

Abstract
B1a cells are an important source of natural antibodies, antibodies directed against T-independent antigens, and are a primary source of IL-10. Bruton's tyrosine kinase (btk) is a cytoplasmic kinase that is essential for mediating signals from the B cell receptor and is critical for development of B1a cells. Consequentially, animals lacking btk have few B1a cells, minimal antibody responses, and can preferentially generate Th1 type immune responses following infection. B1a cells have been shown to aid in protection against infection with attenuated Francisella tularensis but their role in infection mediated by fully virulent F. tularensis is not known. Therefore, we utilized mice with defective btk (XID mice) to determine the contribution of B1a cells in defense against the virulent, F. tularensis ssp. tularensis strain SchuS4. Surprisingly, XID mice displayed increased resistance to pulmonary infection with F. tularensis. Specifically, XID mice had enhanced clearance of bacteria from the lung and spleen and significantly greater survival of infection compared to wild type controls. We revealed that resistance to infection in XID mice was associated with decreased numbers of IL-10 producing B1a cells and concomitant increased numbers of IL-12 producing macrophages and IFN-γ producing NK/NKT cells. Adoptive transfer of wild type B1a cells into XID mice reversed the control of bacterial replication. Similarly, depletion of NK/NKT cells also increased bacterial burdens in XID mice. Together, our data suggest B cell-NK/NKT cell crosstalk is a critical pivot controlling survival of infection with virulent F. tularensis.
INTRODUCTION
Successful defense against pathogens requires the coordinated function of multiple host cells. Among infections with intracellular bacteria, T cells, macrophages, and dendritic cells all play well described, essential, roles in the resolution of infection. In contrast, the contribution B cells make toward control of infection with intracellular bacteria is less well understood. The primary function of B cells in infection is generally attributed to their ability to secrete antibody directed against pathogens that allow for neutralization and/or opsonization of the bacteria (1). However, B cells have a multitude of other functions that may aid in control of infection and/or inflammation including antigen presentation, co-stimulation of T cells, and secretion of both pro- and anti-inflammatory cytokines (as reviewed, (2)). Thus, the relative contribution of B cells in the pathogenesis of disease is complex.
Francisella tularensis is a facultative intracellular, Gram negative bacterium that replicates in the cytoplasm of host cells (3, 4). Virulent subspecies of this bacterium, e.g. F. tularensis ssp tularensis, cause lethal disease following inhalation of as few as 10 bacteria (5). There are no licensed vaccines for F. tularensis and recrudescence following treatment with antibiotics has been noted (6). F. tularensis was also developed and deployed as a biological weapon (7). Thus, this pathogen requires manipulation under BSL-3 laboratory conditions, is classified as a Category A priority pathogen, and is regulated as a select agent in the United States.
Given the high virulence of F. tularensis ssp tularensis and restriction regarding its use, many laboratories have turned to using attenuated subspecies and strains of F. tularensis, e.g. F. tularensis ssp holarctica Live Vaccine Strain (LVS) and F. novicida, as surrogates for their more virulent cousin. Thus, our understanding of the role specific cell types, including B cells, play in F. tularensis infection is largely derived from data generated with attenuated LVS and F. novicida.
Previous work with attenuated strains of F. tularensis has demonstrated that mice completely lacking B cells (μMT−/−) exhibit modest increase in susceptibility to primary infection with LVS and poor resistance to secondary infection (8). Similarly, we have established that μMT−/− exhibit greater susceptibility to infection with virulent F. tularensis ssp tularensis strain SchuS4 than WT animals (9). Thus, B cells as a complete cellular compartment are required to resolve F. tularensis infections.
Since this previous data shows that B cells are important for control of F. tularensis infection and the fact that antibody production is considered one of the primary functions of B cells, several laboratories have explored the efficacy of immune sera and monoclonal antibodies to aid in protection against F. tularensis infection. Passive transfer of immune sera or monoclonal antibodies protects animals against F. tularensis (10–19). Furthermore, passive transfer of hyperimmune serum into humans newly infected with F. tularensis aids in the resolution of infection (20). The specific role of opsonizing IgM and defense against the attenuated F. tularensis vaccine strain (LVS) was highlighted in the study by Cole et al. In that study, animals immunized with LPS purified from LVS are protected from infection with LVS and this protection is largely dependent on antibodies secreted by B1a cells (21). In total, these reports show that antibodies can mediate protection against F. tularensis infection and that antibodies derived specifically from B1a cells are key players in this protection. However, these reports do not address the absolute requirement for antibodies in survival of infection with virulent F. tularensis. Furthermore, the contribution made by alternative B cell functions, i.e. cytokine secretion, toward resolution or exacerbation of disease mediated by virulent F. tularensis has not been explored.
In the report presented herein we demonstrate that neither high titers of antibody directed against F. tularensis ssp tularensis strain SchuS4 nor natural IgM are required for survival of SchuS4 infection. Moreover, we found that B1a cells contribute to the pathogenesis of F. tularensis infection and that this contribution was tightly associated with the interference of early, effective NK/NKT cell responses.
MATERIALS AND METHODS
Mice
Specific-pathogen-free, 6–8 week old CBA/J (wild type; WT) and CBA/CaHN-BtkXID/J (XID) (n = 5–10/group) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were housed in sterile microisolater cages in the BSL-3 facility at the RML. All mice were provided sterile water and food ad libitum and all research involving animals was conducted in accordance with Animal Care and Use guidelines and animal protocols were approved by the Animal Care and Use Committee at RML.
Bacteria
Francisella tularensis ssp. tularensis strain SchuS4 was originally provided by Jeannine Peterson, Ph.D. (Centers for Disease Control, Fort Collins, Colorado). SchuS4 was cultured in modified Mueller-Hinton broth at 37°C with constant shaking overnight, aliquoted into 1 ml samples, frozen at −80°C and thawed just prior to use as previously described (9). Frozen stocks were titered by enumerating viable bacteria from serial dilutions plated on modified Mueller-Hinton (MMH) agar as previously described (22, 23). The number of viable bacteria in frozen stock vials varied less than 1% over a 12 month period.
For generation of killed SchuS4 approximately 1.5 × 109 bacteria were incubated with 50 μg/ml levofloxacin overnight at 37°C. Bacteria were washed once and diluted to the equivalent multiplicity of infection of live organisms in PBS immediately prior to use. Confirmation of efficacy of levofloxacin treatment to obtain 100% dead bacteria was confirmed in preliminary experiments by incubating the entire inoculum onto MMH agar and incubating for 96 hours at 37°C/7%CO2. After this time no colonies, representing viable bacteria, were observed.
Culture and infection of alveolar macrophages and bone marrow derived macrophages (BMM)
Alveolar macrophages were collected as previously described (24). Bone marrow derived macrophages were generated as previously described (22) with following modifications. Progenitor cells isolated from the femurs of the indicated strains of mice were cultured in DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS), 0.2 mM L-glutamine, 1 mM HEPES buffer, and 0.1 mM nonessential amino acids (all from Invitrogen, Carlsbad, CA) (cDMEM) and 10 ng/ml M-CSF (Peprotech) in a T-75cm2 flask. Non-adherent cells were collected and placed in a fresh T-75cm2 flask on day 2 of culture. Medium was replaced on day 2 of culture. Adherent cells were collected on day 5, resuspended at 2 × 105 cells/ml and seeded at 0.5 ml/well into a 48-well tissue culture plate. All cells were used on day 6 for infection. Freshly isolated alveolar macrophages and bone marrow derived macrophages were infected with a MOI = 10 of SchuS4 and assessed for intracellular replication as previously described (22). The infection inoculum was confirmed by plating serial dilutions of stock SchuS4 on modified Mueller-Hinton agar plates immediately prior to addition to cell cultures. As indicated, cells were treated with 1 μg/ml Pam3Csk4 (Invivogen) approximately 24 hours after infection.
Collection and culture of peritoneal exudate cells (PEC)
Resting peritoneal cells were harvested from uninfected mice via peritoneal lavage as previously described (25). Cells were either stained for CD5, CD19 and F4/80 as described below. B1a cells (CD5+CD19+) were collected by fluorescent activated cell sorting (FACS) using an ARIA II and FACS Diva Software (BD Biosciences) or cultured immediately after collection. Sorted B1a cells were greater than 98% pure. All cultured cells (sorted populations and total PEC) were adjusted to 1 × 106 cells/ml in cDMEM and added to a 96-well tissue culture plate at 200 μl/ well. Cells were incubated with the indicated stimulus overnight. Then, supernatants were collected and immediately analyzed for IL-10 or IL-12p40 by ELISA. As indicated, some cultures were supplemented with either 10 μg/ml rat IgG as an isotype control (Ig) or neutralizing anti-IL-10 antibodies (both from R&D Systems).
Adoptive transfer of B1a cells and passive transfer of serum
Freshly isolated B1a cells were washed extensively in PBS and adjusted to 1 × 106/ml in PBS. Immediately following resuspension in PBS, XID mice were injected intraperitoneally with 100 μl (1×105 total B1a cells) of cells one day prior to infection. For passive transfer studies, serum was collected from uninfected WT mice or from WT mice 7 days after infection and passed through a 0.2 μm filter to sterilize. Sterilization of serum was confirmed by plating onto MMH agar plates. XID mice were injected with 200 μl of the indicated serum intraperitoneally one day prior to infection.
Depletion of NK/NKT cells
NK/NKT cells were depleted from XID mice using anti-Asialo GM1 antibodies (WAKO Chemical, Richmond, VA) as previously described (26). Briefly, 2 days prior to infection mice were injected intraperitoneally with 50 μl anti-Asialo GM1 or rabbit immunoglobulin (Jackson ImmunoResearch, West Grove, PA) as a negative control. Mice were injected with anti-Asialo GM1 or rabbit immunoglobulin again on days 1 and 4 and every third day thereafter following infection. Depletion of NK/NKT cells was confirmed by flow cytometry as described below (Supplemental Figure 1). This depletion regime results in greater than 98% depletion of NK/NKT cells.
Infection of mice and treatment with antibiotic
Mice were infected intranasally (i.n.) with F. tularensis SchuS4 as previously described (27). Briefly, bacteria were thawed and diluted in PBS. Mice were anesthetized by intraperitoneal injection of 100 μl of 12.5 mg/ml ketamine + 3.8 mg/ml xylazine. Approximately 50 CFU was administered into the nares of each mouse in a total volume of 25 μl. Actual inoculum concentration was confirmed by plating a portion of the inoculum onto MMH agar plates, incubating plates at 37°C with 5%CO2 and enumerating colonies. As indicated, mice were treated with levofloxacin (LVF) (Ortho-McNeil Pharmaceutical, Raritan, NJ) as previously described (9). Mice were monitored for 30 days or euthanized at the indicated time points after infection. Following infection mice were monitored regularly and euthanized at the first signs of illness. All experiments using animals were performed in accordance with protocols approved by the Animal Care and Use Committee at RML.
Detection of serum antibodies
Serum was collected from mice at the indicated time points after infection. Quantification of total serum IgM was performed by ELISA as previously described (28). Presence of serum IgG and IgM antibodies that recognize antigens in whole cell lysate prepared from SchuS4 and agglutinating serum antibodies directed against viable SchuS4 were detected as previously described (28).
Collection of tissue homogenate and enumeration of bacteria
Bacteria were enumerated from the lungs and spleens as previously described (24, 27). Briefly, organs were aseptically collected and placed in ice cold homogenization buffer (150 mM Tris-HCl, 5 mM EDTA, 10 mM Trizma-base) supplemented with a 1:100 dilution of Phosphatase Inhibitor cocktail I, Phosphatase Inhibitor cocktail II and Proteinase Inhibitor cocktail III (all from AG Scientific, San Diego, CA). Organs were homogenized by grinding tissues through a sterile S/S Type 304 #60 wire mesh screen (Billeville Wire Cloth Co., Cedar Grove, New Jersey) using a 5 ml syringe plunger. A portion of the resulting homogenate was immediately serially diluted in PBS and plated on MMH agar for enumeration of bacterial loads. The remaining homogenate was clarified by centrifuged at 14,000 × g for 30 min at 4°C. The resulting supernatants were sterile filtered through 0.2 μm syringe filters (Millipore Ireland LTD, Cork, Ireland) and stored at −80°C.
Isolation of lung and spleen cells
Lung cells and splenocytes were collected as previously described with the following modifications (9). Lung cells and splenocytes were resuspended in FACS buffer prior to flow cytometric analysis or DMEM supplemented with 10% heat-inactivated fetal calf serum (FCS), 0.2 mM L-glutamine, 1 mM HEPES buffer and 0.1 mM nonessential amino acids (all from Invitrogen, Carlsbad, CA) (cDMEM) prior to addition to tissue cultures. Total live cells from the lungs and spleens were enumerated using trypan blue and a TC10 Automated Cell Counter (Bio-Rad Laboratories, Hercules, CA). A portion of cells were immediately stained for surface receptors as described below. Additional lung and spleen cells were resuspended in cDMEM at 2 × 105 per well in 96-well plates in the presence of 10 μg/ml Brefeldin A, 10 ng/ml phorbol 12-myristate 13-acetate and 1 μg/ml ionomycin (all from Sigma) at 37°C/7% CO2 for 4 hours. Cells were stained for the indicated surface markers and intracellular cytokines as described below.
Flow Cytometry
Lung and splenocyte populations were assessed by flow cytometry as previously (9). Briefly, the following antibodies in various combinations were used for flow cytometric analysis: APC CD4, PerCPCy5.5 CD8, FITC NKp46, PeCy7 B220 and PerCp-Cy5.5 CD11c, PECy7 CD11b, PE Ly6C, FITC Ly6G, APC F4/80, PE MHCII, PerCp-Cy5.5 CD19 and APC CD5 (all from BD Biosciences, San Jose, CA). Staining was performed in FACS buffer at room temperature. Following staining, cells were washed and fixed in 1% paraformaldehyde for 30 minutes at 4°C. Cells were washed a final time, resuspended in FACS buffer and stored at 4°C until analyzed. Samples were collected using a LSRII flow cytometer (BD Biosciences). Analysis gates were set on viable unstained cells and were designed to include all viable cell populations. Approximately 10,000 gated events were analyzed for each sample. Isotype control antibodies were included when analyses and panels were first being performed to assure specificity of staining, but were not routinely included with each experiment. Data was analyzed using FlowJo software (Treestar, Ashland, OR).
Intracellular cytokines were detected by flow cytometry as previously described (9). Following incubation, cells were washed once and resuspended in FACS buffer and stained for CD4, CD8, NKp46, CD19, CD5, F4/80 and CD11b as described above. Then cells were fixed in 2% paraformaldehyde in PBS for 10 min at 37°C/7% CO2 and washed twice more in perm buffer (FACS buffer supplemented with 0.25% saponin [Sigma-Aldrich]). Cells were incubated for 20 min at room temperature with various combinations of PE IFN-γ, APC or Alexa Fluor 488 IL10, PE IL-12p40 (all from BD Biosciences). Cells were washed twice in perm buffer, fixed in 1% paraformaldehyde for 30 min, and then resuspended in FACS buffer and stored at 4°C until analysis. Cells were acquired and analyzed using a LSR II flow cytometer (BD Biosciences) and FlowJo Software (Treestar).
Detection of secreted cytokines
Concentration of TNF-α present in cell culture supernatants was determined using commercially available ELISA kits following manufacturer's instructions (BD Biosciences).
Statistical Analysis
Statistical differences between two groups were determined using an unpaired t test with the significance set at p<0.05. For comparison between three or more groups, analysis was done by non-parametric one-way ANOVA (Kruskal-Wallis Test) followed by Dunn's comparisons test with significance determined at p<0.05. Significance in survival between groups was determined using Log-Rank (Mantel-Cox) test with significance determined at p<0.05.
RESULTS
XID mice show greater survival and control of bacterial replication than wild type mice
To determine the requirement for high titers of anti-SchuS4 antibodies and B1a cells in resolution of pulmonary SchuS4 infection, we first determined if mice lacking B1a cells exhibited differences in survival compared to WT mice. Infection of mice with SchuS4 typically results in death within 5 days of infection (9, 27). This rapid mean time to death often precludes the ability of the host to mount an immune response. Thus, determination of the role specific host cell components make in the pathogenesis of SchuS4 infection among untreated mice is typically not fruitful. Indeed, there was no statistically significant difference among untreated WT and XID mice infected with SchuS4 (Figure 1A). We recently developed a model in which WT mice are treated with low doses of LVF following SchuS4 infection that results in approximately 50% survival of WT mice, thus allowing comparisons of host cell components required for either exacerbation or resolution of infection (9). Using this model, we made the surprising observation that, following treatment with LVF, XID mice exhibited significantly greater survival of SchuS4 infection than WT controls (p=0.034) (Figure 1A). Consistent with our previous observations in C57Bl/6J mice, animals that survived to 30 days after SchuS4 infection did not have detectable numbers of SchuS4 in the lung or spleen (data not shown).
Figure 1. XID mice exhibit enhanced survival and control of SchuS4 replication compared to WT mice.
Mice were intranasally infected with 50 CFU F. tularensis strain SchuS4. As indicated, animals received 5 mg/kg levofloxacin (LVF) on days 3–16 of infection. CBA/J (WT) and CBA/CaHN-BtkXID/J (XID) mice were monitored for survival (A) or euthanized at the indicated time points for assessment of bacterial loads in the lung and spleen (B and C; n=4–5 mice/group/time point). (A) represents the results of two experiments pooled together. (B) is representative of two experiments of similar design. * = significantly less than WT mice (p<0.05). Error bars represent SEM.
WT mice that succumb to SchuS4 infection following treatment with antibiotic fail to control bacterial replication after therapy with LVF has ended (9). Thus, we hypothesized that the increased survival of XID mice treated with LVF would correlate with better control of bacterial replication compared to similarly treated WT animals. We first assessed control of bacterial burdens in among SchuS4 infected animals that had not received LVF. Interestingly, despite no difference in mean time to death, untreated XID mice had significantly fewer bacteria in the lungs (days 1 and 3) and spleen (days 2 and 3) after infection compared to untreated WT animals (Figure 1B). However, by day 4 after infection there were no significant differences in bacterial loads among untreated animals, which is consistent with the similar mean time to death observed in these groups. In contrast, following administration of LVF XID mice had significantly fewer bacteria in the lung on days 7, 10 and 14 after infection compared to LVF treated WT animals (Figure 1B). LVF treated XID mice also had significantly fewer SchuS4 organisms in the spleen on days 7 and 10 after infection compared to WT mice at these time points (Figure 1B). Following cessation of LVF therapy, and 21 days after infection, 3/5 WT mice had detectable bacteria in the lungs and all WT animals had detectable bacteria in the spleen (Figure 1C). In contrast, only 1/5 XID mice had detectable bacteria in the lung and spleen at this time point after infection (Figure 1C). The control of recrudescence of infection among LVF treated XID mice correlated with enhanced survival of these animals following infection with SchuS4 and treatment with LVF. Thus, the absence of functional btk in XID mice is associated with enhanced clearance of bacterial burdens and increased survival of SchuS4 infection.
XID mice have low concentrations of circulating antibodies directed against SchuS4
The immune defect in XID mice that results in attrition of B1a cells and hampered production of IgM and IgG antibodies by conventional B cells is mutation of Bruton's tyrosine kinase (btk). As discussed above, B1a cells contribute to the humoral response via secretion of natural and T-independent IgM antibodies. Furthermore, B1a cells have been shown to be an important source of protective IgM directed against F. tularensis LVS LPS, in that XID mice vaccinated with LVS LPS fail to survive lethal LVS infection (21). Since antibodies, and specifically antibodies derived from B1a cells, contribute to protection against attenuated strains of F. tularensis, we next determined if XID mice truly exhibited differences in their ability to secrete antibodies that recognize SchuS4. As previously established, XID mice had lower titers of total serum IgM antibodies compared to WT controls prior to infection (29), data not shown). Following exposure to SchuS4 and treatment with LVF, XID mice mounted a delayed and weaker IgM and IgG response directed SchuS4 compared to similarly treated WT controls (Figure 2A and B). Moreover, XID mice also had a delayed and lower titer of serum antibodies capable of agglutinating viable SchuS4 compared to WT mice (Figure 2C). Thus, our observation of attenuated antibody production in XID mice following SchuS4 infection and LVF therapy is consistent with previously published reports examining antibody production in these animals following infection with unrelated microorganisms (29–31). Since XID mice have lower levels of antibodies directed against SchuS4 yet exhibit better survival against infection with this bacterium, our data suggest that high, sustained, titers of anti-SchuS4 antibodies are not required to survive SchuS4 infection.
Figure 2. XID mice have delayed and lower titers of antibodies directed against SchuS4.
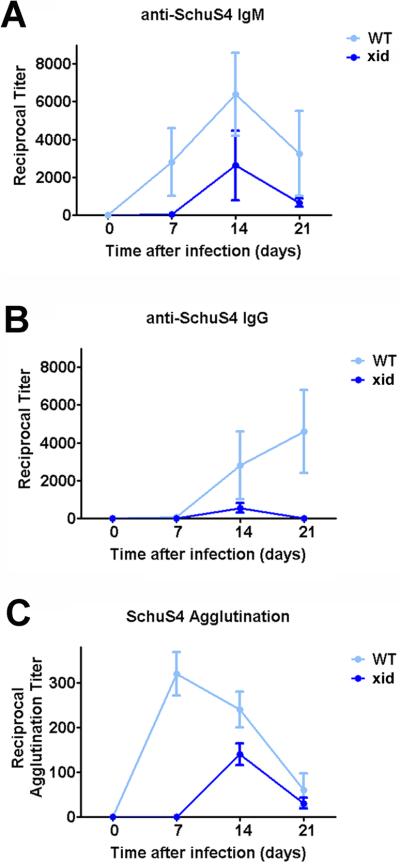
Mice (n= 4–5/group) were intranasally infected with 50 CFU F. tularensis strain SchuS4. As indicated, animals received 5 mg/kg levofloxacin on days 3–16 of infection. At the indicated time points CBA/J (WT) and CBA/CaHN-BtkXID/J (XID) mice were euthanized and serum collected. Serum was serially diluted and assessed for IgM (A) and IgG (B) directed against SchuS4 antigens by ELISA. Diluted serum was also assessed for its ability to agglutinate viable SchuS4 organisms (C). Error bars represent SEM. Data is representative of two experiments of similar design.
XID macrophages are equally susceptible to infection with SchuS4 compared to WT cells in vitro
The dampened antibody response in XID mice is attributed to the lack of functional btk in B cells and the absence of B1a cells in these animals. However, in addition to B cells, btk is also expressed in a variety of myeloid cells including macrophages (32). Among macrophages, btk participates in mediating signals from Toll-Like Receptors (TLR), cell death pathways and production of nitric oxide (33–35). Macrophages represent one of the primary cell types targeted by F. tularensis for intracellular replication. Thus, it was possible that the differences in bacterial burden we observed in vivo was due to impaired replication of SchuS4 in XID macrophages. To test this hypothesis, we compared infection and replication of SchuS4 in freshly isolated alveolar macrophages and bone marrow derived macrophages from XID and WT mice. We did not observe any statistically significant differences in uptake or replication of SchuS4 in XID macrophages compared to WT cells (Figure 3A).
Figure 3. Macrophages from XID and WT mice are not different in their interaction with SchuS4.

Macrophages were collected by bronchoalveolar lavage (alveolar) or differentiated from the bone marrow (Bone Marrow) and infected with SchuS4 at a MOI = 10. (A) At the indicated time points cells were lysed and intracellular bacteria were enumerated by plating lysates on MMH agar. (B) Twenty-four hours after infection cells were treated with Pam3CSK4. Supernatants were harvested 12 hours later and assessed for TNF-α by ELISA. Untreated, uninfected cells served as negative controls. Uninfected cells treated with Pam3CSK4 served as positive controls. *= significantly less than uninfected, Pam3CSk4 treated cells (p<0.05). Error bars represent SEM. Data is representative of three experiments similar design.
Given the involvement of btk in mediating production of inflammatory cytokines following engagement of TLRs, we also tested whether XID macrophages mounted a different inflammatory response following SchuS4 infection compared to WT controls. In agreement with our previous reports, SchuS4 failed to elicit production of TNF-α, IL-6 and IL-12p40 from WT or XID macrophages (Figure 3B and data not shown). SchuS4 also readily inhibits production of inflammatory cytokines and nitric oxide among infected macrophages (36–38). Therefore, an inability of SchuS4 to inhibit this process in XID macrophages may explain the reduced ability to replicate. However, we did not observe any differences in the inhibition of pro-inflammatory cytokines or induction of nitric oxide among XID macrophages compared to WT cells or in tissues of infected animals (Figure 3B and data not shown). Together these data suggest that the impaired control of SchuS4 infection in vivo is not likely due to altered interactions specifically between the bacteria and macrophages alone.
XID and WT mice have similar populations of inflammatory cells following SchuS4 infection
Since there were no observable differences in the interaction between SchuS4 and cells typically targeted for replication, i.e. macrophages, in vitro we next evaluated changes in populations of inflammatory cells following in vivo SchuS4 infection. We and others have previously shown that monocytes are capable of producing inflammatory cytokines associated with protection against various subspecies of Francisella (27, 39, 40). Recruitment of granulocytes, i.e. neutrophils, have also been associated with survival of infection with attenuated subspecies of Francisella (41). Thus, enhanced control of bacterial burdens among XID mice may be attributed to increased numbers of monocytes and/or granulocytes in target organs. However, we did not observe significant differences in the overall cellularity nor specific monocyte (CD11b+/Ly6C+/MHCII−/Ly6G−) or granulocyte (Ly6G+/MHCII−) populations in the lung or spleen on days 4, 7 or 10 after infection (Figure 4 and data not shown).
Figure 4. XID and WT mice do not have differences in recruitment of inflammatory cells following SchuS4 infection.
Mice (n=4–5/group) were intranasally infected with 50 CFU F. tularensis strain SchuS4. As indicated, animals received 5 mg/kg levofloxacin on days 3–6 of infection. Animals were euthanized on day 7 of infection and assessed for the indicated cell populations by flow cytometry. Monocytes were characterized as CD11b+/Ly6C+/MHCII−/Ly6G−. Granulocytes were characterized as Ly6G+/MHCII−. Error bars represent SEM. Data is representative of two experiments of similar design.
Decreased IL-10 producing B1a cells and increased IL-12 producing macrophages in XID mice in vivo
The greater control of SchuS4 infection we observed in XID mice, compared to WT animals, could not be attributed to differences in the primary interaction of bacteria with target macrophages or recruitment of specific inflammatory cells. This suggested the presence or absence of other cell types contribute to control of SchuS4 infection in XID animals. XID mice lack B1a cells that act an important source of IL-10 (42). It is well established that IL-10 can down modulate Th1 responses via inhibition of IL-12 production from macrophages which, in turn, results in weakened IFN-γ production from NK/NKT and T cells (43). IL-12 is essential for survival of SchuS4 infection (9). Given the counterbalance between IL-10 and IL-12 and reported absence of B1a cells capable of producing IL-10 in XID mice, we determined if there were differences in populations of IL-10 and/or IL-12 producing cells in XID and WT mice following SchuS4 infection.
In agreement with previous reports, uninfected WT mice had significantly higher numbers of B1a cells in the lung and spleen compared to XID mice (Figure 5A and (44). Following SchuS4 infection we did not observe significant changes in B1a populations in the lungs of WT mice, nor did we observe significant differences in B1a IL-10+ cells in this organ (Figure 5A). In contrast, WT mice had a significant increase in B1a IL-10+ cells in the spleen following infection. Furthermore, WT animals also had significantly greater numbers of IL-10+ B1a cells in the spleen before and after infection compared to XID mice (Figure 5A).
Figure 5. Control of SchuS4 infection in XID mice is correlated with fewer IL-10+ B1a cells and increased IL-12+ macrophages.
Mice (n=4–5/group) were intranasally infected with 50 CFU F. tularensis strain SchuS4. As indicated, animals received 5 mg/kg levofloxacin on days 3–6 of infection. Animals were euthanized on day 7 of infection and single cell suspensions of the lung and spleen were generated. Cells were incubated with PMA, ionomycin and BFA for 4 hours were stained for specific surface receptors, permeabilized and stained for IL-10 and IL-12 and assessed for specific cell populations by flow cytometry. B1a cells were characterized as CD5Y/CD19Y. Macrophages were characterized as CD11b+/F480Y. Error bars represent SEM. * = significantly greater than uninfected XID (p<0.05). ** = significantly greater than uninfected WT and all XID (p<0.05). *** = significantly greater than all other groups (p<0.05). Data is representative of two experiments of similar design.
IL-12 is a critical cytokine for protection against SchuS4 infection (9, 45). It is well established that IL-10 down regulates IL-12 production from macrophages (43). Thus, we also assessed the ability of macrophages (identified as CD11b+/F480+/Ly6G−) in the lungs and spleens of WT and XID mice to produce IL-10 and IL-12. WT and XID mice had similar numbers of macrophages in the lung and spleen regardless of their infection status (Figure 5B). Furthermore, unlike B1a cells we did not detect significant changes in IL-10+ macrophages among WT and XID mice following SchuS4 infection. Interestingly, and in correlation with decreased numbers of IL-10 producing B1a cells, XID mice had significantly higher numbers of IL-12+ macrophages in the lung and spleen following SchuS4 infection compared to WT animals (Figure 5B). Together these data suggests that decreased IL-10+ B1a cells and increased IL-12 producing macrophages in vivo correlate with enhanced control of SchuS4 infection in XID mice.
SchuS4 induces IL-10 production by B1a cells to inhibit IL-12
To determine if B1a cells could produce IL-10 in response to SchuS4 we isolated B1a, B1b and macrophages from the peritoneal cavity of resting mice and cultured each population of cells in the presence of live or dead SchuS4. In agreement with previous work assessing activity of B1a cells in models of unrelated infectious disease (48–52), B1a cells were the primary source of IL-10 in the peritoneum following co-culture with either live or dead SchuS4 (Supplemental Figure 2 and data not shown). We also confirmed that XID mice lacked B1a cells in the peritoneum and, as expected, PEC from XID mice produced significantly less IL-10 in response to SchuS4 than PEC isolated from wild type animals (Supplemental Figure 2).
IL-10 is a potent inhibitor of IL-12 responses (43). Thus, we hypothesized that the IL-10 produced by B1a cells exposed to SchuS4 would inhibit IL-12 production. Since the peritoneal cavity is comprised primarily of B cells and macrophages we took advantage of this compartment to test our hypothesis. Neutralization of IL-10 in PEC cultures treated with killed SchuS4 significantly enhanced production of IL-12p40 (Figure 6). Interestingly, although live SchuS4 can provoke production of IL-10 from B1a cells, neutralization of IL-10 in those cultures does not significantly increase production of IL-12 (data not shown). However, it is well established that viable SchuS4 readily inhibits production of IL-12. Thus, the absence of IL-12 in cultures stimulated with live SchUS4 where B1a cell derived IL-10 is neutralized is likely due to the suppressive effect viable SchuS4 is exerting on transcription and translation of IL-12. Together, our data demonstrate that B1a cell derived IL-10 can effectively inhibit the ability of macrophages to produce IL-12 in response to dead SchuS4.
Figure 6. B1a cells derived IL-10 inhibits IL-12p40.
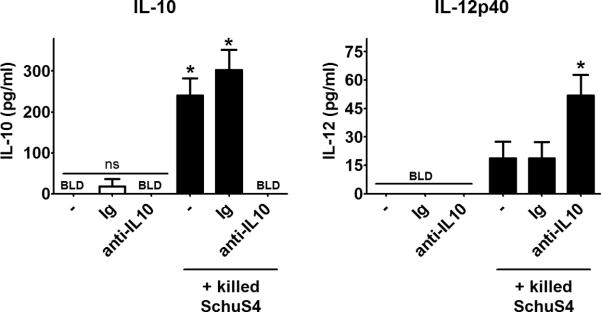
Peritoneal exudate cells were collected from resting WT mice and were cultured with killed SchuS4 overnight. As indicated, some cells were either left untreated (−), or were incubated with rat immunoglobulin (Ig) or neutralizing anti-IL-10 antibodies (both at 10 ug/ml). Cultures were assessed for IL-10 and IL-12p40. * = significantly different untreated and Ig controls (p<0.05). Error bars represent SEM. Data is pooled from three experiments of similar design.
Adoptive transfer of B1a cells exacerbates SchuS4 infection
The data presented above suggested that increased presence of serum antibodies directed against SchuS4 or IL-10 producing B1a cells contributed to exacerbation of SchuS4 infection in CBA/J mice. To determine if this was the case, we first examined the effect of passively transferred serum collected from SchuS4 infected WT mice or uninfected controls on the replication of SchuS4 in XID mice. Passive transfer of serum, regardless of the source, had no effect on the burden of SchuS4 in the lungs and spleens of XID mice (data not shown). We then examined the effect adoptive transfer of B1a cells into XID mice would have on tissue burden of SchuS4. One day prior to infection with SchuS4, XID mice received B1a cells enriched from the peritoneal cavities of WT mice. We confirmed that B1a cells obtained from the peritoneal cavity were a primary source of IL-10 in additional in vitro assays (Supplemental Figure 2). XID mice that received WT B1a cells had significantly higher numbers of SchuS4 in the lung and spleen compared to sham inoculated controls (Figure 7A). Although, adoptive transfer of B1a cells into
Figure 7. B1a cells contribute to exacerbation of SchuS4 infection.

B1a cells were collected from the peritoneal cavities of CBA/J (wild type) mice and enriched by FACS. One day prior to infection XID mice (n=4–5/group) were injected with freshly isolated wild type B1a cells or PBS (untreated). Mice were intranasally infected with 50 CFU F. tularensis strain SchuS4. Animals received 5 mg/kg levofloxacin on days 3–6 of infection. (A) Mice were euthanized on day 7 of infection and bacteria were enumerated from the lung and spleen. (B) Survival of mice receiving B1a cells versus untreated controls. * = significantly different untreated controls (p<0.05). Error bars represent SEM. Data is representative of three experiments of similar design.
XID mice did not reduce the surviving number of animals to the same percent observed in WT mice, adoptive transfer of B1a cells into XID mice decreased survival of XID mice infected with SchuS4 compared to untreated controls (Figure 7B). Moreover, unlike untreated XID mice, survival of XID mice receiving B1a cells was not significantly different from WT animals. Thus, absence of IL-10 producing B1a cells is one facet to the enhanced control of SchuS4 infection among XID mice compared to WT animals.
Increased IFN-γ producing NK/NKT cells in XID mice
IL-10 derived from B1a cells can act on a variety of cell types. Our data suggest that presence of IL-10 producing B1a cells is inversely correlated with IL-12+ macrophages and contribute toward exacerbated SchuS4 infection. Furthermore, we established that IL-10 produced by B1a cells in response to SchuS4 inhibits the ability of cells to produce IL-12. As discussed above, IL-12 is an essential cytokine for the propagation of Th1 responses, namely through its ability to induce production of IFN-γ from NK/NKT and T cells. In addition to IL-12, we have also shown that IFN-γ is critical for survival of SchuS4 infection (9). Thus, we postulated that the increase in IL-12+ macrophages observed in XID mice after SchuS4 infection (Figure 4B) would correlate with increased numbers of IFN-γ+ lymphocytic cells in the lung and spleen. We first determined if there was an increase in the specific lymphocyte populations in the organs of WT and XID mice after SchuS4 infection. We detected expansion of CD4+, CD8+ and NKp46+ cells in the lung and spleen of both WT and XID mice (Figure 8). XID mice had significantly higher numbers of each of these cell types compared to WT controls in the lung and significantly higher numbers of NK/NKT cells in the spleen (Figure 8A). However, despite the overall increase in CD4+ and CD8+ cells observed in XID mice, there were no differences in the number of CD4+IFN-γ+ or CD8+IFN-γ+ compared to WT controls (Figure 8B). In contrast, XID mice had significantly more IFN-γ+ NKp46+ cells compared to WT mice at this time point after infection (Figure 8B).
Figure 8. XID mice have significantly more IFN-γ+ NK/NKT cells after SchuS4 infection compared to WT mice.
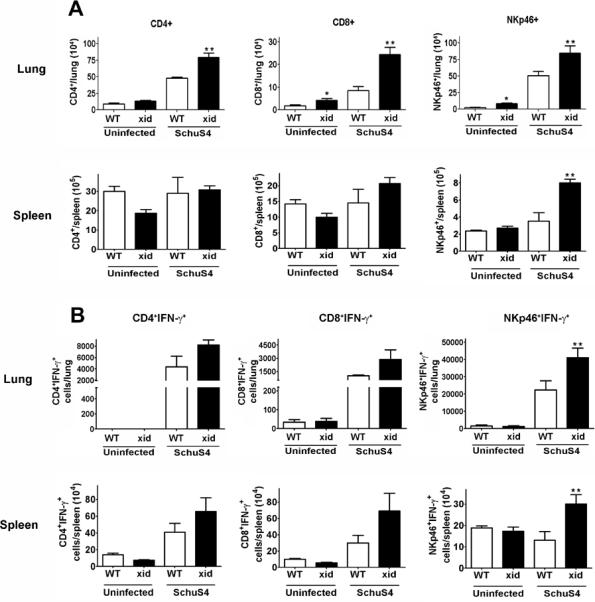
Mice (n=4–5/group) were intranasally infected with 50 CFU F. tularensis strain SchuS4. As indicated, animals received 5 mg/kg levofloxacin on days 3–6 of infection. Animals were euthanized on day 7 of infection and single cell suspensions of the lung and spleen were generated. Cells were incubated with PMA, ionomycin, and BFA for 4 hours, were stained for specific surface receptors, permeabilized and stained for IFN-γ and assessed for specific cell populations by flow cytometry. Error bars represent SEM. * = significantly greater than uninfected WT (p<0.05). ** = significantly greater than all other groups (p<0.05). Data is representative of two experiments of similar design.
NK/NKT cells contribute toward resolution of SchuS4 infection in XID mice
Given the significantly higher numbers of IFN-γ+ NK/NKT cells observed in both the lungs and spleens of XID mice following SchuS4 infection and the critical role IFN-γ plays in resolution of tularemia (Figure 8 and (9)), we next assessed the contribution NK/NKT cells made in control of SchuS4 infection in XID mice. XID mice depleted of NK/NKT cells had significantly higher numbers of bacteria in the spleen compared to animals treated with rabbit immunoglobulin on day 7 after infection (Figure 9). Although not significantly different, depletion of NK/NKT cells in XID mice also increased bacterial burdens in the lungs on day 7 and 21 after infection (Figure 9). Notably, depletion of NK/NKT cells reversed the clearance of SchuS4 in the lungs of XID mice when assessed 21 days after infection (Figure 9). Thus, NK/NKT cells contribute to the control of SchuS4 replication in XID mice.
Figure 9. NK/NKT cells contribute to control of SchuS4 infection in XID mice.

Mice were treated with anti-asialo GM1 (anti-AGM1) antibodies to deplete NK/NKT cells or rabbit immunoglobulin (Ig) as a negative control on days −2, +1, +4, +7, +10. +13, +16, +19 of infection. Mice were intranasally infected with 50 CFU F. tularensis strain SchuS4. Animals received 5 mg/kg levofloxacin on days 3–6 of infection. All mice were euthanized on day 7 and 21 of infection and bacteria were enumerated from the lung and spleen. * = significantly different Ig treated controls (p<0.05). Error bars represent SEM. Data is representative of two experiments of similar design.
DISCUSSION
B cells have a variety of functions during development of immune responses to infection. In addition to serving as antibody producing cells, B cells also act as antigen presenting cells, participate in activation of T cells, and secrete cytokines that may contribute to the control or exacerbation of infection. There is growing evidence and understanding that these functions are often associated with specific subsets of B cells. For example, B1a cells are an important subset B cells since they are the primary producers of natural IgM, IgM antibodies directed against T-independent antigens, and are central source of IL-10 (42, 46, 47). Thus, B1a cells can act in a protective role during infection via their secretion of IgM that aids in opsonization and killing of pathogens, or in a negative role via their production of IL-10 which largely functions to dampen Th1 responses.
In the report presented herein, we demonstrate that B1a cells exacerbate infections mediated by virulent F. tularensis strain SchuS4. Specifically, mice largely deficient for B1a cells and signaling via the B cell receptor (BCR), i.e. XID mice, exhibit better survival following intranasal infection with SchuS4. This survival was correlated with superior control of bacterial loads in the lung and spleen. Additionally, adoptive transfer of B1a cells from WT animals into XID mice resulted in increased bacterial loads and reduced survival following SchuS4 infection (Figure 7). Furthermore, our data also suggests that the increased resistance to SchuS4 infection observed in XID mice is not correlated with intrinsic defect in macrophages found in these mice. For example, both bone marrow derived macrophages and freshly isolated alveolar macrophages obtained from XID mice phagocytosed and supported SchuS4 replication similarly to that observed in WT macrophages. Although we did not directly test the ability of dendritic cells and neutrophils from XID mice to respond to SchuS4 infection, we failed to observe differences in WT and XID mice that could be associated with defects in responses of other cell type in these animals, e.g. there were no differences in the generation of nitric oxide, recruitment of inflammatory cells, or ability of SchuS4 to suppress production of inflammatory cytokines. Thus, the primary difference in XID mice that is associated with survival of SchuS4 infection is the absence of B1a cells. Moreover, our findings point toward an indirect function of B1a cells on other cells via their secretion of IL-10.
Initially, our finding that XID mice were more resistant to infection with SchuS4 was surprising. In our previous study using mice completely deficient for all B cells (μMT−/−), we established that B cells are critical for survival of SchuS4 infection (9). Thus, we predicted that XID mice with their inherent defects in BCR signaling and inability to mount high titers against SchuS4 would be equally susceptible to SchuS4 infection as observed in μMT−/− animals. Furthermore, it has been repeatedly shown that XID mice have increased susceptibility against a wide range of pathogens, including intracellular bacteria (48–52). With regard to infection mediated by attenuated F. tularensis strain LVS, immunization with LVS LPS failed to protect XID mice from infection with LVS (21). The inability of XID mice to develop protective immunity in this report was specifically correlated with the absence of antibody directed against LVS LPS produced by B1a cells. A potential role for natural antibody produced by B1a cells in protection against F. tularensis was also suggested in a recent report (53). Together these data suggested that B1a cells would play a critical role in the early control of F. tularensis infections via their ability to produce antibodies directed against surface antigens.
However, our data suggests that antibodies derived from B1a cells do not play a significant role in the resolution or exacerbation of infection mediated by virulent F. tularensis SchuS4. XID mice have nearly undetectable levels of natural IgM antibodies and develop very poor titers of antibodies directed against SchuS4. Furthermore, passive transfer of serum antibodies from infected WT mice had no impact on bacterial burdens in XID mice (data not shown). Thus, the enhanced clearance of SchuS4 observed in XID mice and the improved survival of these animals compared to WT controls suggests that neither natural IgM antibodies nor antibodies that recognize SchuS4 are required for survival of tularemia.
In contrast to a protective function of B1a cells in tularemia, our data clearly demonstrates that presence of these cells exacerbates infection with F. tularensis SchuS4 resulting in increased mortality under the setting of antibiotic therapy. The importance of B1a cells in mediating inhibitory effects on protection against SchuS4 infection, rather than other cells types affected by the defect in Bruton's tyrosine kinase in XID mice, was confirmed in two ways. First, we established that there were no detectable differences in the ability of macrophages derived from XID mice to support SchuS4 infection or replication compared to WT cells (Figure 3). Second, adoptive transfer of WT B1a cells into XID mice resulted in significant increase in replication of SchuS4 in the lung and spleen (Figure 7). Thus, B1a cells (which are largely absent in XID mice) negatively impact the outcome of SchuS4 infection. Initially, these data appear to be in conflict with our previous observation that the presence of an intact B cell compartment is required to survive SchuS4 infection (9). However, it should be noted that XID mice still retain conventional B cells. Although these cells would be poor producers of antibody in XID mice (due to the lack of signaling through the BCR), these conventional B cells may have other functions that support resolution of SchuS4 infection, e.g. antigen presentation and secretion of protective cytokines (2).
We and others have shown that Th1 type immune responses and IFN-γ are required for survival of F. tularensis infection (9, 54, 55). IL-10 derived from B1a cells is well-established for its ability to interfere with Th1 type immune. Indeed, there are examples of B1a cell IL-10 indirectly limiting IFN-γ mediated protective immune responses against a variety of infections (56–58). Thus, we next addressed if B1a cells may function similarly in our model to inhibit protective immune responses during SchuS4 infection. Our data suggest that the negative role B1a cells play in SchuS4 infection is via indirect modulation of IFN-γ response by NK/NKT via secretion of IL-10. It is important to emphasize that the role for B1a cell derived IL-10 in modulating IFN-γ production is indirect. IL-10 is a potent anti-inflammatory cytokine that can exert its activity on a wide range of host cells including macrophages and dendritic cells (DC) (43), one of which is to limit production of IL-12. IL-12 is an important cytokine for induction of IFN-γ responses in NK/NKT cells. Thus, IL-10 indirectly affects IFN-γ production via its ability to down regulate IL-12 production in macrophages and DC. For this paradigm to be true in our system, one would predict that the absence of IL-10 producing B1a cells should lead to an increase in IL-12 production by macrophages, resulting in an increase in IFN-γ producing NK/NKT and/or T cells. In support of this hypothesis, we first demonstrated that B1a cells respond to killed SchuS4 via production of IL-10. Neutralization of B1a cell derived IL-10 in this in vitro system resulted in increased secretion of IL-12. Similarly, in vivo we found that the absence of IL-10 producing B1a cells in XID mice was inversely correlated with the presence of IL-12+ macrophages in the spleens and lungs of infected animals (Figure 5) and that this increase in IL-12+ macrophages was also correlated with significantly higher numbers of IFN-γ producing NK/NKT cells in XID mice (Figure 8).
The presence of protective and inhibitory cytokines in this model is unlike what occurs in cases of untreated tularemia. When antibiotic treatment is withheld, there is a striking absence of cytokines until the last 24–36 hours of infection, at which time one observes a “cytokine storm” (59–61). The lack of cytokine production during early, critical stages of disease is associated with the impressive ability of virulent strains of SchuS4 to evade and inhibit inflammatory responses in the host (24, 62, 63). How then does one elicit IL-10 and IL-12 following infection with SchuS4 during treatment with antibiotic? We propose that induction of host cytokines after day 4 of infection are a result of both the release of damage associated molecular pattern molecules (DAMPs) from dying host cells and accumulation of lysed and/or dying bacteria. In support of this hypothesis, exposure of B1a cells to SchuS4 killed with levofloxacin readily induces production of IL-10 (Supplemental Figure 2 and Figure 6) by these host cells. The specific host cell receptors and mechanism by which IL-10 is liberated by B1a cells is under intense study in our laboratory.
Together our data suggest the following model (Figure 10). Dead and/or lysing SchuS4 provokes IL-12 from macrophages which then acts on NK/NKT cells to produce IFN-γ. IFN-γ activates newly infected macrophages to kill SchuS4. However, in the presence of B1a cells the dead bacteria provoke a potent IL-10 response. This IL-10 significantly limits IL-12 production from macrophages, resulting in suboptimal production of IFN-γ from local NK/NKT cells. Thus, induction or absence of IL-10 by B1a cells serves as a key pivot point for early control of SchuS4 burdens in target organs and deciding factor for survival of tularemia infection. Finally, our data highlights an important mechanism for cross-talk between a specific subset of B cells, macrophages and NK/NKT cells that significantly impacts the outcome of infection with a highly virulent pathogen.
Figure 10. The role of B1a cells in tularemia.
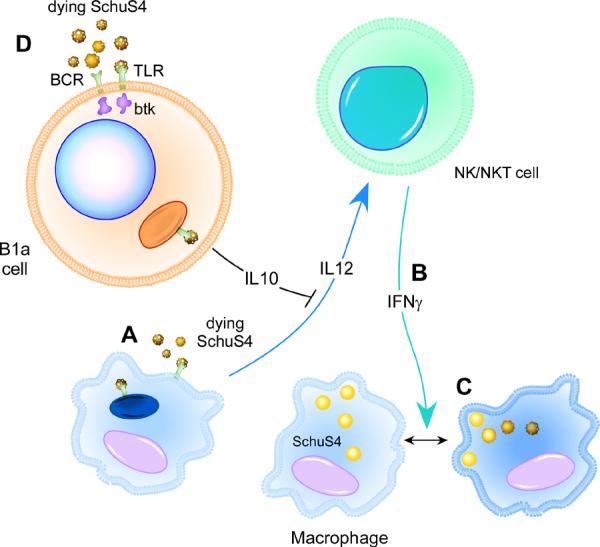
(A) Resident macrophages produce IL-12 in response to dead and/or lysing SchuS4 following antibiotic therapy. (B) IL-12 provokes NK/NKT cells to produce IFN-γ, which then (C) activates newly infected macrophages to kill SchuS4. However, B1a cells secrete IL-10 in response to dead bacteria (D). This IL-10 down regulates IL-12 production from macrophages, which in turn results in suboptimal IFN-γ responses from NK/NKT cells and poor control of SchuS4 replication among infected macrophages.
Supplementary Material
Acknowledgements
The authors wish to thank Mr. Aaron Carmody for his assistance in the FACS isolation of B1a cells, Ms. Anita Mora and Ms. Heather Murphy for generation of model cartoon, and Dr. Jay Carroll for his critical assessment of our manuscript and helpful suggestions.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
REFERENCES
- 1.Swanson CL, Pelanda R, Torres RM. Division of labor during primary humoral immunity. Immunol Res. 2012 doi: 10.1007/s12026-012-8372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lund FE, Randall TD. Effector and regulatory B cells: modulators of CD4(+) T cell immunity. Nat Rev Immunol. 2010;10:236–247. doi: 10.1038/nri2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clemens DL, Lee BY, Horwitz MA. Francisella tularensis phagosomal escape does not require acidification of the phagosome. Infect Immun. 2009;77:1757–1773. doi: 10.1128/IAI.01485-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fortier AH, Leiby DA, Narayanan RB, Asafoadjei E, Crawford RM, Nacy CA, Meltzer MS. Growth of Francisella tularensis LVS in macrophages: the acidic intracellular compartment provides essential iron required for growth. Infect Immun. 1995;63:1478–1483. doi: 10.1128/iai.63.4.1478-1483.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mc CF, Jr., Snyder MJ, Woodward TE. Studies on human infection with Pasteurella tularensis; comparison of streptomycin and chloramphenicol in the prophylaxis of clinical disease. Trans Assoc Am Physicians. 1957;70:74–79. discussion 79–80. [PubMed] [Google Scholar]
- 6.Risi GF, Pombo DJ. Relapse of tularemia after aminoglycoside therapy: case report and discussion of therapeutic options. Clin Infect Dis. 1995;20:174–175. doi: 10.1093/clinids/20.1.174. [DOI] [PubMed] [Google Scholar]
- 7.Harris S. Japanese biological warfare research on humans: a case study of microbiology and ethics. Ann N Y Acad Sci. 1992;666:21–52. doi: 10.1111/j.1749-6632.1992.tb38021.x. [DOI] [PubMed] [Google Scholar]
- 8.Elkins KL, MacIntyre AT, Rhinehart-Jones TR. Nonspecific early protective immunity in Francisella and Listeria infections can be dependent on lymphocytes. Infect Immun. 1998;66:3467–3469. doi: 10.1128/iai.66.7.3467-3469.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crane DD, Scott DP, Bosio CM. Generation of a Convalescent Model of Virulent Francisella tularensis Infection for Assessment of Host Requirements for Survival of Tularemia. PLoS ONE. 2012;7:e33349. doi: 10.1371/journal.pone.0033349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drabick JJ, Narayanan RB, Williams JC, Leduc JW, Nacy CA. Passive protection of mice against lethal Francisella tularensis (live tularemia vaccine strain) infection by the sera of human recipients of the live tularemia vaccine. The American journal of the medical sciences. 1994;308:83–87. doi: 10.1097/00000441-199408000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Hickey AJ, Hazlett KR, Kirimanjeswara GS, Metzger DW. Identification of Francisella tularensis outer membrane protein A (FopA) as a protective antigen for tularemia. Vaccine. 2011;29:6941–6947. doi: 10.1016/j.vaccine.2011.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirimanjeswara GS, Golden JM, Bakshi CS, Metzger DW. Prophylactic and therapeutic use of antibodies for protection against respiratory infection with Francisella tularensis. J Immunol. 2007;179:532–539. doi: 10.4049/jimmunol.179.1.532. [DOI] [PubMed] [Google Scholar]
- 13.Lavine CL, Clinton SR, Angelova-Fischer I, Marion TN, Bina XR, Bina JE, Whitt MA, Miller MA. Immunization with heat-killed Francisella tularensis LVS elicits protective antibody-mediated immunity. Eur J Immunol. 2007;37:3007–3020. doi: 10.1002/eji.200737620. [DOI] [PubMed] [Google Scholar]
- 14.Mara-Koosham G, Hutt JA, Lyons CR, Wu TH. Antibodies contribute to effective vaccination against respiratory infection by type A Francisella tularensis strains. Infect Immun. 2011;79:1770–1778. doi: 10.1128/IAI.00605-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pammit MA, Raulie EK, Lauriano CM, Klose KE, Arulanandam BP. Intranasal vaccination with a defined attenuated Francisella novicida strain induces gamma interferon-dependent antibody-mediated protection against tularemia. Infect Immun. 2006;74:2063–2071. doi: 10.1128/IAI.74.4.2063-2071.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savitt AG, Mena-Taboada P, Monsalve G, Benach JL. Francisella tularensis infection-derived monoclonal antibodies provide detection, protection, and therapy. Clin Vaccine Immunol. 2009;16:414–422. doi: 10.1128/CVI.00362-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stenmark S, Lindgren H, Tarnvik A, Sjostedt A. Specific antibodies contribute to the host protection against strains of Francisella tularensis subspecies holarctica. Microb Pathog. 2003;35:73–80. doi: 10.1016/s0882-4010(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 18.Lu Z, Madico G, Roche MI, Wang Q, Hui JH, Perkins HM, Zaia J, Costello CE, Sharon J. Protective B-cell epitopes of Francisella tularensis O-polysaccharide in a mouse model of respiratory tularaemia. Immunology. 2012;136:352–360. doi: 10.1111/j.1365-2567.2012.03589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Z, Roche MI, Hui JH, Unal B, Felgner PL, Gulati S, Madico G, Sharon J. Generation and characterization of hybridoma antibodies for immunotherapy of tularemia. Immunol Lett. 2007;112:92–103. doi: 10.1016/j.imlet.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foshay L. Tularemia: A summary of certain aspects of the disease including methods for early diagnosis and the results of serum treatment in 600 patients. Medicine. 1940;19:1–83. [Google Scholar]
- 21.Cole LE, Yang Y, Elkins KL, Fernandez ET, Qureshi N, Shlomchik MJ, Herzenberg LA, Vogel SN. Antigen-specific B-1a antibodies induced by Francisella tularensis LPS provide long-term protection against F. tularensis LVS challenge. Proc Natl Acad Sci U S A. 2009;106:4343–4348. doi: 10.1073/pnas.0813411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bosio CM, Elkins KL. Susceptibility to secondary Francisella tularensis live vaccine strain infection in B-cell-deficient mice is associated with neutrophilia but not with defects in specific T-cell-mediated immunity. Infect Immun. 2001;69:194–203. doi: 10.1128/IAI.69.1.194-203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elkins KL, Rhinehart-Jones TR, Culkin SJ, Yee D, Winegar RK. Minimal requirements for murine resistance to infection with Francisella tularensis LVS. Infect Immun. 1996;64:3288–3293. doi: 10.1128/iai.64.8.3288-3293.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosio CM, Bielefeldt-Ohmann H, Belisle JT. Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J Immunol. 2007;178:4538–4547. doi: 10.4049/jimmunol.178.7.4538. [DOI] [PubMed] [Google Scholar]
- 25.Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. Am J Physiol Lung Cell Mol Physiol. 2009;296:L936–946. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warfield KL, Perkins JG, Swenson DL, Deal EM, Bosio CM, Aman MJ, Yokoyama WM, Young HA, Bavari S. Role of natural killer cells in innate protection against lethal ebola virus infection. J Exp Med. 2004;200:169–179. doi: 10.1084/jem.20032141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chase JC, Bosio CM. The presence of CD14 overcomes evasion of innate immune responses by virulent Francisella tularensis in human dendritic cells in vitro and pulmonary cells in vivo. Infect Immun. 2010;78:154–167. doi: 10.1128/IAI.00750-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson RV, Crane DD, Bosio CM. Long lived protection against pneumonic tularemia is correlated with cellular immunity in peripheral, not pulmonary, organs. Vaccine. 2010;28:6562–6572. doi: 10.1016/j.vaccine.2010.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perlmutter RM, Nahm M, Stein KE, Slack J, Zitron I, Paul WE, Davie JM. Immunoglobulin subclass-specific immunodeficiency in mice with an X-linked B-lymphocyte defect. J Exp Med. 1979;149:993–998. doi: 10.1084/jem.149.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pinschewer DD, Ochsenbein AF, Satterthwaite AB, Witte ON, Hengartner H, Zinkernagel RM. A Btk transgene restores the antiviral TI-2 antibody responses of xid mice in a dose-dependent fashion. Eur J Immunol. 1999;29:2981–2987. doi: 10.1002/(SICI)1521-4141(199909)29:09<2981::AID-IMMU2981>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 31.Reale MA, Bona CA, Schulman JL. Isotype profiles of anti-influenza antibodies in mice bearing the xid defect. J Virol. 1985;53:425–429. doi: 10.1128/jvi.53.2.425-429.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Weers M, Verschuren MC, Kraakman ME, Mensink RG, Schuurman RK, van Dongen JJ, Hendriks RW. The Bruton's tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. 1993;23:3109–3114. doi: 10.1002/eji.1830231210. [DOI] [PubMed] [Google Scholar]
- 33.Honda F, Kano H, Kanegane H, Nonoyama S, Kim ES, Lee SK, Takagi M, Mizutani S, Morio T. The kinase Btk negatively regulates the production of reactive oxygen species and stimulation-induced apoptosis in human neutrophils. Nat Immunol. 2012;13:369–378. doi: 10.1038/ni.2234. [DOI] [PubMed] [Google Scholar]
- 34.Jefferies CA, Doyle S, Brunner C, Dunne A, Brint E, Wietek C, Walch E, Wirth T, O'Neill LA. Bruton's tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J Biol Chem. 2003;278:26258–26264. doi: 10.1074/jbc.M301484200. [DOI] [PubMed] [Google Scholar]
- 35.Mangla A, Khare A, Vineeth V, Panday NN, Mukhopadhyay A, Ravindran B, Bal V, George A, Rath S. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood. 2004;104:1191–1197. doi: 10.1182/blood-2004-01-0207. [DOI] [PubMed] [Google Scholar]
- 36.Crane DD, Warner SL, Bosio CM. A novel role for plasmin-mediated degradation of opsonizing antibody in the evasion of host immunity by virulent, but not attenuated, Francisella tularensis. J Immunol. 2009;183:4593–4600. doi: 10.4049/jimmunol.0901655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ireland R, Olivares-Zavaleta N, Warawa JM, Gherardini FC, Jarrett C, Hinnebusch BJ, Belisle JT, Fairman J, Bosio CM. Effective, broad spectrum control of virulent bacterial infections using cationic DNA liposome complexes combined with bacterial antigens. PLoS Pathog. 2010;6:e1000921. doi: 10.1371/journal.ppat.1000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Telepnev M, Golovliov I, Grundstrom T, Tarnvik A, Sjostedt A. Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell Microbiol. 2003;5:41–51. doi: 10.1046/j.1462-5822.2003.00251.x. [DOI] [PubMed] [Google Scholar]
- 39.Butchar JP, Cremer TJ, Clay CD, Gavrilin MA, Wewers MD, Marsh CB, Schlesinger LS, Tridandapani S. Microarray analysis of human monocytes infected with Francisella tularensis identifies new targets of host response subversion. PLoS ONE. 2008;3:e2924. doi: 10.1371/journal.pone.0002924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parsa KV, Ganesan LP, Rajaram MV, Gavrilin MA, Balagopal A, Mohapatra NP, Wewers MD, Schlesinger LS, Gunn JS, Tridandapani S. Macrophage pro-inflammatory response to Francisella novicida infection is regulated by SHIP. PLoS Pathog. 2006;2:e71. doi: 10.1371/journal.ppat.0020071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sjostedt A, Conlan JW, North RJ. Neutrophils are critical for host defense against primary infection with the facultative intracellular bacterium Francisella tularensis in mice and participate in defense against reinfection. Infect Immun. 1994;62:2779–2783. doi: 10.1128/iai.62.7.2779-2783.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O'Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur J Immunol. 1992;22:711–717. doi: 10.1002/eji.1830220314. [DOI] [PubMed] [Google Scholar]
- 43.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 44.Forrester LM, Ansell JD, Micklem HS. Development of B lymphocytes in mice heterozygous for the X-linked immunodeficiency (xid) mutation. xid inhibits development of all splenic and lymph node B cells at a stage subsequent to their initial formation in bone marrow. J Exp Med. 1987;165:949–958. doi: 10.1084/jem.165.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duckett NS, Olmos S, Durrant DM, Metzger DW. Intranasal interleukin-12 treatment for protection against respiratory infection with the Francisella tularensis live vaccine strain. Infect Immun. 2005;73:2306–2311. doi: 10.1128/IAI.73.4.2306-2311.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hayakawa K, Hardy RR, Herzenberg LA. Peritoneal Ly-1 B cells: genetic control, autoantibody production, increased lambda light chain expression. Eur J Immunol. 1986;16:450–456. doi: 10.1002/eji.1830160423. [DOI] [PubMed] [Google Scholar]
- 47.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 48.Al-Qaoud KM, Fleischer B, Hoerauf A. The Xid defect imparts susceptibility to experimental murine filariosis--association with a lack of antibody and IL-10 production by B cells in response to phosphorylcholine. Int Immunol. 1998;10:17–25. doi: 10.1093/intimm/10.1.17. [DOI] [PubMed] [Google Scholar]
- 49.Eguchi M, Kikuchi Y. Binding of Salmonella-specific antibody facilitates specific T cell responses via augmentation of bacterial uptake and induction of apoptosis in macrophages. J Infect Dis. 2010;201:62–70. doi: 10.1086/648615. [DOI] [PubMed] [Google Scholar]
- 50.Kadioglu A, Andrew PW. Susceptibility and resistance to pneumococcal disease in mice. Briefings in functional genomics & proteomics. 2005;4:241–247. doi: 10.1093/bfgp/4.3.241. [DOI] [PubMed] [Google Scholar]
- 51.Lim PL, Choy WF, Chan ST, Leung DT, Ng SS. Transgene-encoded antiphosphorylcholine (T15+) antibodies protect CBA/N (xid) mice against infection with Streptococcus pneumoniae but not Trichinella spiralis. Infect Immun. 1994;62:1658–1661. doi: 10.1128/iai.62.5.1658-1661.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McSorley SJ, Jenkins MK. Antibody is required for protection against virulent but not attenuated Salmonella enterica serovar typhimurium. Infect Immun. 2000;68:3344–3348. doi: 10.1128/iai.68.6.3344-3348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwartz JT, Barker JH, Long ME, Kaufman J, McCracken J, Allen LA. Natural IgM Mediates Complement-Dependent Uptake of Francisella tularensis by Human Neutrophils via Complement Receptors 1 and 3 in Nonimmune Serum. J Immunol. 2012;189:3064–3077. doi: 10.4049/jimmunol.1200816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conlan JW, KuoLee R, Shen H, Webb A. Different host defences are required to protect mice from primary systemic vs pulmonary infection with the facultative intracellular bacterial pathogen, Francisella tularensis LVS. Microb Pathog. 2002;32:127–134. doi: 10.1006/mpat.2001.0489. [DOI] [PubMed] [Google Scholar]
- 55.Lopez MC, Duckett NS, Baron SD, Metzger DW. Early activation of NK cells after lung infection with the intracellular bacterium, Francisella tularensis LVS. Cell Immunol. 2004;232:75–85. doi: 10.1016/j.cellimm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 56.Hoerauf A, Solbach W, Lohoff M, Rollinghoff M. The Xid defect determines an improved clinical course of murine leishmaniasis in susceptible mice. Int Immunol. 1994;6:1117–1124. doi: 10.1093/intimm/6.8.1117. [DOI] [PubMed] [Google Scholar]
- 57.Santos-Lima EC, Vasconcellos R, Reina-San-Martin B, Fesel C, Cordeiro-Da-Silva A, Berneman A, Cosson A, Coutinho A, Minoprio P. Significant association between the skewed natural antibody repertoire of Xid mice and resistance to Trypanosoma cruzi infection. Eur J Immunol. 2001;31:634–645. doi: 10.1002/1521-4141(200102)31:2<634::aid-immu634>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 58.Zhao YX, Abdelnour A, Holmdahl R, Tarkowski A. Mice with the xid B cell defect are less susceptible to developing Staphylococcus aureus-induced arthritis. J Immunol. 1995;155:2067–2076. [PubMed] [Google Scholar]
- 59.Andersson H, Hartmanova B, Kuolee R, Ryden P, Conlan W, Chen W, Sjostedt A. Transcriptional profiling of host responses in mouse lungs following aerosol infection with type A Francisella tularensis. J Med Microbiol. 2006;55:263–271. doi: 10.1099/jmm.0.46313-0. [DOI] [PubMed] [Google Scholar]
- 60.Conlan JW, Zhao X, Harris G, Shen H, Bolanowski M, Rietz C, Sjostedt A, Chen W. Molecular immunology of experimental primary tularemia in mice infected by respiratory or intradermal routes with type A Francisella tularensis. Mol Immunol. 2008;45:2962–2969. doi: 10.1016/j.molimm.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mares CA, Ojeda SS, Morris EG, Li Q, Teale JM. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infect Immun. 2008;76:3001–3010. doi: 10.1128/IAI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bauler TJ, Chase JC, Bosio CM. IFN-{beta} Mediates Suppression of IL-12p40 in Human Dendritic Cells following Infection with Virulent Francisella tularensis. J Immunol. 2011;187:1845–1855. doi: 10.4049/jimmunol.1100377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chase JC, Celli J, Bosio CM. Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect Immun. 2009;77:180–195. doi: 10.1128/IAI.00879-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.