

Abstract
Rodent models are less suitable for predicting drug-drug interactions at the level of the human intestinal mucosa, especially when nuclear receptors like pregnane X receptor (PXR) are involved. Recently, a transgenic mouse model, expressing both human PXR and CYP3A4, was developed and shown to be a better predictor of CYP3A4 induction by xenobiotics in humans as compared to wild-type mice. In the present study, we tested the hypothesis that this mouse model can also predict PXR-mediated induction of intestinal P-gp in humans. By use of the in situ intestinal perfusion technique with mesenteric blood sampling, the effect of oral rifampicin treatment on intestinal permeability for the HIV protease inhibitor darunavir, a dual CYP3A4/P-gp substrate, was investigated. Rifampicin treatment lowered the intestinal permeability for darunavir by 50 % compared to non-treated mice. The P-gp inhibitor GF120918 increased the permeability for darunavir by 400 % in rifampicin-treated mice, while this was only 56 % in mice that were not treated, thus indicating P-gp induction by rifampicin. The non-specific P450 inhibitor aminobenzotriazole (100 μM) did not affect the permeability for darunavir. Quantitative Western blot analysis of the intestinal tissue showed that rifampicin treatment induced intestinal P-gp levels four-fold, while CYP3A4 levels remained unchanged. Oral co-administration of rifampicin with the phytochemical sulforaphane for three days increased the permeability for darunavir by 50 % compared to rifampicin treatment alone. These data show that PXR/CYP3A4-humanized mice can be used to study the inducing effects of xenobiotics on intestinal P-gp.
Keywords: In situ intestinal perfusion, humanized mouse, PXR, P-glycoprotein, rifampicin, darunavir
Introduction
In order to investigate intestinal drug absorption, several techniques are currently being used, including cell culture models, animal intestinal tissue, and in vivo experiments. Drug transport across Caco-2 cells (only for passively transported drugs)1 and rodent intestinal tissue2,3 provides a reliable indication of the fraction absorbed in humans. Unfortunately, these models are less suitable for predicting drug-drug interactions at the level of the human intestinal mucosa, especially when nuclear receptors are involved. For example, the pregnane X receptor (PXR), a xenobiotic receptor, is not expressed in Caco-2 cells4. PXR activation results in upregulation of many P450s, including CYP3A4, CYP3A5, CYP3A7, and the drug transporter P-glycoprotein (P-gp). Human and mouse PXR exhibit different activation profiles in response to xenobiotics5, thus explaining why rodents have limited predictive value with respect to nuclear receptor-mediated drug-drug interactions in humans. For example, rifampicin is a strong activator of human PXR, but is only a poor activator of rodent PXR. Conversely, rat and mouse PXR are activated by pregnenolone 16α-carbonitrile, which does not activate human PXR. Due to the limitations of both Caco-2 cells and rodent intestinal tissue, a better model is required for predicting intestinal drug-drug interactions in humans.
Rifampicin is currently being used as a first-line drug in the treatment of active tuberculosis. Unfortunately, it is often the cause of drug-drug interactions as it induces many P450s and drug transporters6, thereby potentially limiting drug bioavailability. For example, the oral bioavailability of the P-gp substrate digoxin was shown to be significantly lower when co-administered with rifampicin, which can be explained by P-gp induction at the level of the intestine7. Furthermore, HIV/TB patients receiving rifampicin should avoid HIV protease inhibitor (PI)-based regimens because rifampicin affects the oral bioavailability of PIs, which are substrates of CYP3A4 and P-gp, resulting in sub-therapeutic plasma concentrations.
Darunavir is a second generation PI with antiviral efficacy against HIV-1 with multiple resistance mutations to PIs8. Using the in situ intestinal perfusion technique with mesenteric blood sampling9 in P-gp knockout and NMRI mice, P-gp was found to significantly limit the intestinal permeability for darunavir10,11. In these mice, P-gp mediated darunavir transport was inhibited by co-perfusion with the “pharmacokinetic booster” ritonavir, which is generally co-administered with PIs in order to increase their plasma levels.
A transgenic mouse model, expressing both human PXR and CYP3A4, was developed that can serve as a useful tool to study the effect of xenobiotics on the expression of CYP3A4 in humans12. We tested the hypothesis that this mouse model can also be used for predicting the induction of intestinal P-gp by xenobiotics in humans. By performing the in situ intestinal perfusion technique in these PXR/CYP3A4-humanized mice, the effect of oral rifampicin treatment on the intestinal absorption of the PI darunavir was determined, thereby determining the relative contributions of both CYP3A4 and mouse P-gp (mdr1a/1b). The effect of rifampicin treatment on the intestinal CYP3A4 and P-gp protein levels was determined by Western blot analysis.
The phytochemical sulforaphane is formed after hydrolysis of glucoraphanin in many cruciferous vegetables (including broccoli and cabbage)13. Sulforaphane has previously been shown to inhibit CYP3A4 and P-gp induction after rifampicin treatment in primary human hepatocytes, thereby inhibiting PXR-mediated induction of drug clearance14, which would suggest its use to reduce PXR-mediated drug-drug interactions. However, experimental data regarding the use of sulforaphane appear to be inconclusive. In a recent clinical study in humans where rifampicin treatment decreased midazolam AUC by 70 %, concomitant administration of sulforaphane did not reduce the effect of rifampicin15. These investigators suggested that the hepatic sulforaphane concentrations were probably too low to inhibit PXR activation. In the current study, we tested the hypothesis that sulforaphane, which is present at relatively higher concentrations in the intraluminal environment, is able to inhibit a possible effect of rifampicin treatment on the uptake of darunavir in the intestine of the PXR/CYP3A4-humanized mouse.
Materials & methods
Chemicals
Darunavir ethanolate was provided by the NIH AIDS Research and Reference Reagent Program (Germantown, MD). R426857 was kindly donated by Johnson & Johnson (Beerse, Belgium). Rifadine® (rifampicin) was from Sanofi Aventis (Diegem, Belgium). GF120918 (elacridar) was provided by GSK (London, UK). L-Sulforaphane was purchased from Sigma Aldrich (St. Louis, MO). Ketamine (Anesketin) and xylazin (Xyl-M® 2%) were from Eurovet (Heusden, Belgium) and VMD (Arendonk, Belgium), respectively. The protease inhibitor cocktail for use with mammalian cell extracts and albumin from bovine serum (BSA) were purchased from Sigma-Aldrich (St. Louis, MO). Sodium acetate trihydrate and methanol were purchased from VWR International (Leuven, Belgium). Diethyl ether was purchased from Lab-Scan (Gliwice, Poland). Phosphate buffered saline (PBS) was provided by Lonza (Basel, Switzerland). Sodium taurocholic acid practical grade was purchased from MP Biomedicals (Illkirch Cedex, France). Phospholipon 90G (lecithin) was from Nattermann Phospholipid Gmbh (Köln, Germany). All other reagents were used as supplied. Water was purified with a Maxima system (Elga Ltd., High Wycombe Bucks, UK). Stock solutions of rifampicin and sulforaphane were prepared in DMSO.
Animals
Transgenic mice expressing human PXR and CYP3A4 (TgCYP3A4/hPXR) were transferred from the National Cancer Institute (Bethesda Maryland, USA) and housed at KU Leuven (Leuven, Belgium). NMRI (Naval Medical Research Institute) mice were purchased from Janvier (Le Genest Saint Isle, France). Only male mice were used in the experiments. In the rifampicin-treated group, mice were dosed by oral gavage with 10 mg/kg of rifampicin dissolved in PBS during the 3 days prior to the day of the experiment. Sulforaphane was orally administered at 5 mg/kg during the 3 days together with rifampicin. The molar ratio of sulforaphane to rifampicin was 2.3 to 1. Approval for the mouse experiments was granted by the Institutional Ethical Committee for Animal Experimentation of the KU Leuven.
Media
Fasted State Simulated Intestinal Fluid (FaSSIF) was made according to the composition reported by Vertzoni et al. (revised standard FaSSIF with practical grade taurocholate and soybean lecithin)16.
In situ intestinal perfusion
The setup for the in situ perfusion experiments in mice has previously been described by Mols et al.9. The perfusion experiments were performed using an open-loop set-up. A segment of the ileum (between 2 and 3 cm) was perfused at a flow rate of 0.2 ml/min. The exact length of the segment was taken into account when calculating the Papp value (see section permeability calculations). The perfusate consisted of FaSSIF containing darunavir (100 μM) in the absence or presence of GF120918, a specific P-gp inhibitor at 4 μM17, and in the absence or presence of aminobenzotriazole (100 μM), a nonspecific P450 inhibitor. FaSSIF, which contains bile salts and lecithin, was chosen as solvent system to simulate biorelevant conditions. This is important because it was previously shown that compounds present in intestinal fluids may alter the functionality of drug transporters18. Blood was collected continuously from the mesenteric vein for 60 min over 5-min time intervals. After the experiment, the intestine was removed, flushed with ice cold PBS, and stored at - 30 °C until further use.
Tissue homogenization and Western blot analysis
Isolation of enterocytes was performed based on the method previously described by Mohri and Uesawa19. All solutions were used at 4 °C. The frozen intestines were thawed in ice cold PBS. One end was clamped and the intestine was filled with solution A (PBS, pH 7.2, containing 5 mM EDTA, 0.5 mM dithiothreitol, 5 U/ml heparin and protease inhibitor cocktail (1%)). The intestine was tapped gently several times during 10 minutes after which the solution was collected and kept on ice. This procedure was repeated four times. The collected cells were centrifuged at 800 × g for 10 minutes at 4°C and resuspended in solution B (pH 7.8, containing 10 mM HEPES, 250 mM sucrose, 25 mM KCl, 1 mM EDTA and protease inhibitor cocktail (1%)). Next, the cells were added to the glass tube of a Potter-Elvehjem homogenizer (VWR International) and homogenized on ice by 10 strokes with a PTFE pestle rotating at high speed. Protein contents were determined with the method of Lowry20 using BSA as standard. For Western blot analysis, 30 μg of protein from each sample was loaded onto 10% SDS-polyacrylamide gels. The following primary antibodies were used: P-gp [H-241, rabbit polyclonal antibody, Santa Cruz Biotechnology (Heidelberg, Germany)], human CYP3A4 [ab22704, rabbit polyclonal antibody, Abcam (Cambridge, UK)], and β-actin [ab8226, mouse monoclonal antibody, Abcam]. Detection was performed using HRP-labeled secondary antibodies [goat polyclonal anti-rabbit immunoglobulins (P0448, Dako (Heverlee, Belgium)] and the Amersham ECL plus detection kit from GE Healthcare (Diegem, Belgium). Western blots were quantified using the ImageJ 1.45 software (NIH) after scanning of the films.
Analysis of the blood samples
Quantifying darunavir or R426857 in the blood samples required extraction: 100 μl of blood was diluted into 400 μl of a mixture of KH2PO4 (0.1 M, pH 6.0) and methanol (80:20 v/v); subsequently, 100 μl of internal standard solution (butyl-4-hydroxybenzoate, 10 μg/ml) was added. After adding 4 ml of diethyl ether, shaking for 1 minute, and centrifugation (2880 × g, 5 min), the organic layer was transferred to a clean test tube and evaporated to dryness under a gentle stream of air. The residue was dissolved in 150 μl of a solution of water and methanol (50:50 v/v), of which 10 μl was injected into the HPLC system. Darunavir and R426857 were detected with a fluorescence detector. The HPLC system consisted of a Waters Alliance 2695 separations module and a Nova-Pak® C18 Radial-Pak™ (4 μm, 8 × 100 mm) column under radial compression (Waters, Milford, MA). Fluorescence (excitation 268 nm, emission 347 nm) was monitored by a Waters fluorescence detector (W2475). The mobile phase consisted of 25 mM sodium acetate (pH 5.5) and methanol (40:60 v/v); the flow rate amounted to 1.3 ml/min. The retention times of darunavir and the internal standard amounted to 6.3 and 13.4 min, respectively. After elution, the column was flushed with acetonitrile:water (80:20 v/v) for 3 min and re-equilibrated with mobile phase during 3 min. The calibration curve was linear over the concentration range of 0.63 – 20 μM. The assessment of interday repeatability of darunavir, determined at 5 μM, resulted in a relative standard deviation of 2.0% (n = 5). The deviation from the theoretical concentration amounted to −4.6 %.
Permeability calculations
By measuring the amount of darunavir in the mesenteric blood, the apparent permeability (Papp) was determined, which reflects the overall intestinal absorption process including passive diffusion, the contribution of transporters, as well as intestinal metabolism. Papp values were calculated from the cumulative amount of darunavir appearing in the blood between 40–60 minutes according to the following equation:
where Q is the cumulative amount of drug appearing in the mesenteric blood, A is the surface area of the perfused cylindrical intestinal segment, and Cdonor is the drug concentration in the perfusate. The surface area was calculated with the following equation:
Where r is the radius of the intestinal segment (1 mm for the mouse ileum) and ℓ is the length of the perfused cylindrical intestinal segment.
Statistics
Statistical analysis was performed using an unpaired t-test or one-way ANOVA followed by Dunnett's test, as specified in the legends of the figures. P-values of less than 0.05 are considered as statistically significant.
Results
System suitability
A mouse intestinal perfusion model with mesenteric blood sampling using a closed-loop setup was previously described9. Recently, we reported the use of an open-loop setup10,11, which has the advantage that a constant Cdonor is being maintained during the entire experiment (Cdonor was measured at the beginning and the end of each experiment). The cumulative transport of darunavir, corrected for the length of the perfused intestinal segment, is shown as a function of time as the mean of 3 individual experiments in NMRI mice (Figure 1). During transport experiments, the steady-state darunavir transport was typically reached before 30 min, after which the transport rate was fairly constant.
Figure 1.
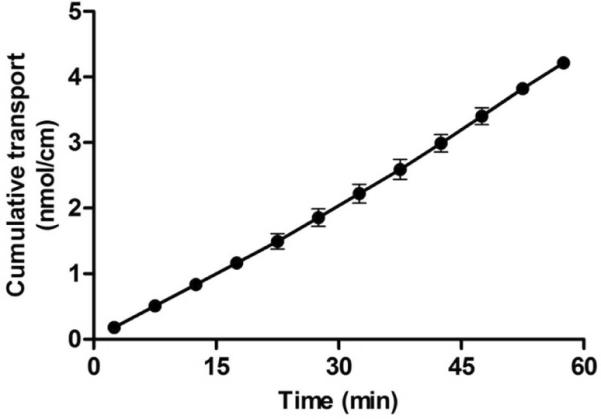
Transepithelial transport of darunavir as a function of time using the mouse in situ perfusion model. The ileum of NMRI mice was perfused with FaSSIF containing darunavir (100 μM). Cumulative transport values, corrected for the length of the intestinal segment, are shown as the mean ± SD (n=3).
The effect of rifampicin on the intestinal absorption of darunavir in NMRI mice
In the first set of experiments, the effect of oral rifampicin treatment on the intestinal transport of darunavir in NMRI mice was determined. NMRI mice were orally treated with rifampicin at pharmacological doses (10 mg/kg) for three days prior to the experiment. On the fourth day, we performed in situ intestinal perfusion studies with mesenteric blood sampling where the perfusate consisted of darunavir (100 μM) dissolved in FaSSIF. As a result of oral rifampicin treatment, a 66% increase in intestinal permeability was observed for darunavir that however, did not reach statistical significance (Figure 2).
Figure 2.
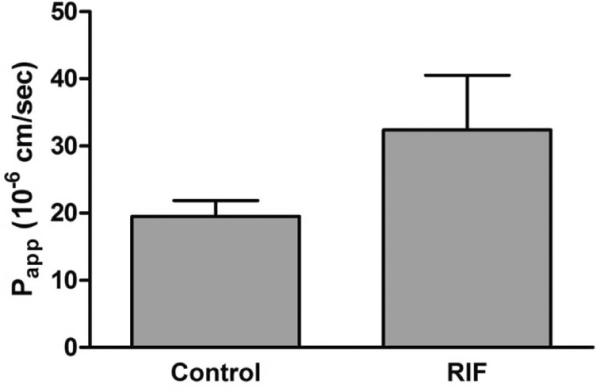
Apparent permeability values of the ileum of NMRI mice for darunavir (100 μM) using FaSSIF as perfusate medium in mice that were not treated with rifampicin (control) and mice orally treated with rifampicin (10 mg/kg) for 3 days. Bars represent the mean ± SD. (n = 3). Statistical significance between the control and rifampicin-treated mice was evaluated using an unpaired t-test. Control condition was not statistically different from rifampicin-treated condition.
The effect of rifampicin on the intestinal absorption of darunavir in TgCYP3A4/hPXR mice
We tested the hypothesis that the TgCYP3A4/hPXR mouse model is suitable to study the induction of intestinal P-gp by rifampicin at pharmacological doses. We first investigated whether this transporter has a modulatory effect on the intestinal permeability for darunavir in these mice. When the intestine of the humanized mice (which had not been treated with rifampicin) was perfused with darunavir and the P-gp inhibitor GF120918 (4 μM), the intestinal permeability for darunavir was increased by 56 % (p < 0.05), indicating that P-gp limits the intestinal permeability for darunavir (Figure 3). Next, we tested the hypothesis that rifampicin treatment affects the intestinal absorption of darunavir. Oral dosing of rifampicin (10 mg/kg) during three days prior to the intestinal perfusion experiment caused a decrease of 50 % in the intestinal permeability for darunavir compared to untreated mice. This observation may be explained by an increased biochemical barrier function of the intestinal mucosa, either by an induction of intestinal CYP3A4, P-gp, or both. To investigate whether intestinal P-gp expression was induced by rifampicin treatment, GF120918 (4 μM) was added to the intestinal perfusate, which increased the intestinal permeability for darunavir by 400 % (p<0.05). When we compared the effect of GF120918 in the untreated mice with the rifampicin-treated mice, there was a strong indication of P-gp induction. To investigate whether intestinal CYP3A4 expression was induced by rifampicin treatment, the non-specific P450 inhibitor aminobenzotriazole (100 μM) was included in the perfusate. The use of aminobenzotriazole in rifampicin-treated mice did not increase the intestinal permeability for darunavir, thus suggesting that rifampicin treatment has no effect on the intestinal CYP3A4 expression or functionality.
Figure 3.
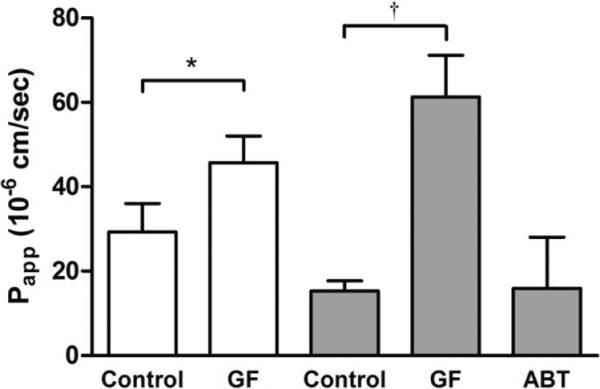
Apparent permeability values of the ileum of TgCYP3A4/hPXR mice for darunavir (100 μM) using FaSSIF as perfusate medium. Experiments were performed in mice that were not treated with rifampicin (open bars) and mice orally treated with rifampicin (10 mg/kg) during 3 days (gray bars), in the absence and presence of the P-gp inhibitor GF120918 (GF, 4 μM) or the non-specific P450 inhibitor aminobenzotriazole (ABT, 100 μM). Bars represent the mean ± SD. (n = 3). Statistical significance between the different conditions in the group that was not treated with rifampicin was evaluated using an unpaired t-test. *, significantly different from control condition (p < 0.05). Statistical significance between the different conditions in the rifampicin -treated group was evaluated using one-way ANOVA followed by Dunnett′s test. †, significantly different from control condition (p < 0.05).
Intestinal metabolism of darunavir in TgCYP3A4/hPXR mice
Darunavir is mainly metabolized by CYP3A4 in humans, with R426857 being the most prominent metabolite present in the plasma21. To investigate whether intestinal metabolism limits the absorption of darunavir, the mesenteric blood that was collected during the in situ intestinal perfusion experiments was analyzed for the presence of R426857. Indeed, the intestinal metabolism of darunavir was very low in the TgCYP3A4/hPXR mice, where the concentration of R426857 was less than 0.8 % of this of darunavir. The relative amount of R426857 to darunavir was higher in the mice that were perfused with GF120918, being 1.6 and 3.1 % for the untreated and rifampicin-treated mice, respectively.
The effect of rifampicin on the intestinal protein expression levels of CYP3A4 and P-gp in TgCYP3A4/hPXR mice
Western blot analysis of the intestinal tissue of the TgCYP3A4/hPXR mice was performed to investigate the effect of rifampicin treatment on CYP3A4 and P-gp protein levels. Rifampicin treatment did not increase the intestinal expression levels of CYP3A4, while P-gp expression was increased four fold (Figure 4). Since the TgCYP3A4/hPXR mice also express murine Cyp3a isoforms in addition to CYP3A4, we investigated whether there was cross reactivity of the CYP3A4 antibodies with rodent Cyp3a. Using rat intestinal and liver homogenate, we found that the CYP3A4 antibodies did not react with rat Cyp3a, which corresponded with the manufacturers data (data not shown).
Figure 4.

The effect of rifampicin on intestinal CYP3A4 and P gp protein expression levels in TgCYP3A4/hPXR mice. Intestinal homogenates were analyzed by Western blotting. CYP3A4 antibodies recognize human CYP3A4 but not mouse Cyp3a. Protein expression levels were quantified in mice orally treated with rifampicin (10 mg/kg) for 3 days (gray bars, n=9) and mice that were not treated with rifampicin (open bars, n=6). Bars represent the mean ± SEM of CYP3A4 and P-gp normalized to β-actin. Statistical significance between the different conditions was evaluated using an unpaired t-test. *, significantly different from control condition (p < 0.05).
The effect of sulforaphane on the intestinal absorption of darunavir
We tested the hypothesis that the PXR antagonist sulforaphane can neutralize the limiting effect of rifampicin on the intestinal absorption of darunavir. Oral co-administration of sulforaphane (5 mg/kg) with rifampicin (10 mg/kg) during 3 days prior to the intestinal perfusion experiments resulted in a 50 % increase in permeability for darunavir compared to mice treated with rifampicin alone (Figure 5). Although this difference was not statistically significant, these data suggest that the limiting effect of rifampicin on the intestinal permeability for darunavir was partially reversed by co-administration of sulforaphane.
Figure 5.
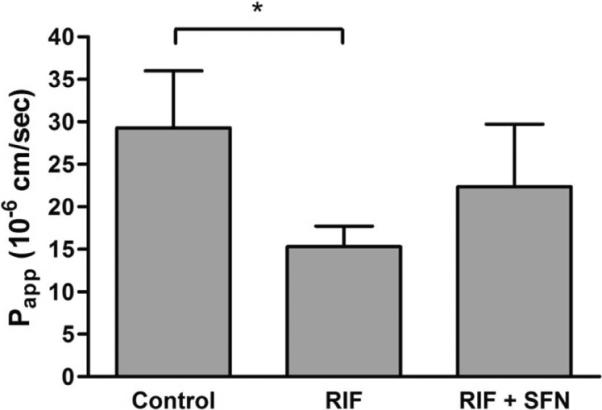
Apparent permeability values of the ileum of TgCYP3A4/hPXR mice for darunavir (100 μM) using FaSSIF as perfusate medium. Experiments were performed in control mice, mice orally treated with rifampicin (10 mg/kg) for 3 days and mice orally treated with rifampicin (10 mg/kg) + sulforaphane (5 mg/kg) for 3 days. Statistical significance between the different conditions was evaluated using one way ANOVA followed by Dunnett′s test. *, significantly different from control condition (p < 0.05). Parts of these data have been shown in Figure 3.
Discussion
The aim of this study was to evaluate whether TgCYP3A4/hPXR mice can be used to predict the effect of xenobiotics on the intestinal expression levels of P-gp in humans. The use of wild-type mice in nuclear receptor-mediated drug-drug interaction studies may have only limited predictive value towards the human situation, because significant species differences exist in the ligand binding domain of PXR5. In order to circumvent this problem, a transgenic mouse containing the human PXR gene (on the Pxr-null background), along with the CYP3A4 and CYP3A7 genes (TgCYP3A4/hPXR mice), was generated and used to demonstrate rifampicin-PI interactions, which could not be seen in mice without human PXR12. Besides inducing many of the P450 enzymes in humans, oral rifampicin treatment was also shown to increase intestinal P-gp expression levels7. The current study has investigated whether the TgCYP3A4/hPXR mouse model allows us to make the same observations. Using the in situ intestinal perfusion technique with mesenteric blood sampling, the effect of rifampicin treatment on the intestinal absorption of the PI darunavir was investigated. Rifampicin was orally administered at a dose of 10 mg/kg, which corresponds to the recommended dose in humans (600 mg per day). Darunavir was chosen as model compound because it is a substrate of both CYP3A4 and P-gp, which allows us to investigate the effect of rifampicin treatment at the level of both drug transporters and metabolism.
In a first set of experiments, the standard NMRI mice containing mouse PXR were used to investigate the effect of oral rifampicin treatment on the intestinal permeability for darunavir. As expected, rifampicin did not decrease the intestinal permeability for darunavir because rifampicin is not an activator of mouse PXR. Instead, rifampicin caused a 66 % increase in darunavir permeability, which may be explained by an inhibition of P-gp by rifampicin22 still present in the enterocytes.
In a second set of experiments, TgCYP3A4/hPXR mice were dosed with rifampicin, after which a 50% decrease was observed in the intestinal permeability for darunavir compared to TgCYP3A4/hPXR mice that were not treated with rifampicin. Because this observation may be attributed to an induction of intestinal CYP3A4, P-gp, or both, we further investigated each hypothesis. In rifampicin-treated TgCYP3A4/hPXR mice, perfusion with the P-gp inhibitor GF120918 caused an increase in darunavir permeability of 400%, while in the non-treated mice, GF120918 only increased darunavir permeability by 56%. These data are consistent with rifampicin-mediated induction of intestinal P-gp. Furthermore, it was remarkable that GF120918 tended to increase the apparent permeability for darunavir to a higher level in the rifampicin-treated mice (Papp = 61.3 ± 9.9 × 10−6 cm/sec) as compared to the non-treated mice (Papp = 45.7 ± 6.3 × 10−6 cm/sec). This difference was however not statistically significant. Use of the non-specific P450 inhibitor aminobenzotriazole had no impact on darunavir permeability, thus suggesting no effect of rifampicin on intestinal CYP3A4 functionality in this mouse model. Western blot analysis of the intestinal tissues confirmed our observations obtained from the in situ intestinal perfusion experiments: P-gp expression levels had increased four-fold after rifampicin treatment, while CYP3A4 expression levels remained unchanged.
Since the TgCYP3A4/hPXR mice were not humanized for P-gp (MDR1), activation of human PXR appears to induce the expression of mouse P-gp (mdr1a/1b). This observation was previously reported in other studies, where rifampicin caused an induction of mouse P-gp in the brain of transgenic hPXR mice23. Comparing our data with the literature, an induction of intestinal CYP3A4 protein levels was observed after rifampicin treatment12; however, in this study rifampicin was administered for six days at a dose of 100 mg/kg, which was 10 times higher than the dose we applied. Another study in humans showed a significant increase in intestinal CYP3A4 mRNA and protein levels after 10 days of oral rifampicin administration at a dose of 600 mg/day24. The reason why we did not observe any induction of CYP3A4 remains unclear; however, literature data suggest that CYP3A4 is highly constitutively expressed in the intestine of TgCYP3A4/hPXR mice, and thus the extent of induction is generally lower compared to robust CYP3A4 induction in the liver, where the constitutive expression of CYP3A4 is lower25. After analyzing the mesenteric blood for the most prominent metabolite of darunavir present in human plasma (R426857), intestinal darunavir metabolism appeared to be very low. This was surprising, since intestinal CYP3A4 protein levels were readily detectable. Nevertheless, it is possible that darunavir inhibits its own intestinal metabolism since it was found to be an inhibitor of CYP3A4 with an inhibitory constant (Ki) value of 0.40 μmol/L, which is well within the range of clinically relevant concentrations26. The darunavir concentrations at the level of the intestine are expected to be even higher. Also, the much lower intestinal P450 levels relative to the liver may further contribute to an inhibition of the available enzymes. Using human intestinal microsomes, we found that darunavir (5 μM) partially inhibits the metabolism of lopinavir (5 μM), while darunavir itself was not metabolized, indicating that darunavir inhibits CYP3A4-mediated metabolism (data not shown). The amount of darunavir metabolites measured in the mesenteric blood was highest when using the P-gp inhibitor GF120918, suggesting that most of the darunavir metabolites that are formed in the enterocytes are effluxed into the intestinal lumen by P-gp instead of being taken up into the blood via the basolateral membrane. This could however, not be verified experimentally, since we did not measure the appearance of metabolites in the perfusate.
In a final set of experiments, the effect of the phytochemical sulforaphane, a PXR antagonist, on the drug-drug interaction between rifampicin and darunavir at the level of the intestine was investigated. Although sulforaphane was previously shown to reduce CYP3A4 and P-gp induction by rifampicin in primary human hepatocytes14, a recent clinical trial showed that sulforaphane was unable to reduce CYP3A4 induction by rifampicin in humans15. It should be noted that the sulforaphane levels in the liver were probably too low to inhibit PXR activation by rifampicin. We found that co-administration of sulforaphane (5 mg/kg) with rifampicin (10 mg/kg) for three days prior to our experiments increased the intestinal permeability for darunavir by 50 % compared to rifampicin treatment alone. This indicates that, for compounds that are a substrate for P-gp, and for which first-pass elimination is not primarily mediated by hepatic metabolism, sulforaphane may (partially) inhibit the effects of rifampicin on PXR. It also needs to be mentioned that the probability of an effect at the level of the intestine may be higher than an effect in the liver in view of the much higher local sulforaphane concentrations in the intestine.
Conclusion
We conclude that, in TgCYP3A4/hPXR mice, oral rifampicin treatment significantly decreased the intestinal permeability for darunavir by induction of P-gp; CYP3A4 protein levels remained unchanged. Because this could not be observed in conventional mice, PXR/CYP3A4-humanized mice are a promising tool to study PXR/P-gp mediated drug-drug interactions at the level of the intestine.
Acknowledgements
We would like to thank Lies Pauwels for performing the Western blot analysis. We would also like to thank the NIH AIDS Research and Reference Reagent Program for providing darunavir and Johnson & Johnson for providing R426857. This research was funded by grants from: (1) Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT), (2) Fund for Scientific Research in Flanders (FWO), and (3) `Onderzoeksfonds' of the KU Leuven in Belgium.
Abbreviations used
- PXR
pregnane X receptor
- P450
cytochrome P450
- FaSSIF
Fasted State Simulated Intestinal Fluid
- PBS
phosphate buffered saline
- Papp
apparent permeability coefficient
- P-gp
P-glycoprotein
- PI
HIV protease inhibitor
- GF120918
N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide
References
- (1).Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Deliv. Rev. 2012 doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- (2).Chiou WL, Barve A. Linear correlation of the fraction of oral dose absorbed of 64 drugs between humans and rats. Pharm. Res. 1998;15:1792–1795. doi: 10.1023/a:1011981317451. [DOI] [PubMed] [Google Scholar]
- (3).Fortuna A, Alves G, Falcão A, Soares-da-Silva P. Evaluation of the permeability and P-glycoprotein efflux of carbamazepine and several derivatives across mouse small intestine by the Ussing chamber technique. Epilepsia. 2012;53:529–538. doi: 10.1111/j.1528-1167.2012.03409.x. [DOI] [PubMed] [Google Scholar]
- (4).Thummel KE, Brimer C, Yasuda K, Thottassery J, Senn T, Lin Y, Ishizuka H, Kharasch E, Schuetz J, Schuetz E. Transcriptional control of intestinal cytochrome P-4503A by 1alpha,25-dihydroxy vitamin D3. Mol. Pharmacol. 2001;60:1399–1406. doi: 10.1124/mol.60.6.1399. [DOI] [PubMed] [Google Scholar]
- (5).Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NC, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. The pregnane X receptor: a promiscuous xenobiotic receptor that has diverged during evolution. Mol. Endocrinol. 2000;14:27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- (6).Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42:819–850. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- (7).Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J. Clin. Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tremblay CL. Combating HIV resistance - focus on darunavir. Ther Clin Risk Manag. 2008;4:759–766. doi: 10.2147/tcrm.s1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mols R, Brouwers J, Schinkel AH, Annaert P, Augustijns P. Intestinal perfusion with mesenteric blood sampling in wild-type and knockout mice: evaluation of a novel tool in biopharmaceutical drug profiling. Drug Metab. Dispos. 2009;37:1334–1337. doi: 10.1124/dmd.109.026591. [DOI] [PubMed] [Google Scholar]
- (10).Holmstock N, Mols R, Annaert P, Augustijns P. In situ intestinal perfusion in knockout mice demonstrates inhibition of intestinal p-glycoprotein by ritonavir causing increased darunavir absorption. Drug Metab. Dispos. 2010;38:1407–1410. doi: 10.1124/dmd.110.032771. [DOI] [PubMed] [Google Scholar]
- (11).Holmstock N, Annaert P, Augustijns P. Boosting of HIV Protease Inhibitors by Ritonavir in the Intestine: The Relative Role of Cytochrome P450 and P-Glycoprotein Inhibition Based on Caco-2 Monolayers versus In Situ Intestinal Perfusion in Mice. Drug Metab. Dispos. 2012;40:1473–1477. doi: 10.1124/dmd.112.044677. [DOI] [PubMed] [Google Scholar]
- (12).Ma X, Cheung C, Krausz KW, Shah YM, Wang T, Idle JR, Gonzalez FJ. A double transgenic mouse model expressing human pregnane X receptor and cytochrome P450 3A4. Drug Metab. Dispos. 2008;36:2506–2512. doi: 10.1124/dmd.108.022723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liang H, Yuan QP, Dong HR, Liu YM. Determination of sulforaphane in broccoli and cabbage by high-performance liquid chromatography. J. Food Compos. Anal. 2006;19:473–476. [Google Scholar]
- (14).Zhou C, Poulton E-J, Grün F, Bammler TK, Blumberg B, Thummel KE, Eaton DL. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol. Pharmacol. 2007;71:220–229. doi: 10.1124/mol.106.029264. [DOI] [PubMed] [Google Scholar]
- (15).Poulton E-J, Levy L, Lampe J, Shen D, Tracy J, Shuhart M, Thummel KE, Eaton DL. Sulforaphane is not an effective antagonist of the human Pregnane X-Receptor in vivo. Toxicol Appl Pharmacol. doi: 10.1016/j.taap.2012.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Vertzoni M, Fotaki N, Kostewicz E, Stippler E, Leuner C, Nicolaides E, Dressman J, Reppas C. Dissolution media simulating the intralumenal composition of the small intestine: physiological issues and practical aspects. J. Pharm. Pharmacol. 2004;56:453–462. doi: 10.1211/0022357022935. [DOI] [PubMed] [Google Scholar]
- (17).Matsson P, Pedersen JM, Norinder U, Bergström CAS, Artursson P. Identification of novel specific and general inhibitors of the three major human ATP-binding cassette transporters P-gp, BCRP and MRP2 among registered drugs. Pharm. Res. 2009;26:1816–1831. doi: 10.1007/s11095-009-9896-0. [DOI] [PubMed] [Google Scholar]
- (18).Deferme S, Tack J, Lammert F, Augustijns P. P-glycoprotein attenuating effect of human intestinal fluid. Pharm. Res. 2003;20:900–903. doi: 10.1023/a:1023891320858. [DOI] [PubMed] [Google Scholar]
- (19).Mohri K, Uesawa Y. Enzymatic activities in the microsomes prepared from rat small intestinal epithelial cells by differential procedures. Pharm. Res. 2001;18:1232–1236. doi: 10.1023/a:1010951732288. [DOI] [PubMed] [Google Scholar]
- (20).Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- (21).Vermeir M, Lachau-Durand S, Mannens G, Cuyckens F, van Hoof B, Raoof A. Absorption, metabolism, and excretion of darunavir, a new protease inhibitor, administered alone and with low-dose ritonavir in healthy subjects. Drug Metab. Dispos. 2009;37:809–820. doi: 10.1124/dmd.108.024109. [DOI] [PubMed] [Google Scholar]
- (22).Reitman ML, Chu X, Cai X, Yabut J, Venkatasubramanian R, Zajic S, Stone JA, Ding Y, Witter R, Gibson C, Roupe K, Evers R, Wagner JA, Stoch A. Rifampin′s acute inhibitory and chronic inductive drug interactions: experimental and model-based approaches to drug drug interaction trial design. Clin. Pharmacol. Ther. 2011;89:234–242. doi: 10.1038/clpt.2010.271. [DOI] [PubMed] [Google Scholar]
- (23).Bauer B, Yang X, Hartz AMS, Olson ER, Zhao R, Kalvass JC, Pollack GM, Miller DS. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up regulation. Mol. Pharmacol. 2006;70:1212–1219. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- (24).Glaeser H, Drescher S, Eichelbaum M, Fromm MF. Influence of rifampicin on the expression and function of human intestinal cytochrome P450 enzymes. Br J Clin Pharmacol. 2005;59:199–206. doi: 10.1111/j.1365-2125.2004.02265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cheung C, Yu A-M, Chen C-S, Krausz KW, Byrd LG, Feigenbaum L, Edwards RJ, Waxman DJ, Gonzalez FJ. Growth hormone determines sexual dimorphism of hepatic cytochrome P450 3A4 expression in transgenic mice. J. Pharmacol. Exp. Ther. 2006;316:1328–1334. doi: 10.1124/jpet.105.094367. [DOI] [PubMed] [Google Scholar]
- (26).Brown KC, Paul S, Kashuba ADM. Drug interactions with new and investigational antiretrovirals. Clin Pharmacokinet. 2009;48:211–241. doi: 10.2165/00003088-200948040-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]