 1992;51(4):687–700. [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, Sandberg MA, Berson EL. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343(6256):364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Ferreira PA, Nakayama TA, Pak WL, Travis GH. Cyclophilin-related protein RanBP2 acts as chaperone for red/green opsin. Nature. 1996;383(6601):637–640. doi: 10.1038/383637a0. [DOI] [PubMed] [Google Scholar]
- Govardovskii VI, Fyhrquist N, Reuter T, Kuzmin DG, Donner K. In search of the visual pigment template. Vis Neurosci. 2000;17(4):509–528. doi: 10.1017/s0952523800174036. [DOI] [PubMed] [Google Scholar]
- Haim M, Fledelius HC, Skarsholm X-linked myopia in Danish family. Acta Ophthalmol (Copenh) 1988;66(4):450–456. doi: 10.1111/j.1755-3768.1988.tb04039.x. [DOI] [PubMed] [Google Scholar]
- Hood DC, Zhang X, Ramachandran R, Talamini CL, Raza A, Greenberg JP, Sherman J, Tsang SH, Birch DG. The inner segment/outer segment border seen on optical coherence tomography is less intense in patients with diminished cone function. Invest Ophthalmol Vis Sci. 2011;52(13):9703–9709. doi: 10.1167/iovs.11-8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insinna C, Daniele LL, Davis JA, Larsen DD, Kuemmel C, Wang J, Nikonov SS, Knox BE, Pugh EN., Jr An S-opsin knock-in mouse (F81Y) reveals a role for the native ligand 11-cis-retinal in cone opsin biosynthesis. J Neurosci. 2012;32(23):8094–8104. doi: 10.1523/JNEUROSCI.0131-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagla WM, Jägle H, Hayashi T, Sharpe LT, Deeb SS. The molecular basis of dichromatic color vision in males with multiple red and green visual pigment genes. Hum Mol Genet. 2002;11(1):23–32. doi: 10.1093/hmg/11.1.23. [DOI] [PubMed] [Google Scholar]
- Jägle H, de Luca E, Serey L, Bach M, Sharpe LT. Visual acuity and X-linked color blindness. Graefes Arch Clin Exp Ophthalmol. 2006;244(4):447–453. doi: 10.1007/s00417-005-0086-4. [DOI] [PubMed] [Google Scholar]
- Karnik SS, Sakmar TP, Chen HB, Khorana HG. Cysteine residues 110 and 187 are essential for the formation of correct structure in bovine rhodopsin. Proc Natl Acad Sci U S A. 1988;85(22):8459–8463. doi: 10.1073/pnas.85.22.8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmi MA, Sakmar TP, Ostrer H. Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stability. Invest Ophthalmol Vis Sci. 1997;38(6):1074–1081. [PubMed] [Google Scholar]
- Kosmaoglou M, Kanuga N, Aguila M, Garriga P, Cheetham ME. A dual role for EDEM1 in the processing of rod opsin. J Cell Sci. 2009;122(Pt 24):4465–4472. doi: 10.1242/jcs.055228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmor MF, Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Arch Ophthalmol. 1993;111(5):601–604. doi: 10.1001/archopht.1993.01090050035023. [DOI] [PubMed] [Google Scholar]
- Marmor MF, Zrenner E. Standard for clinical electroretinography. 1994 update. Doc Ophthalmol. 1995;89(3):199–210. doi: 10.1007/BF01203373. [DOI] [PubMed] [Google Scholar]
- McClements ME, Neitz M, Moore AT, Hunt DM. Bornholm Eye Disease arises from a specific combination of amino acid changes encoded by exon 3 of the L/M cone opsin gene. ARVO Meeting Abstracts. 2010:Abstract 2609. [Google Scholar]
- Merbs SL, Nathans J. Absorption spectra of human cone pigments. Nature. 1992a;356(6368):433–435. doi: 10.1038/356433a0. [DOI] [PubMed] [Google Scholar]
- Merbs SL, Nathans J. Absorption spectra of the hybrid pigments responsible for anomalous color vision. Science. 1992b;258(5081):464–466. doi: 10.1126/science.1411542. [DOI] [PubMed] [Google Scholar]
- Merbs SL, Nathans J. Role of hydroxyl-bearing amino acids in differentially tuning the absorption spectra of the human red and green cone pigments. Photochem Photobiol. 1993;58(5):706–710. doi: 10.1111/j.1751-1097.1993.tb04956.x. [DOI] [PubMed] [Google Scholar]
- Michaelides M, Johnson S, Bradshaw K, Holder GE, Simunovic MP, Mollon JD, Moore AT, Hunt DM. X-linked cone dysfunction syndrome with myopia and protanopia. Ophthalmology. 2005;112(8):1448–1454. doi: 10.1016/j.ophtha.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Mizrahi-Meissonnier L, Merin S, Banin E, Sharon D. Variable retinal phenotypes caused by mutations in the X-linked photopigment gene array. Invest Ophthalmol Vis Sci. 2010;51(8):3884–3892. doi: 10.1167/iovs.09-4592. [DOI] [PubMed] [Google Scholar]
- Mollon JD, Astell S, Reffin JP. In: Drum B, Moreland JD, Serra A, editors. A minimalist test of colour vision; Colour Vision Deficiencies X: Proceedings of the Tenth Symposium of the International Research Group on Colour Vision Deficiencies; Cagliari, Italy. 25–28 June 1989; Dordrecht, The Netherlands: Kluwer Academic Publishers; 1991. pp. 59–67. [Google Scholar]
- Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS. Molecular genetics of inherited variation in human color vision. Science. 1986;232(4747):203–210. doi: 10.1126/science.3485310. [DOI] [PubMed] [Google Scholar]
- Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986;232(4747):193–202. doi: 10.1126/science.2937147. [DOI] [PubMed] [Google Scholar]
- Neitz J, Wagner-Schuman M, Dubra A, Sjoberg SA, Moore AT, Young TL, Neitz M, Carroll J, Michaelides M. Cone Mosaic Disruption Caused by L/M Opsin Mutations in Bornholm Eye Disease. ARVO Meeting Abstracts. 2011;52:4896. [Google Scholar]
- Neitz M, Carroll J, Renner A, Knau H, Werner JS, Neitz J. Variety of genotypes in males diagnosed as dichromatic on a conventional clinical anomaloscope. Vis Neurosci. 2004;21(3):205–216. doi: 10.1017/s0952523804213293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitz M, Neitz J. Numbers and ratios of visual pigment genes for normal red-green color vision. Science. 1995;267(5200):1013–1016. doi: 10.1126/science.7863325. [DOI] [PubMed] [Google Scholar]
- Neitz M, Neitz J. Molecular genetics of color vision and color vision defects. Arch Ophthalmol. 2000;118(5):691–700. doi: 10.1001/archopht.118.5.691. [DOI] [PubMed] [Google Scholar]
- Oda S, Ueyama H, Nishida Y, Tanabe S, Yamade S. Analysis of L-cone/M-cone visual pigment gene arrays in females by long-range PCR. Vision Res. 2003;43(5):489–495. doi: 10.1016/s0042-6989(02)00679-x. [DOI] [PubMed] [Google Scholar]
- Pokorny J, Smith VC, Verriest G, Pinckers AJLG. Congenital and Acquired Color Vision Defects. Grune & Stratton; New York: 1979. [Google Scholar]
- Regan BC, Reffin JP, Mollon JD. Luminance noise and the rapid determination of discrimination ellipses in colour deficiency. Vision Res. 1994;34(10):1279–1299. doi: 10.1016/0042-6989(94)90203-8. [DOI] [PubMed] [Google Scholar]
- Rha J, Schroeder B, Godara P, Carroll J. Variable optical activation of human cone photoreceptors visualized using a short coherence light source. Opt Lett. 2009;34(24):3782–3784. doi: 10.1364/OL.34.003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrer B, Lou HR, Humphries P, Redmond TM, Seelinger MW, Crouch RK. Cone Opsin Mislocalization in Rpe65−/− Mice: A Defect That Can Be Corrected by 11-cis Retinal. Invest Ophthalmol Vis Sci. 2005;46(10):3876–3882. doi: 10.1167/iovs.05-0533. [DOI] [PubMed] [Google Scholar]
- Stamnes MA, Shieh BH, Chuman L, Harris GL, Zuker CS. The cyclophilin homolog ninaA is a tissue-specific integral membrane protein required for the proper synthesis of a subset of Drosophila rhodopsins. Cell. 1991;65(2):219–227. doi: 10.1016/0092-8674(91)90156-s. [DOI] [PubMed] [Google Scholar]
- Tanna H, Dubis AM, Ayub N, Tait DM, Rha J, Stepien KE, Carroll J. Retinal imaging using commercial broadband optical coherence tomography. Br J Ophthalmol. 2010;94(3):372–376. doi: 10.1136/bjo.2009.163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama H, Kuwayama S, Imai H, Oda S, Nishida Y, Tanabe S, Shichida Y, Yamade S. Analysis of L-cone/M-cone visual pigment gene arrays in Japanese males with protan color-vision deficiency. Vision Res. 2004;44(19):2241–2252. doi: 10.1016/j.visres.2004.04.011. [DOI] [PubMed] [Google Scholar]
- Ueyama H, Li YH, Fu GL, Lertrit P, Atchaneeyasakul LO, Oda S, Tanabe S, Nishida Y, Yamade S, Ohkubo I. An A-71C substitution in a green gene at the second position in the red/green visual-pigment gene array is associated with deutan color-vision deficiency. Proc Natl Acad Sci U S A. 2003;100(6):3357–3362. doi: 10.1073/pnas.0637437100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winderickx J, Sanocki E, Lindsey DT, Teller DY, Motulsky AG, Deeb SS. Defective colour vision associated with a missense mutation in the human green visual pigment gene. Nat Genet. 1992;1(4):251–256. doi: 10.1038/ng0792-251. [DOI] [PubMed] [Google Scholar]
- Young TL, Deeb SS, Ronan SM, Dewan AT, Alvear AB, Scavello GS, Paluru PC, Brott MS, Hayashi T, Holleschau AM, Benegas N, Schwartz M, Atwood LD, Oetting WS, Rosenberg T, Motulsky AG, King RA. X-linked high myopia associated with cone dysfunction. Arch Ophthalmol. 2004;122(6):897–908. doi: 10.1001/archopht.122.6.897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Amino acid alignment of the N-terminal region of vertebrate visual opsins representing the four cone (LWS, RH2, SWS2 and SWS1) and rod (RH1) classes. Human LWS, NM_020061; Human MWS, NG_011606; Macaque LWS, AF158968; Marmoset LWS, XM_002763410; Cow LWS, NM_174566; Mouse LWS, NM_008106; Wallaby LWS, AY286018; Dunnart LWS, EU232013; Honey possum LWS, AY772470; Platypus LWS, EU624413; Pigeon LWS, AF149243; Anolis LWS, ACU08131; Zebrafish LWS, NM_131175; Lamprey LWS, LWSAY366491; Pigeon RH2, AF149232; Anolis RH2, S79167; Zebrafish RH2, NM_131253; Lamprey RHA, AY366493; Lamprey RHB, AY366494; Platypus SWS2, EU624412; Pigeon SWS2, AF149238; Anolis SWS2, AF133907; Xenopus SWS2, NM_001089142; Zebrafish, SWS2, NM_131192; Lamprey SWS2, AY366492; Human SWS1, NM_001708; Macaque SWS1, AF158976; Cow SWS1, NM_174567; Mouse SWS1, NM_007538; Wallaby SWS1, AY286017; Honey Possum SWS1, AY772472; Dunnart SWS1, AY442173; Pigeon SWS1, AF149234; Anolis SWS1, AF134192; Zebrafish SWS1, NM_131319; Lamprey SWS1, AY366495; Human RH1, NM_000539; Macaque RH1, S76579; CowRH1NM_001014890; Mouse RH1, NM_145383; Dunnart RH1, AY159786; Platypus RH1, EF050076; Pigeon RH1, AF149230; Xenopus RH1, NM_001087048; Zebrafish RH1, BC045288.