

Abstract
Background and Purpose
The apelin receptor (APJ) is often co-expressed with the angiotensin II type-1 receptor (AT1) and acts as an endogenous counter-regulator. Apelin antagonizes Ang II signalling, but the precise molecular mechanism has not been elucidated. Understanding this interaction may lead to new therapies for the treatment of cardiovascular disease.
Experimental Approach
The physical interaction of APJ and AT1 receptors was detected by co-immunoprecipitation and bioluminescence resonance energy transfer (BRET). Functional and pharmacological interactions were measured by G-protein-dependent signalling and recruitment of β-arrestin. Allosterism and cooperativity between APJ and AT1 were measured by radioligand binding assays.
Key Results
Apelin, but not Ang II, induced APJ : AT1 heterodimerization forced AT1 into a low-affinity state, reducing Ang II binding. Likewise, apelin mediated a concentration-dependent depression in the maximal production of inositol phosphate (IP1) and β-arrestin recruitment to AT1 in response to Ang II. The signal depression approached a limit, the magnitude of which was governed by the cooperativity indicative of a negative allosteric interaction. Fitting the data to an operational model of allosterism revealed that apelin-mediated heterodimerization significantly reduces Ang II signalling efficacy. These effects were not observed in the absence of apelin.
Conclusions and Implications
Apelin-dependent heterodimerization between APJ and AT1 causes negative allosteric regulation of AT1 function. As AT1 is significant in the pathogenesis of cardiovascular disease, these findings suggest that impaired apelin and APJ function may be a common underlying aetiology.
Linked Article
This article is commented on by Goupil et al., pp. 1101–1103 of this issue. To view this commentary visit http://dx.doi.org/10.1111/bph.12040
Keywords: GPCR, APJ receptor, AT1 receptor, heterodimerization, apelin, angiotensin II, allosterism
Introduction
The apelin receptor (APJ) and angiotensin II type-1 receptor (AT1) are GPCRs, the largest family of cell surface receptor proteins. GPCRs regulate virtually all physiological processes in response to extracellular stimuli (Kroeger et al., 2003; Bai, 2004; Gurevich and Gurevich, 2008; Klabunde et al., 2009; Rovira et al., 2009); and approximately half of all prescription drugs act directly or indirectly through GPCRs (Drews, 1996). Indeed, AT1 is the molecular target of angiotensin II receptor blockers (ARBs), a widely used class of drugs for cardiovascular disease. Although GPCRs were initially thought to function as monomers; many GPCRs can form dimers (Bouvier, 2001), and heterodimerization among GPCRs can alter receptor function by modulating ligand binding, receptor activation, or desensitization (Kaupmann et al., 1998; White et al., 1998; Jordan and Devi, 1999; Maggio et al., 1999; George et al., 2000; Gines et al., 2000; Rocheville et al., 2000a,b; Gomes et al., 2001; Pfeiffer et al., 2001; Lavoie et al., 2002; Pfeiffer et al., 2002; Stanasila et al., 2003; Xu et al., 2003).
Apelin and APJ are endogenous counter-regulators of the renin–angiotensin–aldosterone system (RAAS). APJ and AT1 are co-expressed in a number of cardiovascular tissues and cell types under both normal and diseased conditions (Ishida et al., 2004; Zhong et al., 2005; Ashley et al., 2006; Iwanaga et al., 2006). We and others have shown that apelin antagonizes the effects of Ang II, the primary effector molecule of the RAAS in the vasculature (Chun et al., 2008; Siddiquee et al., 2011; Sun et al., 2011). This antagonism could be mediated through the convergence of two independent intracellular signalling pathways or via the direct physical interaction of APJ with AT1 to form a GPCR heterodimer. AT1 heterodimerizes with GPCRs that regulate cardiovascular function, including the Ang II type 2 receptor (AT2) (AbdAlla et al., 2001); the β2-adrenergic receptor (β2AR) (Barki-Harrington et al., 2003); the bradykinin (B2) receptor (Quitterer et al., 2004) and APJ (Chun et al., 2008; Sun et al., 2011), leading to significant changes in AT1 function.
Here, we determined the mechanism by which apelin signalling antagonizes AT1. We confirmed heterodimerization of endogenously expressed APJ and AT1 GPCRs using co-immunoprecipitation and of heterologously expressed receptors using both co-immunoprecipitation and BRET. Furthermore, we demonstrated that Ang II signalling is antagonized through apelin-dependent heterodimerization and APJ-mediated negative allosteric modulation of AT1 function. This work establishes a new paradigm for cross-regulation of GPCR signalling, with implications for approaches to pharmacological interventions targeting this important class of receptors.
Methods
Materials
Unlabelled apelin-13 (Ap13), the endogenous agonist for APJ, and Ang II peptides were purchased from Bachem (King of Prussia, PA). [125I]-Glp65, Nle75, Tyr77-Ap13, [125I]–Tyr4-Ang II, and [125I]-Sar2, Ile8-Ang II were purchased from Perkin Elmer (Waltham, MA). All other chemicals and reagents were from Sigma-Aldrich (St Louis, MO). All drug and molecular target nomenclature follows Alexander et al. (2011).
Cell culture
HEK293 cells were cultured in minimum essential medium (MEM) (Invitrogen, Carlsbad, CA) supplemented with 10% FBS and penicillin, streptomycin and amphotericin (Sigma, St Louis, MO). CHO-K1 cells stably expressing APJ (CHO-K1-APJ) and/or AT1 (CHO-K1-APJ : AT1, CHO-K1-AT1) with β-arrestin/β-galactosidase enzyme fragment complementation constructs (DiscoveRx, Freemont, CA) were cultured in HAM's F-12 medium (Hyclone, Logan, UT) supplemented with 10% FBS and 200 μg mL−1 hygromycin (EMD Biosciences, San Diego, CA) and 600 μg mL−1 Geneticin (Cellgro, Manassas, VA). All cells were incubated at 37°C (5% CO2, 95% relative humidity) and maintained at less than 70% to 80% confluence (approximately 75 000 cells per cm2). Radioligand binding studies confirmed equivalent cell surface expression of APJ and AT1 in CHO-K1-APJ : AT1 (Table S1).
Immunoprecipitation and Western blot analyses
HEK-293 cells (5.0x106 cells per 10 cm dish) were lysed in RIPA buffer (10 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1.0% Triton X100, 1.0% sodium deoxycholate, 1 mM EDTA and 1X mini-complete protease inhibitor cocktail; Roche, Indianapolis, IN) and subjected to immunoprecipitation using either mouse monoclonal anti-AT1 or rabbit polyclonal anti-APJ antibody (R&D, Minneapolis, MN). Cross-linking was performed on the cell lysates either before or after the immunoprecipitation using 3,3′-dithiobis-sulfosucciniimidyl propionate (DTSSP) (ThermoScientific, Rockford, IL). In a similar experiment, HEK-293 cells (5.0 × 106 cells per 10 cm dish) were transfected with equivalent molar masses of individual plasmids, one encoding haemagglutinin (HA)-tagged APJ (pHA-APJ) and another encoding Flag-tagged AT1 (pFlag-AT1). Forty-eight hours post transfection, cellular membranes were isolated from these cells using established protocols (George et al., 1998), and DTSSP was used to cross-link the isolated cell membranes (ThermoScientific). Isolated cell membranes were subjected to immunoprecipitation using either mouse monoclonal anti-HA or rabbit polyclonal anti-Flag antibody (Sigma).
Total cell lysates, immunoprecipitates, or membrane preparations were loaded onto a precast 4–10% NuPAGE gel (Invitrogen), separated by electrophoresis and transferred to PVDF membrane (Invitrogen). Immunoreactivity was detected by Western blot using mouse anti-HA, rabbit anti-Flag, rabbit anti-APJ or mouse anti-AT1 antibodies as appropriate. Immunoreactive proteins on the membrane were visualized using goat anti-mouse antibody conjugated with IRDye800, rabbit anti-Flag or goat anti-rabbit antibody conjugated with IRDye680 and quantified using the Odyssey scanner (Li-Cor Biosciences, Lincoln, NE). Fluorescence intensity of each band was determined using the Image Studio software (Li-Cor Biosciences).
Bioluminescence resonance energy transfer (BRET)
BRET experiments and analysis were performed following established protocols for optimization (Pfleger et al., 2006). Custom plasmids encoding C-terminally tagged Renilla luciferase (RLuc) and enhanced-yellow fluorescent protein (eYFP) forms of the human APJ receptor (designated pAPJ-RLuc and pAPJ-eYFP) and of the human AT1 receptor (designated pAT1-RLuc, pAT1-eYFP) were designed in-house, synthesized by Genecopeia (Gaithersburg, MD), and the DNA sequence verified prior to their use. The donor to acceptor pairs and molar ratio were optimized using various concentrations of each plasmid and, once optimized, remained constant throughout the study. For non-saturating BRET studies, HEK293 cells were transiently transfected with 2.75 μg of pAT1-eYFP and 8.25 μg of pAPJ-RLuc (ratio of 1:3 acceptor to donor plasmid) using FuGENE-6 transfection reagent (Roche) in OptiMEM medium (Invitrogen). When saturation BRET studies were performed, the ratio of acceptor to donor plasmids was varied accordingly. After transfection, cells were dissociated from the transfection plate using non-enzymatic dissociation buffer (Sigma), pooled and dispensed into a white opaque 96-well plate at a cell seeding density of 50 000 cells per well. To initiate BRET, cells were incubated with Ap13 (100 nM), Ang II (100 nM) or Ap13 plus Ang II (100 nM each) for 1 h. After, coelenterazine h (5 μM) in PBS was added to each well. The resulting BRET signals were measured using the Flexstation3 Multimode Plate Reader (Molecular Device, Sunnyvale, CA) with sequential integration of both the RLuc signal (480 nm) and the eYFP signal (530 nm). The BRET signal is determined by calculating the ratio of the light intensity emitted by AT1-eYFP over the light intensity emitted by the APJ-RLuc. netBRET is determined by subtracting the BRET ratio obtained under unstimulated (vehicle control) conditions from the BRET ratio obtained when cells were exposed to ligands (Ap13, Ang II alone or in combination) (Pfleger et al., 2006).
Radioligand binding
APJ binding assays were performed with the APJ agonist [125I]-Glp65, Nle75, Tyr77-Ap13 (0.2–4 nM) in 25 mM HEPES pH 7.4, 10 mM MgCl2, 1 mM CaCl2, 0.5% BSA. AT1 binding assays were performed with either the agonist [125I]–Tyr4-Ang II (0.2–4 nM) or the antagonist [125I]-Sar1, Ile8-Ang II (0.2 nM) in 50 mM Tris–HCl pH 7.4, 5 mM MgCl2, 0.5% BSA. All ligand-binding experiments were performed on membrane fractions collected from cell lines stably expressing APJ and AT1, either alone or together (Smith et al., 2001). Assays were performed at room temperature for 60 min before termination by vacuum filtration through a pre-wet 96-well GF/C filter plate (MultiScreen Harvest plate, Millipore Corp., Billerica, MA, USA) and washed five times with 200 μL of ice-cold wash buffer (50 mM Tris–HCl pH 7.4, 0.5% BSA). Harvest plates were pre-coated with 0.3% polyethyleneimine or 0.5% BSA (Sigma) to reduce nonspecific binding of apelin or Ang II respectively. After filtration and washing, plates were air-dried overnight and 20 μL of scintillation liquid (MultiScint 20, Perkin Elmer) was added to each well. The bound radioactivity was measured using a TopCount NXT (Perkin Elmer) microplate scintillation and luminescence counter. Non-specific binding was determined in parallel reactions in the presence of an excess of unlabelled ligand (10 μM), and specific binding was defined as the difference between total and non-specific binding. Non-specific binding accounted for 20–30% of the total binding.
Inositol-1-Phosphate (IP1) assay
IP1 production was measured using the IP-one homogeneous time resolved fluorescence (HTRF) kit (Cisbio, Bedford, MA) according to the manufacturer's protocol (Trinquet et al., 2011). Briefly, CHO-K1-APJ : AT1 cells were seeded at a density of 10 000 cells per well in a 384-well low volume white, tissue culture-treated plate (Corning, Manassas, VA) containing serum-free HAM's F-12 medium. After 8 h, the culture medium was removed from the plate by centrifugation at 41× g for 30 s, and 7 μL of assay buffer (20 mM HEPES + 0.1% BSA in HBSS buffer, pH 7.4) was added into each well. Next, an additional 7 μL of assay buffer containing Ang II at a range of concentrations (0.0–2.0 μM), with or without Ap13 (1000, 100 and 10 nM) was added to each well, and the plate was sealed. The plate was incubated for 30 min at 37°C, after which an additional 3 μL of detection reagent (lysis buffer containing 2.5% Eu3+-anti-IP1 antibody and 2.5% IP1-d2) was added, and the plate was incubated for 1h at room temperature. The plate was read each hour for 3 h using the Flexstation3 Multimode Plate Reader (Molecular Devices, Sunnyvale, CA). The wells were excited with light at 340 nm and emitted light was measured at 615 and 665 nm. The time resolved-fluorescence resonance energy transfer (TR-FRET) 665 nm/615 nm ratio, which is inversely proportional to the IP1 accumulation, was used to measure the amount of IP1 produced.
β-arrestin recruitment assay
Receptor activation was measured using the receptor specific PathHunter™ β-arrestin eXpress Assay kit (DiscoveRx). Two days prior to assay, frozen aliquots of the PathHunter CHO-K1-AT1 β-arrestin EFC cell line were thawed, resuspended in cell plating reagent (CP2) and reverse-transfected by dispensing 20 000 cells per well into a 96-well plate containing pHA-APJ (250 ng) or the empty vector pcNDA3.1 (250 ng) pre-complexed with lipofectamine2000 transfection reagent (Invitrogen) in OptiMem serum-free media and incubated at 37°C and 5% CO2. After 5 h, the transfection media was replaced with complete culture medium, and the cells were returned to the incubator for another 43 h. The day of the assay, 50 μL of Ang II alone or in combination with Ap13 was suspended in assay buffer (1X HBSS, 20 mM HEPES and 0.01% BSA) and added to the wells at the indicated concentrations. Cells were incubated with peptides for 1.5 h at 37°C, after which 100 μL of PathHunter detection reagent was added to each well. In the reciprocal experiment, CHO-K1-APJ β-arrestin EFC cells were transfected with pFlag-AT1 or pcDNA3.1 and exposed to Ap13 at a range of concentrations alone or in combination with Ang II. In both experiments, the EFC reaction was developed for 90 min at room temperature in the dark. Receptor activation, as indicated by EFC-dependent chemiluminescence, was measured using a SpectraMax M5 Multimode Plate Reader (Molecular Devices) with 1.0 s per well integration time.
Statistical analysis and curve fitting
All data are presented as mean ± SEM. Statistical analysis and curve fitting of the data were performed using GraphPad Prism version 5 (GraphPad Software, San Diego, CA).
[125I]-Tyr4-Ang II saturation binding data in the absence and presence of Ap13 were globally fit to the equation
| (1) |
where Y represents [125I]-Tyr4-Ang II specific binding; [X] is the concentration of [125I]- Tyr4-Ang II; Bmax denotes the maximal density of binding sites recognized by [125I]-Tyr4-Ang II; and KD represents the radioligand equilibrium dissociation constant.
Competition binding analysis for the agonist [125I]-Tyr4-Ang II by unlabelled Ang II in the absence and presence of Ap13 (100 nM) according to the equation (GraphPad Prism version 5):
| (2) |
where Y represents specific binding of the radioligand, Top is the specific binding of the radioligand in the absence of competing ligand, Bottom is the specific binding of the radioligand equivalent to nonspecific binding, IC50 is the concentration of competing ligand that produces radioligand binding halfway between the Top and Bottom, and X is the logarithm of the concentration of the competing ligand. For the antagonist [125I]-Sar1,Ile8-Ang II, the competition binding by unlabelled Ang II in the absence and presence of Ap13 (1 μM) was preferentially fit, as determined by an extra sum-of-squares F-test, to one-site (Eq.2) or the following two-site inhibition mass-action curve respectively:
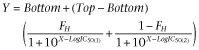 |
(3) |
The Cheng and Prusoff 1973) equation was used to convert IC50 estimates to equilibrium dissociation constants.
Whole cell interaction studies investigating the influence of Ap13 on Ang II-mediated IP1 formation and β-arrestin recruitment to the AT1 receptor were globally fit to the following operational model of allosterism to derive functional estimates of affinity and cooperativity:
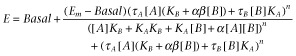 |
(4) |
where Em is the maximal system response for the pathway; n is the transducer slope factor; KA and KB are the equilibrium dissociation constants of Ang II (A) and Ap13 (B), respectively; α is the cooperative factor describing the allosteric modulation of ligand binding; β denotes the allosteric effect of Ap13 on the efficacy of Ang II; and τA and τB are the operational measures of Ang II and Ap13 efficacy respectively. Within the analysis, LogτB was constrained to equal −100 due to the lack of Ap13 efficacy; α was constrained to equal 1, as radioligand-binding studies indicated that Ang II and Ap13 have neutral binding cooperatively; and Em was constrained to less than 150% as Ang II, in the absence of an interacting ligand, is considered to be a full agonist.
Results
Formation of APJ : AT1 heterodimers
Natively expressed APJ and AT1 were individually detected in lysates from untransfected HEK293 cells that were precipitated using antibodies directed against human AT1 or APJ. The immunoreactivity of each receptor was observed at the appropriate molecular weight (∼46 and 49 kDa for APJ and AT1, respectively) after being subjected to polyacrylamide gel electrophoresis under denaturing conditions (Figure 1A, B, lane 1). Cross-linking the proteins with DTSSP both before and after immunoprecipitation resulted in the appearance of a higher molecular weight (∼98 kDa) protein complex that exhibited immunoreactivity with both native anti-APJ and anti-AT1 antibodies. This high molecular weight complex was observed in samples that were precipitated with either antibody, indicating that APJ and AT1 physically interact in the cell membrane (Figure 1A, B, lanes 2, 3). Homodimers of both APJ and AT1 were observed in untransfected HEK-293 cell lysates by immunoprecipitation and cross-linking between the receptors (Figure S1A, B).
Figure 1.
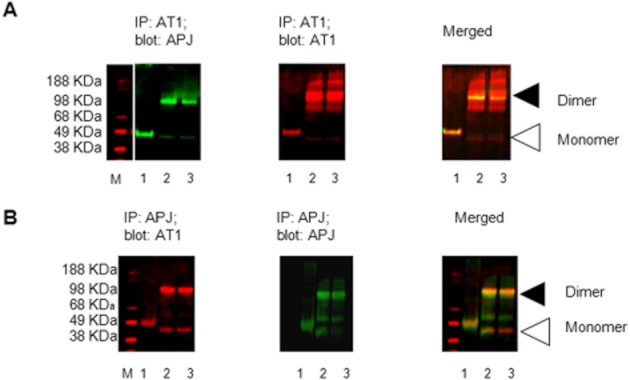
Physical interaction between endogenous APJ and AT1 receptors in HEK293 cells. (A) Immunoprecipitation of AT1 using α-AT1 antibody followed by Western blot with α-APJ antibody; lane 1: whole cell lysate immunoprecipitated with α-AT1; lane 2: whole cell lysates subjected to precipitation with AT1 antibody followed by exposure to the chemical cross-linker DTSSP; lane 3: whole cell lysate subjected to precipitation with AT1 antibody prior to cross-linking. Left panel, APJ immunoreactivity, Middle panel shows AT1 immunoreactivity; right panel, merged of left and middle panels. M: Marker. (B) In the reciprocal experiment, APJ receptor immunoprecipitated with α-APJ antibodies, and co-enriched AT1 receptor was detected by Western blot; lane 1: whole cell lysate immunoprecipitated with α-APJ; lane 2: whole cell lysates subjected to precipitation with APJ antibody followed by exposure to the chemical cross-linker DTSSP; lane 3: whole cell lysate subjected to precipitation with APJ antibody prior to cross-linking. Left panel, AT1 immunoreactivity; middle panel, APJ immunoreactivity; right panel, merged of left and middle panels. Triangles indicate the position at which monomers and dimers migrated through the gel.
We next sought to confirm these initial findings and more thoroughly characterize the interaction between APJ and AT1 using overexpressed, tagged forms of these receptors. First, HEK-293T cells were transiently transfected with equal masses of either pFlag-AT1 or pHA-APJ. As before, heterodimerization between these receptors was determined by co-immunoprecipitation followed by Western blotting. HA-APJ immunoreactivity was detected in cell lysates precipitated with anti-Flag antibody (Figure 2A, lane 2). In the reciprocal experiment, Flag-AT1 immunoreactivity was detected in cell lysates precipitated with anti-HA antibodies (Figure 2A, lane 4). In lysates from cells lacking one of the individual protomers of the APJ : AT1 dimer, neither of the cognate protomers was detected after immunoprecipitation, indicating that the interaction between the two is specific (Figure 2A, lanes 1, 3).
Figure 2.
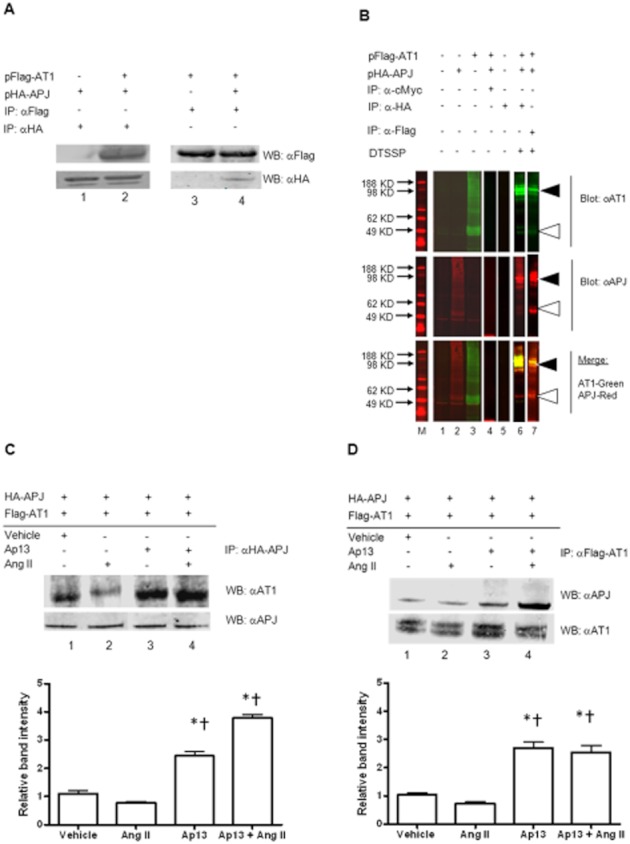
Specific physical interaction between APJ and AT1. (A) Immunoprecipitation of tagged APJ using α-HA antibody followed by Western blot with α-Flag antibody to detect tagged AT1 revealed a co-enrichment of the two receptors (lanes 1, 2). In the reciprocal experiment, AT1 receptors were precipitated with α-Flag antibodies, and co-enriched APJ receptors were detected by Western blot (lanes 3, 4) with α-HA antibodies. (B) Control precipitation and cross-linking studies reveals specificity of the APJ interaction with AT1. Top panel, AT1 immunoreactivity using antibodies directed at the native receptor. Middle panel, APJ immunoreactivity using antibodies directed at the native receptor. Bottom panel, top and middle panels merged. M: Marker; lane 1: negative control, untransfected HEK293 cell lysate; lane 2: whole cell lysate from cells transfected with pHA-APJ alone; lane 3: whole cell lysate from cells transfected with pFlag-AT1 alone; lane 4: IP control, lysate from cells transfected with pHA-APJ and pFlag-AT1 and precipitated with α-c-Myc; lane 5: untransfected cell lysate precipitated with α-HA; lane 6: whole cell lysate from cells transfected with pHA-APJ and pFlag-AT1 and subjected precipitation with α-HA antibodies followed by cross-linking with DTSSP; lane 7: whole cell lysate from cells transfected with pHA-APJ and pFlag-AT1 and subjected precipitation with α-Flag antibodies followed by cross-linking with DTSSP. The use of antibodies directed against the native APJ and AT1 reveal trace levels of endogenous APJ and AT1 in lanes 1, 2, 3. Triangles indicate the position at which monomers and dimers migrated through the gel. Lanes 1–5 are from the same blot. Lanes 6, 7 are from a separate experiment. All lanes are presented as discrete panels to delineate the different conditions under which each sample was prepared and to facilitate comparison between the cross-linking samples and those that were obtained without cross-linking. Ap13, but not Ang II, enhances the interaction between APJ and AT1. Immunoprecipitation and Western blot of HEK293T cells transfected with pHA-APJ and pFlag-AT1 and incubated with to Ap13 (100 nM) or Ang II (100 nM), alone or in combination. (C) Immunoprecipitation of HA-tagged APJ using α-HA antibody followed by Western blot with α-AT1 antibodies revealed a co-enrichment of the two receptors under vehicle (PBS)-treated conditions (lane 1). Immunoreactivity for AT1 was slightly decreased in the presence of Ang II (lane 2), but significantly increased in the presence of Ap13 alone (lane 3) and in combination with Ang II (lane 4). (D) Immunoprecipitation of Flag-tagged AT1 using α-Flag antibody followed by Western blot with α-APJ antibodies revealed a similar co-enrichment of the two receptors under vehicle (PBS)-treated conditions (lane 1). Immunoreactivity for APJ was slightly decreased in the presence of Ang II (lane 2), but significantly increased in the presence of Ap13 alone (lane 3) and in combination with Ang II (lane 4).Quantification of individual band intensity by optical density analysis is presented below each panel. *P < 0.01 when compared with control, *P < 0.001 when compared with Ang II as determined by anova with Bonferroni's multiple comparison test. IP, immunoprecipitation; WB, Western blot.
To confirm the specificity of the interaction and rule out the possibility that the observed dimerization of the APJ with AT1 is a consequence of transmembrane protein overexpression or non-specific interaction/aggregation between affinity tagged-proteins, we performed a number of control experiments. First, we repeated our immunoprecipitation studies using the chemical cross-linker DTSSP to stabilize the interaction between the APJ and AT1 receptors, and to limit the interactions to proteins within 12.0 angstroms, the length of the DTSSP linker arm. Membranes from cells transfected with both pHA-APJ and pFLAG-AT1 were incubated with DTSSP prior to immunoprecipitation, followed by Western blotting. As before, endogenous APJ and AT1 monomers were each detected in lysates from transfected HEK-293 cells at the expected molecular weight (Figure 2B, lane 1). Similarly, immunoreactivity for both APJ and AT1 was observed in cells transfected with pHA-APJ or pFlag-AT1 but at a greater intensity, reflecting the increased expression of the receptors by these plasmids (Figure 2B, lanes 2, 3). A high-molecular weight band (∼96 kDa) that displayed immunoreactivity for both APJ and AT1 was detected in cross-linked samples. The appearance of the higher molecular weight complex coincided with reduced immunoreactivity for lower molecular weight bands representing the individual receptor monomers (Figure 2B, lanes 6, 7).
We then attempted to precipitate the dimer using antibodies against the unrelated affinity tag, cMyc. Anti-cMyc antibodies failed to precipitate any protein that reacted with either receptor antibody (Figure 2B, lane 4). Immunoprecipitation using anti-HA antibodies in lysates from untransfected cells similarly failed to detect either the individual receptors or the heterodimeric complexes of APJ with AT1 (Figure 2B, lane 5). These results, together with the experiment using endogenous receptors and native antibodies, indicate that the co-precipitation of APJ with AT1 was specific and did not result from the random co-precipitation of integral membrane proteins or the aggregation of solubilized hydrophilic proteins in solution.
We next examined the effect of receptor activation on heterodimerization between APJ and AT1 by exposing cells to the APJ agonist, Ap13, and the AT1 agonist, Ang II, individually and in combination prior to immunoprecipitation. Increased AT1 immunoreactivity was observed in lysates from HEK-293T cells expressing HA-APJ and Flag-AT1 incubated with Ap13 or Ap13 + Ang II and subsequently precipitated with anti-HA antibodies (Figure 2C), indicating that Ap13 increased the physical interaction between APJ and AT1. In contrast, Ang II incubation did not significantly affect formation of the heterodimer. Results were similar when the reciprocal experiment was performed (Figure 2D).
We next measured receptor heterodimerization using BRET assay in cells transfected with the fusion proteins, AT1-Rluc and APJ-eYFP, that had been functionally verified using Ca2+ or cAMP assays (Figure S2A, B). BRET assays involved the addition of Coelenterazine h to cells expressing APJ-RLuc and AT1-eYFP, which generated the expected bioluminescence signal with a primary emission peak at 480 nm and an additional peak at 530 nm. This second peak represents the energy transfer from RLuc to eYFP and is indicative of the physical proximity of two labelled constructs. No luminescence was detected in cells expressing AT1-eYFP alone (Figure S3). Co-transfection of the unrelated melanocortin 4 receptor fused to eYFP (hMC4R-eYFP) with APJ-RLuc did not modify the fluorescence signal, indicating the specificity of the APJ : AT1 BRET interaction (Figure S3). The ratio of BRET donor and acceptor plasmids was systematically optimized by increasing the acceptor plasmid mass while holding the donor plasmid mass constant. Increasing the acceptor plasmid mass elicited a rise in BRET signal that depended upon the mass of acceptor plasmid (Figure S4A, B). Using non-saturating, optimized ratios of donor-to-acceptor plasmids, Ap13 (100 nM) increased netBRET 83-fold over saline controls (0.3% ± 0.2% vs. 25.0% ± 2.0% netBRET; P < 0.001). Results were similar upon concomitant addition of Ap13 + Ang II (each at 100 nM), but Ang II alone did not affect netBRET (Figure 3A). We next performed BRET under saturating conditions to demonstrate the specificity of the interaction between the BRET pairs, and to further characterize the effect of Ap13 and Ang II ligands on the dimerization of APJ with AT1. At a range of donor-to-acceptor ratios, Ap13 (100 nM) saturated the BRET signal. In contrast Ang II (100 nM) significantly reduced basal BRET relative to Ap13 and vehicle (max BRET Ang II = 47.3% ± 1.8%, saline = 58.6% ± 2.0%, Ap13 = 107.3% ± 4.5%; P < 0.0001 by extra sum-of-squares F test) (Figure 3B).
Figure 3.
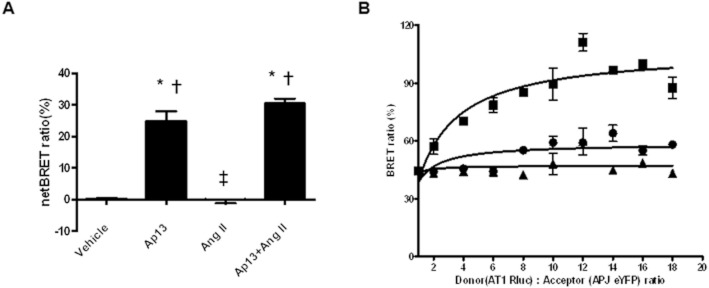
Ap13 but not Ang II induces APJ : AT1 heterodimerization in living cells. (A) HEK-293 cells transfected with APJ-Rluc and AT1-eYFP treated with either Ap13 (100 nM), Ang II (100 nM), Ap13 + Ang II (100 nM each). *P < 0.001 when compared with Ang II, ‡P < 0.001 when compared with Ap13 as determined by anova with Bonferroni's multiple comparison test. (B) Saturation of donor to acceptor BRET ratio in the presence of vehicle, Ap13 (100 nM) and Ang II (100 nM). Data presented are from two or three independent experiments each with an n = 3.The peak light intensities of APJ-RLuc and AT1-eYFP were measured at 480 and 530 nM respectively. The BRET signal is determined by calculating the ratio of the light intensity emitted by AT1-eYFP over the light intensity emitted by the APJ-RLuc, and the netBRET ratio is calculated by subtracting the vehicle induced BRET ratio from the ligand induced BRET ratio.
Allosteric modulation of AT1 by Ap13 and APJ
Equilibrium radioligand binding studies were performed to determine the effect of Ap13/APJ on the binding of Ang II at AT1. Membranes from CHO-K1 cells stably expressing AT1 alone or co-expressed with APJ exhibited a modest, statistically insignificant, decrease in affinity (KD AT1 alone = 0.282 ± 0.076 nM vs. KD AT1 + APJ = 0.390 ± 0.069 nM) and Bmax for [125I]-Tyr4-Ang II (Bmax AT1 alone = 2800.0 ± 245.2 fmol mg−1 protein vs. Bmax AT1 + APJ = 2472.0 ± 153.0 fmol mg−1 protein) (Figure 4A, B, Table 1). Unlabelled Ap13 enhanced this effect in a concentration-dependently decreased [125I]-Tyr4-Ang II binding to membranes from cells co-expressing AT1 and APJ (Figure 4C, Table 2A). In the reciprocal experiment, unlabelled Ang II did not affect the KD or the Bmax of [125I]-Glp65, Nle75, Tyr7-Ap13 for APJ in membranes from cells co-expressing APJ and AT1 (Figure 4D, Table 3B). Membranes from CHO cells stably expressing AT1, but not APJ, did not exhibit specific binding for [125I]-Glp65, Nle75, Tyr7-Ap13 (Figure S5). In competitive binding assays using membranes with APJ and AT1, excess unlabelled Ap13 (100 nM) significantly reduced the affinity of [125I]-Tyr4-Ang II for AT1 (pIC50 AT1 + APJ = 8.74 ± 0.04 with vehicle vs. pIC50 AT1 + APJ = 8.91 ± 0.06 with Ap13, p < 0.05 as determined by an extra sum-of-squares F-test) (Figure 5A). The pIC50 for Ang II was unaltered by Ap13 in plasma membranes from CHO cells expressing only the AT1 receptor (pIC50 AT1 = 9.41 ± 1.85 with vehicle vs. pIC50 AT1 = 9.54 ± 1.34 with Ap13) (Figure S6). We next examined the effect of Ap13/APJ on the affinity state of AT1. As expected, competition of binding of the AT1 antagonist, [125I]-Sar1, Ile8-Ang II (0.03 nM), by unlabelled Ang II revealed the classic biphasic inhibition isotherm in CHO cells co-expressing AT1 and APJ. The logarithm of equilibrium dissociation constant (LogKA) for the high- and low-affinity states, determined by global nonlinear regression analysis, was −9.6 ± 0.2 and −7.5 ± 0.2, respectively. Adding Ap13 (1 μM) shifted the inhibition isotherm from a biphasic to a monophasic isotherm with an estimated LogKA of −7.8 ± 0.1, which corresponds to the low-affinity state AT1 observed in the absence of Ap13 (Figure 5B, Table 4).
Figure 4.
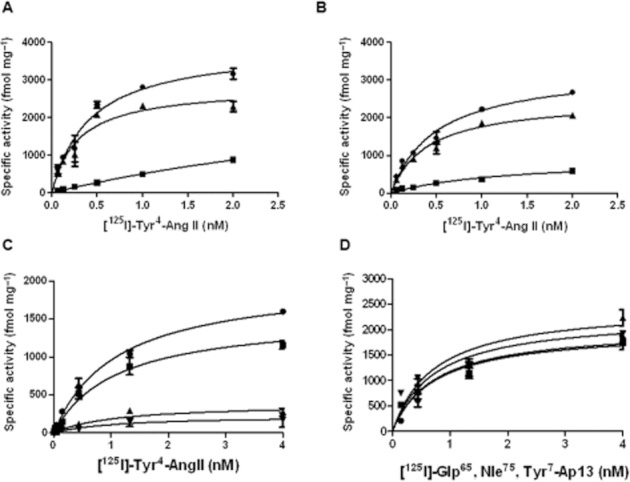
Effects of Ap13/APJ on Ang II binding. APJ alone has no significant effect on [125I]-Tyr4-Ang II binding to membranes from cells expressing AT1 receptor alone (A), or from cells expressing AT1 and APJ (B). Six different concentrations of [125I]-Tyr4-Ang II were incubated with 0.5 μg of membranes for 1 h at room temperature before quenching the reaction by rapid filtration to separate bound Ang II from free Ang II. Nonspecific binding was determined by using excess unlabelled Ang II (10 μM). Specific binding was calculated by subtracting nonspecific binding from total binding. KD and Bmax values are reported in Table 1. Ap13-activated APJ decreases binding of [125I]-Tyr4-Ang II to AT1. (C) Increasing concentrations of apelin reduced the Bmax of Ang II to the AT1 receptor when co-expressed with APJ. An extra sum-of-squares F test determined that estimates of KD were not significantly different, and therefore were shared, across data sets. In contrast, the Bmax was significantly different across data sets (P < 0.05). (D) Increasing concentrations of Ang II had no effect on the Bmax of Ap13 to APJ when co-expressed with AT1. An extra sum-of-squares F test determined that estimates of KD were not significantly different and therefore were shared, across data sets. Similarly, the Bmax was not significantly different across data sets (P > 0.05). Each symbol represents the mean specific activity ± SEM (n = 3) from three independent experiments. Curves superimposed on the data represent the best fit of the data from equation 1 obtained by nonlinear regression analysis performed using GraphPad Prism 5. Details of the analysis are provided in the data analysis section of Methods.
Table 1.
Specific binding [125I]-Tyr4-Ang II to membranes from cells expressing AT1 or AT1 + APJ receptors
| Receptors | ||
|---|---|---|
| Binding parameter | AT1 | AT1 + APJ |
| Bmax (fmol mg protein−1) | 2800 ± 245.2 | 2472.0 ± 153.0 |
| KD (nM) | 0.282 ± 0.0762 | 0.39 ± 0.069 |
Bmax: the maximal density of binding sites; KD: radioligand equilibrium dissociation constant. The values represent the means ± SEM of at least three independent experiments.
Table 2A.
Effect of apelin on the saturation binding of [125I]-Tyr4-Ang II to membranes from cells expressing AT1 with APJ
| Apelin (nM) | Ang II Bmax (fmol mg protein−1) |
|---|---|
| 0 | 2004 ± 131.2 |
| 10 | 1286 ± 213.1 |
| 100 | 834.7 ± 312.6 |
| 1000 | 373.1 ± 71.22 |
Calculated Bmax values obtained from saturation isotherm [125I]-Tyr4-Ang II in the presence or absence of various concentration of Ap13. The values represent the mean ± SEM of at least three independent experiments.
Table 2B.
Effect of Ang II on the saturation binding of [125I]- Glp65, Nle75, Tyr7–Ap13 to membranes from cells expressing AT1 with APJ
| Ang II (nM) | Ap13 Bmax (fmol mg protein−1) |
|---|---|
| 0 | 2413 ± 179.7 |
| 10 | 1929 ± 212.4 |
| 100 | 2625 ± 265.2 |
| 1000 | 1874 ± 185.0 |
Calculated Bmax values obtained from saturation isotherm of [125I]- Glp65, Nle75, Tyr7–Ap13 (nM) in the presence or absence of various concentration of Ang II. The values represent the mean ± SEM of at least three independent experiments.
Figure 5.
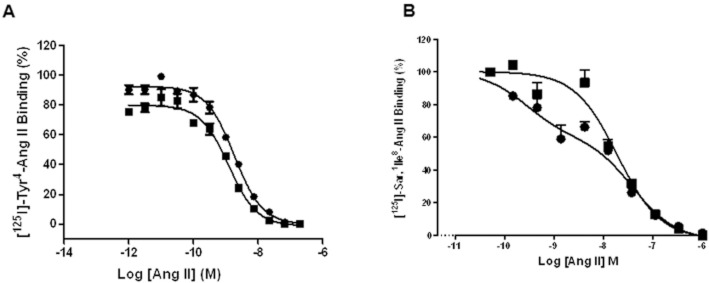
Ap13-activated APJ modulates the affinity state of AT1. (A) When AT1 is co-expressed with APJ, Ap13 decreases the affinity of [125I]-Tyr4-Ang II for AT1. Competition binding assays were performed on membranes from cells expressing both APJ and AT1 by using 0.28 nM of [125I]-Tyr4–Ang II in the presence of increasing concentrations of unlabelled Ang II or in the presence of increasing concentrations of unlabelled Ang II plus 100 nM of unlabelled Ap13. GraphPad Prism5 was used to calculate IC50 using equation 2 as described in the Methods section. (B) Competitive binding between [125I]-Sar2, Ile8-Ang II and unlabelled Ang II in the absence and presence of Ap13 (1 μM). The curve was preferentially fit using equations 3 and 2, as determined by an extra sum-of-squares F test, to a two- and one-site inhibition mass–action curve respectively. Each symbol represents the mean percentage of specific binding ± SEM (n = 4–6) from two independent experiments.
Table 3.
Effect of Apelin on the affinity state of AT1 co-expressed with APJ
| Receptor (AT1 + APJ) | LogKA (high affinity) | LogKA (low affinity) |
|---|---|---|
| Vehicle | −9.6 ± 0.2 | −7.5 ± 0.2 |
| Apelin (1 μM) | – | −7.8 ± 0.1 |
LogKA: The logarithm of equilibrium dissociation constant for the high- and low- affinity states. The values represent the means ± SE of at least three independent experiments.
APJ : AT1 heterodimerization antagonizes AT1 function
To determine the functional consequence of APJ heterodimerization with AT1, we measured IP1 generation and β-arrestin recruitment to AT1. In cells expressing equivalent levels of AT1 and APJ, Ang II (3.47 pM–2.5 μM) dose-dependently stimulated the formation of IP1. Apelin (10, 100 and 1000 nM) dose-dependently decreased the maximal Ang II response. This depression approached a limit, the magnitude of which was governed by the cooperativity, indicative of a negative allosteric interaction between APJ and AT1 (Figure 6A, B). The results of global fitting of IP1 accumulation data and β-arrestin recruitment data to an operational model of allosterism are presented in Table 5. The β-values for IP1 accumulation and β-arrestin recruitment suggest that Ap13 reduces Ang II signalling by 1.4- and 2.5-fold, respectively, in the presence of APJ : AT1 heterodimerization. Apelin did not affect AT1 activity in CHO-K1-AT1 cells lacking APJ (Figure S7A). Ang II did not affect APJ function in cells expressing both APJ and AT1 (Figure S7B).
Figure 6.
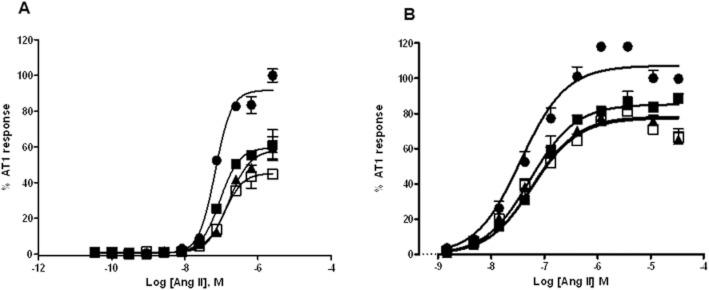
APJ : AT1 heterodimerization antagonizes AT1 function. (A) Inhibition of Gαq signalling pathway of AT1 by Ap13-activated APJ. Inositol-1-Phosphate (IP1) production was measured using CHO-K1-APJ : AT1 cells stably and equivalently expressing AT1 and APJ receptors in the presence of Ang II (0–2.5 μM) alone or in the presence of a fixed concentration of Ap13. (B) Inhibition of β-arrestin recruitment to AT1 by Ap13-activated APJ. PathHunter AT1 β-Arrestin EFC cells were transfected with pHA-APJ and incubated with Ang II (0.0–100 μM) alone or with a fixed concentration of Ap13. Each symbol represents the mean percent activation of AT1 ± SEM (n = 6) by Ang II at the indicated concentration. In both panels, curves represent the best global fit of the operation model of allosterism as determined using Equation 4.
Table 4.
The operational parameters for the model of allosteric interaction between AT1 and APJ receptors
| Allosteric modulator: Ap13 | ||
|---|---|---|
| Parameters | IP1 assay | β-Arrestin assay |
| LogKA | −7.5 ± 0.2 | −6.9 ± 0.1 |
| LogτA | 0.12 ± 0.05 | 0.38 ± 0.2 |
| LogKB | −8.5 ± 0.08 | −8.6 ± 0.1 |
| Logβ | −0.15 ± 0.06 | −0.35 ± 0.07 |
LogKA: The logarithm of equilibrium dissociation constant for the agonist (Ang II); LogKB: The logarithm of equilibrium dissociation constant for the allosteric modulator(AP13); LogτA: the operational measures of agonist (Ang II) efficacy; Logβ: denotes the allosteric effect of modulator (Ap13) on the efficacy of agonist (Ang II). The values represent the means ± SE of at least three independent experiments.
Discussion
We previously reported that apelin antagonizes Ang II-induced hypertension and cardiac fibrosis by inhibiting Ang II signalling through AT1 (Siddiquee et al., 2011). Here, we used biochemical, biophysical, and pharmacological approaches to determine the molecular basis of this inhibition. Immunoprecipitation and Western blot revealed that APJ and AT1 physically interact in cells that express these receptors both endogenously and heterologously. Adding the APJ agonist, Ap13, increased the relative abundance of APJ : AT1 dimers twofold over baseline, whereas adding the AT1 agonist, Ang II, did not affect APJ : AT1 heterodimerization. Ap13 dose-dependently increased the APJ : AT1 interaction as measured by BRET and netBRET, whereas Ang II did not. The interaction between APJ and AT1 displays characteristics associated with allosteric modulation, including inhibition of orthosteric AT1 ligand binding and non-competitive inhibition of orthosteric AT1 agonist efficacy. In equilibrium binding assays, Ap13-activated APJ decreased Ang II binding to AT1, without direct competition, by inducing dimerization of APJ with AT1 to induce a low-affinity state in AT1. In two distinct cell-based assays of AT1 function (IP1 production and β-arrestin recruitment), Ap13 saturably but incompletely blocked Ang II-mediated activation of AT1. These data clearly demonstrate that Ap13 induces the formation of a specific heterodimer complex between APJ and AT1, and that this formation disrupts Ang II binding to its receptor, thus inhibiting AT1 signal transduction.
The physical interaction between APJ and AT1 has significant consequences for AT1 function and pharmacology. Equilibrium binding experiments revealed that Ap13 dose-dependently decreased the Bmax of radiolabelled-Ang II in membranes from cells expressing both receptors. Competition-binding studies revealed that Ap13 decreases Ang II high-affinity binding at the AT1 receptor. Because neither Ap13 nor Ang II bind or act as competitive inhibitors of each other at their cognate receptors, induction of a low-affinity state for AT1 must result from negative allosteric modulation (Lazareno and Birdsall, 1995). Consistent with this interpretation, analysis of these functional data using the best global fit of the operational model of allosterism (Price et al., 2005) revealed that, in the presence of APJ : AT1 heterodimerization, Ap13 reduces Ang II signalling efficacy (Figure 6). Taken together, these data establish the molecular basis by which apelin antagonizes Ang II. It is interesting to note that data from the IP1 and β-arrestin recruitment assays suggest an allosteric interaction that modulates the activation state of a receptor, causing a global modulation of receptor function rather than only the primary signalling pathway. Future studies are required to more fully explore this potentially novel mode of allosteric interaction.
Our results indicate that APJ : AT1 heterodimerization is ligand-dependent. While this interpretation is consistent with other reports of ligand-induced heterodimerization of GPCRs (Rocheville et al., 2000a,b; Patel et al., 2002; Percherancier et al., 2005), it conflicts with studies showing that GPCR dimerization is a prerequisite for receptor ontogeny and that GPCR dimers are assembled into functional units in the endoplasmic reticulum (Percherancier et al., 2005; Pfleger and Eidne, 2006; Pfleger et al., 2006). Although we observed some basal interaction between APJ and AT1 in the BRET experiments, this interaction can be attributed to the use of overexpressed and tagged forms of the receptors. BRET changes in response to Ap13 could be attributed to a rearrangement of BRET donor and acceptor proteins (RLuc, eYFP) in a pre-formed APJ : AT1 heterodimer. While possible, the inverse effects of Ap13 and Ang II on BRET observed in both the saturation and optimized BRET experiments argue against this interpretation. Moreover, the ability of Ap13 to increase the amount of co-precipitated AT1 further supports our proposed model. Finally, Ap13 dose-dependently antagonized AT1 activity, but co-expression of APJ alone did not. Thus, the converging evidence from these complimentary experimental strategies indicates that APJ : AT1 dimerization is related to the APJ R and R* equilibria, which determines the extent of receptor activation (Samama et al., 1993). Therefore, we conclude that trans-inhibition of AT1 depends upon the activation state of the APJ protomer, a mechanism of action consistent with other models of Class A GPCR dimerization (Pin et al., 2005; Sohy et al., 2007; Vilardaga et al., 2008; Han et al., 2009). Furthermore, the magnitude of modulation by Ap13 was governed by the cooperativity between the modulator, Ap13-activated APJ, and the target, AT1. Figure 7 is a schematic diagram of this interpretation and is consistent with a model proposed to describe the interaction between the orexin-1 receptor and cannabinoid CB1 receptor (Ellis et al., 2006; Milligan and Smith, 2007).
Figure 7.
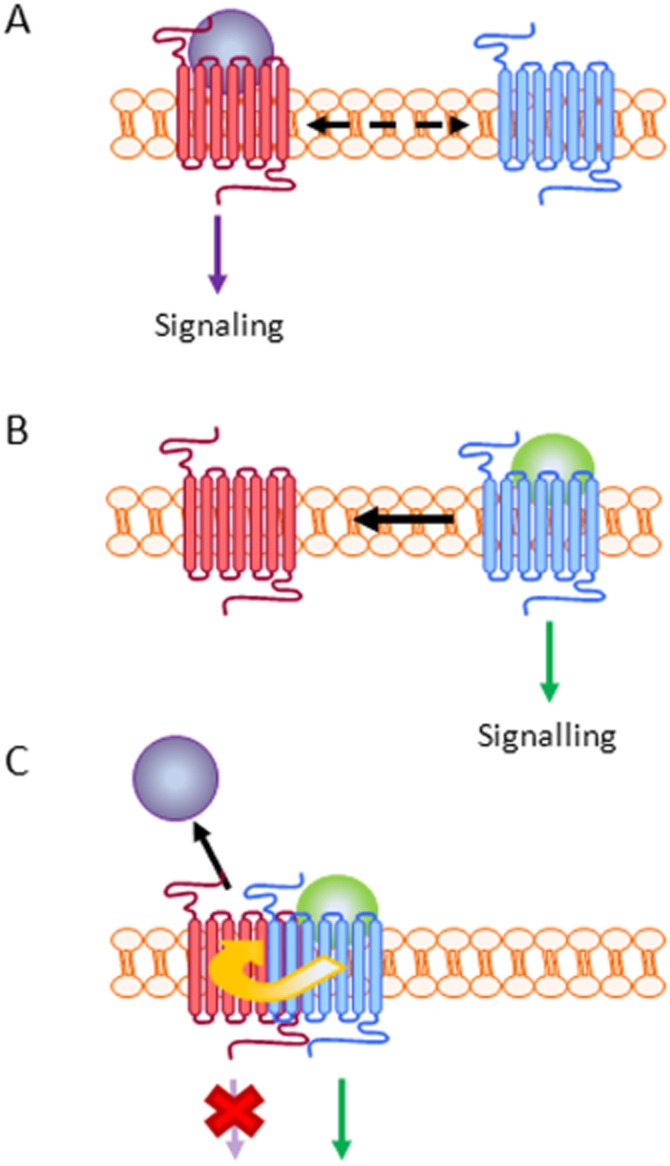
Apelin-dependent formation of an APJ : AT1 heterodimer results in negative allosteric modulation of AT1. (A) In the absence of Ap13, Ang II binds to the AT1 receptor (seven-transmembrane spanning receptor), activating the receptor and the subsequent transduction of signals through G-protein-dependent and -independent pathways. Even in the absence of Ap13, some APJ receptor may exist in a confirmation that favours heterodimerization with AT1 (as demonstrated in Figures 1 and 2). This is indicated by the dashed arrow in panel A. (B) Ap13 binding to APJ (seven-transmembrane spanning receptor) stimulates both APJ signalling and the formation of the APJ : AT1 heterodimer; a mechanism that is consistent with the role of apelin and APJ as counter-regulators of Ang II signalling via AT1. (C) The physical interaction of APJ and AT1 results in the transduction of an antagonist effect from APJ through AT1 to inhibit Ang II binding and AT1 signalling (red X). Thus, Ap13 functions allosterically (via APJ) to alter the binding and function of AT1.
Chun et al. (2008) demonstrated that Ang II stimulated dimerization between APJ and AT1, whereas Ap13 had only a modest effect, if any. Sun et al. (2011) reported that apelin-mediated activation of APJ opposed the formation of an APJ : AT1 heterodimer. Our data indicate that Ap13 drives the formation of APJ : AT1 heterodimers, a mechanism consistent with reports in which apelin antagonizes the physiological effects of Ang II in vivo, and not the reverse (Chun et al., 2008; Siddiquee et al., 2011). However, given the propensity for AT1 to form both homodimers and heterodimers, the relative cell surface receptor densities of APJ and AT1 are important in the formation and functional consequences of APJ : AT1 dimer formation. Indeed, our results suggest that the ability of apelin to oppose Ang II signalling depends both upon the activation state of APJ and the number of APJ receptors available to physically interact with AT1. In our experiments, the stoichiometric ratio between APJ and AT1 receptor density, as determined by equilibrium binding studies, was roughly equivalent. Previous studies did not examine the relative cell surface densities of APJ and AT1. In addition, the convergence of various technical approaches showed that apelin, not Ang II, consistently stimulated the formation of an APJ : AT1 dimer at physiological doses. Furthermore, much of our work was done in membrane preparations, which eliminates the effects of receptor internalization. Ultimately, the contradictory findings of these three studies reflect a greater complexity of the interaction between these two physiologically important systems. Future studies should explore the relative expression levels of APJ and AT1 in tissues to determine how receptor density modulates APJ : AT1 heterodimer formation and function.
In summary, apelin stimulates APJ to form a heterodimer with AT1 and antagonize Ang II signalling. These findings are consistent with the role of apelin and APJ to counter-regulate Ang II; the findings also extend previously reported observations (Chun et al., 2008; Siddiquee et al., 2011) by describing the underlying molecular mechanism. Collectively, the fundamental mechanism by which apelin counter-regulates the RAAS may be through ligand-dependent negative allosteric modulation of AT1. One aetiology of primary hypertension and other cardiovascular diseases characterized by a hyperactive RAAS may include impaired apelin and APJ function, possibly as a result of genetic variations (Niu et al., 2010). Moreover, given the clinical utility of AT1 receptor antagonists [also known as angiotensin receptor blockers (ARBs)], APJ agonists that induce APJ : AT1 dimerization and antagonize AT1 could represent a novel therapeutic strategy for the treatment of cardiovascular disease.
Acknowledgments
We thank Dr Arthur Christopoulos for his critical review of our allosteric modelling and subsequent conclusions and Pam Hurley, PhD, for her editorial critique of the manuscript, which was supported by the Sanford Burnham Medical Research Institute. This work was supported in part by the Florida Department of Health, James and Esther King Biomedical Research Program (grant 06-NIR-09 to LHS).
Glossary
- β2AR
β2-adrenergic receptor
- Ang II
angiotensin II
- Ap13
apelin-13
- APJ
apelin receptor
- APJ : AT1
APJ heterodimer complex with AT1
- AT1
angiotensin II type 1 receptor
- AT2
angiotensin II type 2 receptor
- B2
bradykinin B2 receptor
- BRET
bioluminescence energy resonance transfer
- CPM
counts per minute
- EFC
enzyme fragment complementation
- eYFP
enhanced-yellow fluorescent protein
- HA
haemagglutinin
- MEM
minimum essential medium
- RAAS
renin–angiotensin–aldosterone system
- RLuc
Renilla luciferase
Conflict of interest
The authors report no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Detection of native APJ and AT1 homodimers in HEK293 cell lysates. (A) Immunoprecipitation of AT1 using α-AT1 antibody followed by Western blot with α-AT1 antibody; lane 1: whole cell lysate immunoprecipitated with α-AT1 without cross-linking; lane 2: whole cell lysates cross-linked with DTSSP and followed by precipitation with AT1 antibody. (B) Immunoprecipitation of APJ with α-APJ antibodies followed by Western blot with α-APJ antibody; lane 1: whole cell lysate immunoprecipitated with α-APJ without cross-linking; lane 2: whole cell lysates cross-linked and followed by precipitation with APJ antibody. Triangles indicate the position at which monomers and dimers migrated through the gel.
Figure S2 AT1-eYFP and APJ-RLuc BRET constructs are functional receptors. (A) HEK-293T cells transiently expressing AT1-eYFPwas stimulated with Ang II (0.0–2.5 0μM) to mobilize Ca2+. (B) HEK-293T transiently cells expressing APJ-RLuc stimulated with a range of Ap13 (0.0–2.5 μM) to reduce forskolin-induced cAMP production. Each symbol represents the mean percentage of specific binding ± SEM (n = 3). Curves superimposed on the data represent the best fits using equation 2. Details are given in supplemental Methods section of the manuscript.
Figure S3 Determination of BRET specificity. BRET was detected in HEK-293 cells co-expressing APJ-RLuc and AT1-eYFP as indicated by an increase in signal intensity between 500 and 550 nm, whereas cells expressing either the APJ-RLuc or the AT1-eYFP alone did not exhibit changes in the signal intensity between 500 and 550 nm. When APJ-RLuc was co-expressed with the human melanocortin 4 receptor-eYFP (hMC4R-eYFP), no BRET was observed, confirming the specificity of the BRET between APJ-RLuc and AT1-eYFP.
Figure S4 Optimization of donor to acceptor ratio for BRET (A and B). To determine the optimum ratio between APJ and AT1 receptors for BRET, HEK-293T cells were transiently transfected with fixed amount of donor plasmid (pAPJ-RLuc or pAT1-RLuc) and various amount of acceptor plasmid (pAT1-eYFP or pAPJ-eYFP). The signal intensities at 480 and 530 nm were determined for RLuc and eYFP by using the Flexstation3 Multimode Plate Reader. The BRET signal is obtained by calculating the ratio of the light intensity emitted by AT1-eYFP over the light intensity emitted by the APJ-RLuc, and the net BRET ratio is calculated by subtracting the only donor BRET ratio from donor to acceptor BRET ratio.
Figure S5 Binding of [125I]-Glp65, Nle75, Tyr7–Ap13 to AT1. Membranes (0.5 μg of protein) from cells expressing AT1 receptor alone were incubated with six different concentrations of [125I]-Glp65,Nle75,Tyr7-Ap13 with or without 10 μM unlabelled Ap13 for 1 h at room temperature before rapid filtration was performed to separate bound from free Ap13. Nonspecific and specific binding were determined as described. Each symbol represents the mean counts per minute (CPM) ± SEM (n = 6). The lack of binding makes it impossible to determine the KD and Bmax values. Und, Undetermined.
Figure S6 Ap13 has no effect on the affinity of [125I-]Tyr4-Ang II for AT1 in membranes containing AT1 receptor expressed alone Competition binding assays were performed by using 0.28 nM of [125I]-Tyr4-Ang II in the presence of increasing concentrations of unlabelled Ang II or in the presence of increasing concentrations of unlabelled Ang II plus 100 nM of unlabelled Ap13. Each symbol represents the mean percentage of specific binding ± SEM (n = 2) from two independent experiments. GraphPad Prism5 was used to calculate IC50 using equation 2 as described in the Methods section
Figure S7 Ap13/APJ modulation of Ang II/AT1 is unidirectional. (A) Effects of Ang II on AT1 function in the absence of Ap13 and/APJ. CHO-K1-AT1 cells were incubated with Ang II (0.0–100 μM) to stimulate recruitment of β-arrestin. Each symbol represents the mean percent activation of AT1 ± SEM (n = 6) by Ang II at the indicated concentration. Curves superimposed on the data were generated by non-linear regression analysis [Log (agonist) vs. Normalized response-variable slope] using GraphPad Prism 5. (B) Effects of Ang II and AT1 on APJ function. PathHunter β-Arrestin CHO-K1 cells expressing APJ (CHO-K1-APJ) and transfected with pFlag-AT1 were stimulated with Ap13 (0.0–100 μM) or were exposed to a fixed concentration of Ang II and a range of concentrations of Ang II13 (0.0–100 μM) exhibit similar affinity for APJ. Each symbol represents the mean percent activation of APJ ± SEM (n = 6) by Ap13 at the indicated concentration. Curves superimposed on the data represent the best global fit by equation 4.
Table S1 Specific binding of [125I]-Tyr4-Ang II and [125I]-Glp65, Nle75, Tyr7-Ap13 to membranes from cells expressing AT1 + APJ.
References
- AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276:39721–39726. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley E, Chun HJ, Quertermous T. Opposing cardiovascular roles for the angiotensin and apelin signaling pathways. J Mol Cell Cardiol. 2006;41:778–781. doi: 10.1016/j.yjmcc.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Bai M. Dimerization of G-protein-coupled receptors: roles in signal transduction. Cell Signal. 2004;16:175–186. doi: 10.1016/s0898-6568(03)00128-1. [DOI] [PubMed] [Google Scholar]
- Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of beta-adrenergic and angiotensin II receptors by a single antagonist: a functional role for receptor-receptor interaction in vivo. Circulation. 2003;108:1611–1618. doi: 10.1161/01.CIR.0000092166.30360.78. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of an inhibitor which causes 50 per cent inhibition (Im) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118:3343–3354. doi: 10.1172/JCI34871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews J. Genomic sciences and the medicine of tomorrow. Nat Biotechnol. 1996;14:1516–1518. doi: 10.1038/nbt1196-1516. [DOI] [PubMed] [Google Scholar]
- Ellis J, Pediani JD, Canals M, Milasta S, Milligan G. Orexin-1 receptor-cannabinoid CB1 receptor heterodimerization results in both ligand-dependent and -independent coordinated alterations of receptor localization and function. J Biol Chem. 2006;281:38812–38824. doi: 10.1074/jbc.M602494200. [DOI] [PubMed] [Google Scholar]
- George SR, Lee SP, Varghese G, Zeman PR, Seeman P, Ng GY, et al. A transmembrane domain-derived peptide inhibits D1 dopamine receptor function without affecting receptor oligomerization. J Biol Chem. 1998;273:30244–30248. doi: 10.1074/jbc.273.46.30244. [DOI] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, et al. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- Gines S, Hillion J, Torvinen M, Le Crom S, Casado V, Canela EI, et al. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci U S A. 2000;97:8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes I, Jordan BA, Gupta A, Rios C, Trapaidze N, Devi LA. G protein coupled receptor dimerization: implications in modulating receptor function. J Mol Med. 2001;79:226–242. doi: 10.1007/s001090100219. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Gurevich EV. How and why do GPCRs dimerize. Trends Pharmacol Sci. 2008;29:234–240. doi: 10.1016/j.tips.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Moreira IS, Urizar E, Weinstein H, Javitch JA. Allosteric communication between protomers of dopamine class A GPCR dimers modulates activation. Nat Chem Biol. 2009;5:688–695. doi: 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida J, Hashimoto T, Hashimoto Y, Nishiwaki S, Iguchi T, Harada S, et al. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J Biol Chem. 2004;279:26274–26279. doi: 10.1074/jbc.M404149200. [DOI] [PubMed] [Google Scholar]
- Iwanaga Y, Kihara Y, Takenaka H, Kita T. Down-regulation of cardiac apelin system in hypertrophied and failing hearts: possible role of angiotensin II-angiotensin type 1 receptor system. J Mol Cell Cardiol. 2006;41:798–806. doi: 10.1016/j.yjmcc.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Klabunde T, Giegerich C, Evers A. Sequence-derived three-dimensional pharmacophore models for G-protein-coupled receptors and their application in virtual screening. J Med Chem. 2009;52:2923–2932. doi: 10.1021/jm9001346. [DOI] [PubMed] [Google Scholar]
- Kroeger KM, Pfleger KD, Eidne KA. G-protein coupled receptor oligomerization in neuroendocrine pathways. Front Neuroendocrinol. 2003;24:254–278. doi: 10.1016/j.yfrne.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Lavoie C, Mercier JF, Salahpour A, Umapathy D, Breit A, Villeneuve LR, et al. Beta 1/beta 2-adrenergic receptor heterodimerization regulates beta 2-adrenergic receptor internalization and ERK signaling efficacy. J Biol Chem. 2002;277:35402–35410. doi: 10.1074/jbc.M204163200. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol. 1995;48:362–378. [PubMed] [Google Scholar]
- Maggio R, Barbier P, Colelli A, Salvadori F, Demontis G, Corsini GU. G protein-linked receptors: pharmacological evidence for the formation of heterodimers. J Pharmacol Exp Ther. 1999;291:251–257. [PubMed] [Google Scholar]
- Milligan G, Smith NJ. Allosteric modulation of heterodimeric G-protein-coupled receptors. Trends Pharmacol Sci. 2007;28:615–620. doi: 10.1016/j.tips.2007.11.001. [DOI] [PubMed] [Google Scholar]
- Niu W, Wu S, Zhang Y, Li W, Ji K, Gao P, et al. Validation of genetic association in apelin-AGTRL1 system with hypertension in a larger Han Chinese population. J Hypertens. 2010;28:1854–1861. doi: 10.1097/HJH.0b013e32833b1fad. [DOI] [PubMed] [Google Scholar]
- Patel RC, Kumar U, Lamb DC, Eid JS, Rocheville M, Grant M, et al. Ligand binding to somatostatin receptors induces receptor-specific oligomer formation in live cells. Proc Natl Acad Sci U S A. 2002;99:3294–3299. doi: 10.1073/pnas.042705099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percherancier Y, Berchiche YA, Slight I, Volkmer-Engert R, Tamamura H, Fujii N, et al. Bioluminescence resonance energy transfer reveals ligand-induced conformational changes in CXCR4 homo- and heterodimers. J Biol Chem. 2005;280:9895–9903. doi: 10.1074/jbc.M411151200. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Koch T, Schroder H, Klutzny M, Kirscht S, Kreienkamp HJ, et al. Homo- and heterodimerization of somatostatin receptor subtypes. Inactivation of sst(3) receptor function by heterodimerization with sst(2A) J Biol Chem. 2001;276:14027–14036. doi: 10.1074/jbc.M006084200. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Koch T, Schroder H, Laugsch M, Hollt V, Schulz S. Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J Biol Chem. 2002;277:19762–19772. doi: 10.1074/jbc.M110373200. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Eidne KA. Illuminating insights into protein-protein interactions using bioluminescence resonance energy transfer (BRET) Nat Methods. 2006;3:165–174. doi: 10.1038/nmeth841. [DOI] [PubMed] [Google Scholar]
- Pfleger KD, Seeber RM, Eidne KA. Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat Protoc. 2006;1:337–345. doi: 10.1038/nprot.2006.52. [DOI] [PubMed] [Google Scholar]
- Pin JP, Kniazeff J, Liu J, Binet V, Goudet C, Rondard P, et al. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005;272:2947–2955. doi: 10.1111/j.1742-4658.2005.04728.x. [DOI] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Quitterer U, Lother H, Abdalla S. AT1 receptor heterodimers and angiotensin II responsiveness in preeclampsia. Semin Nephrol. 2004;24:115–119. doi: 10.1016/j.semnephrol.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Rocheville M, Lange DC, Kumar U, Patel SC, Patel RC, Patel YC. Receptors for dopamine and somatostatin: formation of hetero-oligomers with enhanced functional activity. Science. 2000a;288:154–157. doi: 10.1126/science.288.5463.154. [DOI] [PubMed] [Google Scholar]
- Rocheville M, Lange DC, Kumar U, Sasi R, Patel RC, Patel YC. Subtypes of the somatostatin receptor assemble as functional homo- and heterodimers. J Biol Chem. 2000b;275:7862–7869. doi: 10.1074/jbc.275.11.7862. [DOI] [PubMed] [Google Scholar]
- Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the beta 2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- Siddiquee K, Hampton J, Khan S, Zadory D, Gleaves L, Vaughan DE, et al. Apelin protects against angiotensin II-induced cardiovascular fibrosis and decreases plasminogen activator inhibitor type-1 production. J Hypertens. 2011;29:724–731. doi: 10.1097/HJH.0b013e32834347de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, McCall D, Timmermans P. Characterization of Angiotensin II Receptors. Curr Protoc Pharmacol. 2001 doi: 10.1002/0471141755.ph0110s01. [Google Scholar]
- Sohy D, Parmentier M, Springael JY. Allosteric transinhibition by specific antagonists in CCR2/CXCR4 heterodimers. J Biol Chem. 2007;282:30062–30069. doi: 10.1074/jbc.M705302200. [DOI] [PubMed] [Google Scholar]
- Stanasila L, Perez JB, Vogel H, Cotecchia S. Oligomerization of the alpha 1a- and alpha 1b-adrenergic receptor subtypes. Potential implications in receptor internalization. J Biol Chem. 2003;278:40239–40251. doi: 10.1074/jbc.M306085200. [DOI] [PubMed] [Google Scholar]
- Sun X, Iida S, Yoshikawa A, Senbonmatsu R, Imanaka K, Maruyama K, et al. Non-activated APJ suppresses the angiotensin II type 1 receptor, whereas apelin-activated APJ acts conversely. Hypertens Res. 2011;34:701–706. doi: 10.1038/hr.2011.19. [DOI] [PubMed] [Google Scholar]
- Trinquet E, Bouhelal R, Dietz M. Monitoring Gq-coupled receptor response through inositol phosphate quantification with the IP-One assay. Expert Opin Drug Discov. 2011;6:981–994. doi: 10.1517/17460441.2011.608658. [DOI] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol. 2008;4:126–131. doi: 10.1038/nchembio.64. [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, et al. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Rovira X, Pin JP, Giraldo J. The asymetric / symmertic activation of GPCR dimer as a possible mechanistic rationale for multiple signalling pathways. Trends Pharmacol Sci. 2009;31:15–21. doi: 10.1016/j.tips.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Xu J, He J, Castleberry AM, Balasubramanian S, Lau AG, Hall RA. Heterodimerization of alpha 2A- and beta 1-adrenergic receptors. J Biol Chem. 2003;278:10770–10777. doi: 10.1074/jbc.M207968200. [DOI] [PubMed] [Google Scholar]
- Zhong JC, Huang DY, Liu GF, Jin HY, Yang YM, Li YF, et al. Effects of all-trans retinoic acid on orphan receptor APJ signaling in spontaneously hypertensive rats. Cardiovasc Res. 2005;65:743–750. doi: 10.1016/j.cardiores.2004.10.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.