

Abstract
Dimers of GPCRs have held the imagination of researchers for almost 20 years. However, only recently has their value as potentially novel drug targets been increased significantly, and primarily, in the context of GPCR heterodimers. The view of receptor heterodimers as allosteric machines has transformed the way we understand structural and functional asymmetries inherent in their organization. These asymmetries alter both signalling output and how they might be targeted pharmacologically. The paper in this issue of BJP by Siddiquee and colleagues (2013) highlights our growing understanding of such asymmetries and their implications. They show that heterodimers of the angiotensin II AT1 receptor and the apelin receptor recognize and respond to their respective ligands in distinct ways from the parent receptors expressed alone. Further, they demonstrate asymmetric allosteric effects in the context of the heterodimer that may have significant implications for our understanding of such receptor complexes.
Linked Article
This article is a commentary on the research paper by Siddiquee et al., pp. 1104–1117 of this issue. To view this paper visit http://dx.doi.org/10.1111/j.1476-5381.2012.02192.x
Keywords: GPCRs, dimers, allosterism, signalling, drug discovery
GPCR heterodimerization is now a widely accepted means by which new signalling entities can be created with respect to ligand binding, G-protein and effector coupling and receptor trafficking [reviewed in Smith and Milligan, (2010)]. However, our understanding of such heterodimers as potential drug targets has lagged far behind. This was likely due to two factors: the lack of tools to study such dimers in homologous systems which did not rely on receptor overexpression, and secondly, a lack of understanding as to how their unique architecture might be targeted pharmacologically. The best examples of GPCR heterodimers remain class C receptors such as the GABA-B receptor and the taste receptors [reviewed in (Kniazeff et al., 2011)]. The idea of heterodimers naturally suggests a potential division of labour with respect to ligand binding or interaction with signalling partners (such as G-proteins), elegantly demonstrated for GABA-B receptors (Kniazeff et al., 2011) or the MT1/MT2 melatonin receptor heterodimer (Maurice et al., 2010). There is likely a spectrum of organizational paradigms operating for interactions between GPCR signalling systems, ranging from classic molecular cross-talk between receptors (alone or in complexes without direct interactions between them), to strict heterodimers operating via allosteric mechanisms arranged in a cell-specific fashion (Figure 1). This organizational complexity will need to be understood for future drug discovery efforts.
Figure 1.
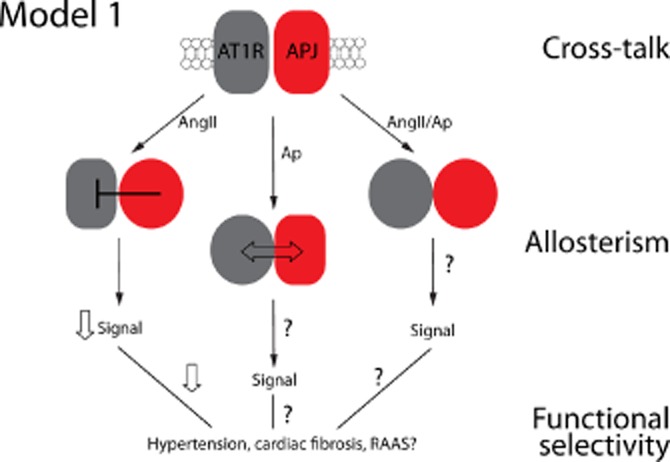
Interactions between APJ and AT1R, as described by Siddequee et al. shows how dimerization of AT1R and APJ protomers can lead to biased signalling responses downstream, via molecular cross-talk. In their model, Ap13 binding increases the affinity of each receptor for each other. The APJ protomer then inhibits, probably through conformational change, the AT1R protomer – resulting in attenuated Ang II-mediated responses – via allosteric modulation. The consequences of Ap, or concomitant Ang II/Ap binding to the dimer on downstream signalling remains to be determined. Moreover, the consequences of such allosterically biased signals on more complex phenotypic responses (hypertension, cardiac fibrosis, RAAS function, etc.), remain unknown.
AT1R are social animals- they interact with other GPCRs
A number of research groups have previously shown physiologically relevant interactions between apelin receptors (APJ) and the Ang II AT1 receptor (AT1R) subtype in controlling function of the latter in the vasculature (Chun et al., 2008; Siddiquee et al., 2011). Such regulation likely depends on the relative abundance of the two receptors in a given tissue. Indeed, others have shown the AT1R is likely to be a signalling hub whose functions are controlled by a number of partner receptors such as the β2AR (Barki-Harrington et al., 2003), the B2 bradykinin receptor (Quitterer et al., 2004) and the apelin receptor (Chun et al., 2008; Siddiquee et al., 2011; Sun et al., 2011).
In the present study, Siddequee and his colleagues confirm this previous work showing that APJ and AT1R dimerize using a number of biochemical and biophysical techniques. They show that this dimer forms with endogenous receptors in HEK 293 cells by using an interesting cross-linking approach, which limits the distance between the two protomers. This approach would likely also have worked in a more relevant physiological setting, such as vascular smooth muscle cells. This, while interesting, is not the most important finding of the paper. The AT1R/APJ interaction described here provides a glimpse into the true allosteric nature of receptor heterodimers, in that depending on how they are arranged, striking asymmetries may be detected in how each protomer interacts with ligands and further, how it might signal in a manner distinct from the parent monomeric or homodimeric receptors. Previous studies have demonstrated that activated APJ modulates AT1R signalling in HEK 293 cells (Sun et al., 2011) and in primary vascular smooth muscle cells (Chun et al., 2008; Siddiquee et al., 2011). In one study, there were striking differences detected in the effects of unoccupied and ligand-occupied APJ on AT1R signalling, suggesting distinct allosteric interactions depending on the state of the system (Sun et al., 2011). The idea that unoccupied receptors might play a key role in allosteric modulation of dimeric partners was elegantly demonstrated in vivo for the ghrelin receptor/D2 dopamine receptor heterodimer, in brain regions where ghrelin itself is undetected (Kern et al., 2012). It has also been suggested that many orphan GPCRs might actually be such ‘ligand-free’ allosteric modulators for known GPCRs (Levoye et al., 2006).
Unequal partnerships – functional aymmetries in GPCR heterodimers
Here, the authors also demonstrated a number of asymmetries in that the physical interaction between the AT1R and APJ was altered upon treatment with Ap13, but not Ang II. They demonstrated that Ap13, but not Ang II increased the ability of the receptors to be immunoprecipitated together. Further, decreased Ang II affinity for AT1R was noted when AJP was occupied by Ap13 but again not the converse (Figure 1), which the authors demonstrate is a manifestation of negative cooperativity. Finally, Ap13 decreased Ang II-mediated signalling although the converse was not tested directly and remains an open question. The authors demonstrated that canonical signalling of AT1R through Gαq and β-arrestin recruitment were both affected by Ap13, which may suggest that both signalling states of the AT1R were effected equally. However, it may also suggest a common receptor state where both pathways are modulated by APJ. This may argue for the G-protein dependence of so-called G–protein-independent signalling events and may be better thought of as ‘post'–G-protein signalling mechanisms. More direct measures of proximal G-protein activation in the present study might have helped to resolve this apparent dilemma.
Interestingly, although the authors prefer a different interpretation, claiming that the interaction itself is stimulated by ligand, their data clearly shows constitutive heterodimer formation measured via co-immunoprecipitation or BRET. It is difficult to imagine allosteric effects in membrane preparations in the absence of constitutive heterodimers (i.e. more high-affinity binding if the dimer breaks apart). Thus, the ligand effects likely represent cooperative, allosteric changes in the heterodimer conformation reflected by altered stability of the complex in immunoprecipitation experiments. Their BRET experiments show that neither Ap13 nor Ang II affected BRET50 values even though they had opposing effects on the magnitude of BRET with the dimer complex, again suggesting conformational changes in the constitutively formed dimer.
The new world of structural asymmetries – opportunities and challenges for drug discovery
Importantly, a recent study demonstrated that the two-receptor equivalents, in the context of a D2 dopamine receptor homodimer, were organized asymmetrically with respect to their G-protein partners (Han et al., 2009), such that occupation of one receptor protomer activates the receptor to facilitate downstream signalling, and occupation of the other protomer modulates signalling allosterically without inducing a signal of its own. This study also has tremendous implications for the formation of receptor heterodimers, in that multiple asymmetrical arrangements become possible, depending on the relative orientation of each monomer in dimer to the G-protein and possibly effector molecules. Thus, in one arrangement, protomer A signals and protomer B acts as the allosteric modulator that does not necessarily generate a signalling output of its own. Reciprocally, the converse is true when the system is organized the other way around. This greatly increases the potential organizational complexity of GPCR signalling pathways, suggesting that determinants of signalling complex assembly will be of paramount importance in initially defining signalling specificity in a given tissue, cellular or subcellular compartment. Further, it suggests perhaps why heterodimers may have been difficult to detect in vivo because one receptor might in fact be silent with respect to signalling and thus missed in standard drug screens. That arrangement can be reversed if the complex is assembled or arranged differently – i.e. even with the same set of interacting partners, signalling output will be quite distinct. Not only are these considerations likely to be important for therapeutic efficacy, but may also predict and explain numerous off-target effects of currently used drugs.
This notion becomes even more important given that recent findings have revealed that GPCRs do not act as simple switches that turn single signalling pathways ‘on’ or ‘off’. Instead, individual receptors engage multiple signalling cascades and individual ligands can have differential efficacies towards specific subsets of these signalling effectors. This phenomenon, known as ligand-biased signalling or functional selectivity, offers interesting opportunities to identify and develop compounds with increased selectivity and improved safety profiles (Kenakin, 2012). It has been demonstrated that some β-adrenoceptor and AT1R antagonists used to treat heart failure, are not simply receptor antagonists (as defined for signalling pathways studied), but may also activate cardioprotective pathways, thus acting as biased ligands. However, the mechanistic basis of biased signalling through GPCRs remains unknown. It has been assumed that different receptors ‘select’ downstream signalling pathways in response to different ligands and how they might occupy the ligand-binding site and selectively alter or stabilize unique receptor conformations. It could be suggested that assembly of receptor homo- and hetero-dimeric/oligomeric complexes is a more likely basis for distinct cellular responses to particular ligands. In the future, we must better understand the connections, if any, between asymmetric dimers, which may be ‘silent’ in conventional experiments in vivo, as discussed earlier and the ability for receptors to respond to biased ligands. The present study by Siddequee and colleagues represents a good start to this effort. Understanding dimers in different cellular contexts will also be critical as the asymmetries detected here may in fact be cell-context specific. It will be important to examine other AT1R dimer partners in light of the present study. There are interesting days ahead.
References
- Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of β-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation. 2003;108:1611–1618. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118:3343–3354. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol. 2009;5:688–695. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin TP. Biased signalling and allosteric machines: new vistas and challenges for drug discovery. Br J Pharmacol. 2012;165:1659–1669. doi: 10.1111/j.1476-5381.2011.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron. 2012;73:317–332. doi: 10.1016/j.neuron.2011.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeff J, Prezeau L, Rondard P, Pin JP, Goudet C. Dimers and beyond: the functional puzzles of class C GPCRs. Pharmacol Ther. 2011;130:9–25. doi: 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Levoye A, Dam J, Ayoub MA, Guillaume JL, Jockers R. Do orphan G-protein-coupled receptors have ligand-independent functions? New insights from receptor heterodimers. EMBO Rep. 2006;7:1094–1098. doi: 10.1038/sj.embor.7400838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice P, Daulat AM, Turecek R, Ivankova-Susankova K, Zamponi F, Kamal M, et al. Molecular organization and dynamics of the melatonin MT(1) receptor/RGS20/G(i) protein complex reveal asymmetry of receptor dimers for RGS and G(i) coupling. EMBO J. 2010;29:3646–3659. doi: 10.1038/emboj.2010.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quitterer U, Lother H, Abdalla S. AT1 receptor heterodimers and angiotensin II responsiveness in preeclampsia. Semin Nephrol. 2004;24:115–119. doi: 10.1016/j.semnephrol.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Siddiquee K, Hampton J, Khan S, Zadory D, Gleaves L, Vaughan DE, et al. Apelin protects against angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J Hypertens. 2011;29:724–731. doi: 10.1097/HJH.0b013e32834347de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiquee K, Hampton J, McAnally D, May LT, Smith LH. The apelin receptor inhibits the angiotensin II type 1 receptor via allosteric trans-inhibition. Br J Pharmacol. 2013;168:1104–1117. doi: 10.1111/j.1476-5381.2012.02192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NJ, Milligan G. Allostery at G protein-coupled receptor homo- and heteromers: uncharted pharmacological landscapes. Pharmacol Rev. 2010;62:701–725. doi: 10.1124/pr.110.002667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Iida S, Yoshikawa A, Senbonmatsu R, Imanaka K, Maruyama K, et al. Non-activated APJ suppresses the angiotensin II type 1 receptor, whereas apelin-activated APJ acts conversely. Hypertens Res. 2011;34:701–706. doi: 10.1038/hr.2011.19. [DOI] [PubMed] [Google Scholar]