

Abstract
Background and Purpose
KCNQ2-5 channels are voltage-gated potassium channels that regulate neuronal excitability and represent suitable targets for the treatment of hyperexcitability disorders. The effect of Chlor-N-(6-chlor-pyridin-3-yl)-benzamid was tested on KCNQ subtypes for its ability to alter neuronal excitability and for its anticonvulsant potential.
Experimental Approach
The effect of 4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid was evaluated using whole-cell voltage-clamp recordings from CHO cells and Xenopus laevis oocytes expressing different types of KCNQ channels. Epileptiform afterdischarges were recorded in fully amygdala-kindled rats in vivo. Neuronal excitability was assessed using field potential and whole cell recording in rat hippocampus in vitro.
Key Results
4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid caused a hyperpolarizing shift of the activation curve and a pronounced slowing of deactivation in KCNQ2-mediated currents, whereas KCNQ3/5 heteromers remained unaffected. The effect was also apparent in the Retigabine-insensitive mutant KCNQ2-W236L. In fully amygdala-kindled rats, it elevated the threshold for induction of afterdischarges and reduced seizure severity and duration. In hippocampal CA1 cells, 4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid strongly damped neuronal excitability caused by a membrane hyperpolarization and a decrease in membrane resistance and induced an increase of the somatic resonance frequency on the single cell level, whereas synaptic transmission was unaffected. On the network level, 4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid caused a significant reduction of γ and θ oscillation peak power, with no significant change in oscillation frequency.
Conclusion and Implications
Our data indicate that 4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid is a potent KCNQ activator with a selectivity for KCNQ2 containing channels. It strongly reduces neuronal excitability and displays anticonvulsant activity in vivo.
Keywords: CA1 pyramidal cell, KCNQ, potassium channels, M-current, Kv7 channel opener, resonance, oscillations, epilepsy, amygdala kindling
Introduction
Neuronal KCNQ (Kv7) channels are voltage-gated K+-channels that slowly activate at depolarized membrane potentials. The subunits KCNQ2, KCNQ3 and KCNQ5 are predominantly expressed and widely distributed in the brain, including the hippocampus (Jentsch et al., 2000; Dalby-Brown et al., 2006). These subunits mediate the muscarine-modulated M-currents (IM), which profoundly contribute to transient afterhyperpolarization following action potentials, thereby influencing the firing rate in many neurons. They also affect action potential threshold and resting membrane potential (Marrion, 1997; Delmas and Brown, 2005; Hu et al., 2007; Shah et al., 2008). Potentiation of IM has been proposed as a way to attenuate neuronal excitability and could be therefore beneficial in treating diseases characterized by neuronal hyperexcitability, such as epilepsy, neuropathic pain or bipolar disorders (Jentsch, 2000; Rogawski and Bazil, 2008; Wickenden and McNaughton-Smith, 2009; Kristensen et al., 2012). A well-known IM activator is Retigabine (RTG) (Rundfeldt, 1997), which effectively suppresses epileptiform activity in vitro (Armand et al., 1999; 2000) in a dose- and developmental-dependent manner (Forcelli et al., 2012), and in a broad range of animal models of epilepsy (Rostock et al., 1996) and pain in several preclinical pain models (Blackburn-Munro and Jensen, 2003; Munro et al., 2007). It has also undergone a number of clinical phase II and III trials demonstrating its efficacy (Porter et al., 2007; Brodie et al., 2010; French et al., 2011), although the subunit specificity of RTG is quite low and displays side effects on synaptic transmission at higher concentrations (Rundfeldt and Netzer, 2000). RTG was recently approved for treatment by the European Medicines Agency and by the United States Food and Drug Administration. During the last decade, additional compounds activating KCNQ channel have been characterized (Xiong et al., 2008), including a number of benzamide derivates (Wickenden et al., 2008; Gao et al., 2010), with improved selectivity for KCNQ2/3 over KCNQ4 or KCNQ3/5 channels (Wickenden et al., 2008).
IM is active in the voltage range of action potential initiation and therefore particularly important in regulating neuronal excitability, action potential firing and neurotransmitter release (Marrion, 1997; Delmas and Brown, 2005; Hu et al., 2007; Shah et al., 2008). Especially in the hippocampus, axo-somatic Kv7 channels are crucial for setting the spike frequency (Yue and Yaari, 2004; Otto et al., 2006; Shah et al., 2008), the action potential threshold, and contribute to the medium afterhyperpolarization in pyramidal cells (Gu et al., 2005; Peters et al., 2005). Block or reduction of IM leads to a pronounced afterdepolarization and bursting of hippocampal pyramidal cells, whereas its augmentation causes regular firing in bursting neurons (Yue and Yaari, 2004). Given that IM is regulated by multiple neurotransmitters including ACh, it may provide a substrate for modulation of neuronal input/output relations (Delmas and Brown, 2005), which can modulate oscillatory activity and therefore local field potential oscillations. Indeed, an involvement of IM in the generation of hippocampal θ frequency oscillations is supported by genetic (Peters et al., 2005), and pharmacological experiments showing that intrinsic oscillations such as resonance of individual pyramidal cells (Hu et al., 2002) and pharmacologically induced hippocampal network oscillations (Boehlen et al., 2009) are reduced during IM modulation.
In this study, we demonstrate that 4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid (ICA-110381) predominantly acts on KCNQ2 containing channels in CHO cells and shows anticonvulsive properties in fully amygdala-kindled rats in vivo. In CHO cells, the effect of ICA-110381 is predominantly caused by prolonged time course of deactivation of KCNQ2- and KCNQ2/3-mediated currents. In CA1 pyramidal cells, the increase of IM in the presence of ICA-110381 resulted in a damping of neuronal excitability by reducing the input resistance and shifting the resting membrane potential in hyperpolarizing direction. In rat hippocampal slices, network oscillations in the θ and γ frequency range were highly sensitive to ICA-110381, although glutamatergic and GABAergic postsynaptic currents were unaffected.
Methods
Preparations, drugs and solutions
ICA-110381 (4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid) was synthesized at Elbion AG (Radebeul, Germany) as a reference compound based on patent information of the originator ICAgen (WO 0110381), and to this stage, the development status is unknown. The identity was confirmed by NMR spectroscopy and elemental analysis. Based on the structural information, it is expected that the compound is not charged at physiological pH (7.4). ICA-110381 and RTG (Elbion AG) were dissolved in DMSO (final DMSO concentration ≤ 0.1%) and applied via bath perfusion.
CHO cells were transiently transfected with hKCNQ2, hKCNQ3, hKCNQ4 and hKCNQ5 cDNA plasmids (0.5 μg). Co-transfection with a GFP served for identification of successfully transfected cells. The cDNA ratio for transfections was 1:0.5 (KCNQ2 : GFP, KCNQ4 : GFP), 1:1:0.5 (KCNQ2 : KCNQ3 : GFP) and 1:1:0.5 (KCNQ3 : KCNQ5 : GFP). Fugene (Roche, Germany) was used as transfection reagent. Cells were grown at 37°C and 5% CO2 in Ham's F12 medium with 10% fetal calf serum and 1% penicillin–streptomycin. Cells were split and plated on glass coverslips 6–24 h after transfection and used for recording after a further 16–72 h.
For oocyte experiments, KCNQ2 constructs and capped cRNAs were generated as described previously (Schenzer et al., 2005). Individual stage V to VI oocytes were obtained from anaesthetized frogs and isolated by collagenase treatment; 10 ng of total KCNQ cRNA was injected into oocytes. Following injection, oocytes were kept at 17°C in ND96 solution (in mM) (96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4).
Acute 400 μm-thick brain slices were obtained from 5–8 week-old Wistar rats of either sex (n = 32) as previously described (Boehlen et al., 2009), in full compliance with the European Commission and the Animal Ethics Committee Berlin (T0068/02). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Slices were prepared (∼4°C), stored (room temperature) and recorded from (32 ± 1°C perfused at 1.8–2.4 mL min−1) in artificial cerebrospinal fluid (aCSF) composed of (in mM) 129 NaCl, 21 NaHCO3, 3 KCl, 1.6 CaCl2, 1.8 MgSO4, 1.25 NaH2PO4 and 10 glucose. The pH was buffered to 7.4 by continuous flow of 5% CO2 and 95% O2.
For somatic whole-cell recordings from pyramidal cells in hippocampal slices, pipettes (4–6 MΩ) were filled with an intracellular solution containing (in mM) K-gluconate 140, MgCl2 2, phosphocreatine 10, Na2ATP 2, NaGTP 0.4, and HEPES 10 or CsCl 130, MgCl2 2, Na2ATP 2, NaGTP 0.3, HEPES 10, EGTA 2. In a subset of experiments, 100 nM kainic acid ((2S,3S,4R)-Carboxy-4-(1-methylethenyl)-3-pyrrolidineacetic acid), 10 μM DNQX, 5 μM SR-95531 hydrobromide, 5 μM bicuculline (all from Tocris, Bristol, UK), 60 μM DL-APV or 1 μM TTX (both from Sigma-Aldrich, Steinheim, Germany) were added to the aCSF. For CHO cell recordings, patch pipettes (3–5 MΩ) were filled with an intracellular solution containing (in mM) 130 KCl, 1 MgCl2, 5 K2ATP, 5 EGTA, 10 HEPES (290 mOsm kg−1, pH 7.3); the bath solution contained (in mM) 140 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, adjusted with NaOH to pH 7.4 (310 mOsm kg−1). In some experiments, NaCl was partly replaced by KCl (25 mM).
For in vitro experiments, all pharmacological agents were applied via continuous bath perfusion, ICA-110381, Retigabine ([2-amino-4-(4-fluoro-benzylamino)-phenyl]-carbamic acid ethyl ester; Elbion AG; 1 μM), XE991 ((10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone dihydrochloride; Tocris Bioscience, Bristol, UK; 10 μM) was dissolved in DMSO and diluted in ACSF to the final concentration (final DMSO concentration ≤0.1%). All drug-containing solutions were freshly prepared prior to the experiment. For in vivo experiments, ICA-110381 was suspended in 0.5% hydroxyethyl cellulose in water and administered in a volume of 5 mL kg−1 orally.
Kindling acquisition
In vivo experiments were approved by the Committee on Animal Care and Use of the Federal State of Saxony and carried out following the German Law on the Protection of Animals. Female Wistar rats (Charles River, Germany) weighing 200–300 g were kept in groups of five under controlled environmental conditions (room temperature at 22–24°C, humidity 55 ± 15%, 12 h/12 h light/dark cycle) with free access to water and standard diet (ssniff M/R 15, Spezialdiäten GmbH, Soest/Westfalen, Germany).
The experiments were carried out as previously described (Tober et al., 1996). Briefly, anaesthetized rats received a stereotaxic implantation of one bipolar electrode in the right basolateral amygdala according to the surgery methods described in the atlas (Paxinos et al., 1986). Coordinates for electrode implantation were AP –2.2, L –4.7, V –8.7 measured from bregma. The size, shape and resistance of the bipolar electrode were the same for all kindled rats used for the present study. A skull screw, positioned over the contralateral parietal cortex, served as the indifferent reference electrode. Bipolar and reference electrodes were connected to plugs, and the electrode assembly and anchor screws were held in place with dental acrylic cement applied to the exposed skull surface. After implantation of electrodes, the rats were housed in individual cages.
After at least 2 weeks of recovery, electrical stimulation of the amygdala was initiated using an electrical stimulator (Stimulator Typ 215/I, Hugo Sachs, March Hugstetten, Germany). Constant current stimulations (500 μA, 1 ms monophasic square-wave pulses, 50 s−1 for 1 s) were delivered to the amygdala once daily until 10 consecutive stage V seizures (Racine et al., 1972) were elicited. Only fully kindled rats with reproducible severity and duration of seizures were used.
Electrical activity of the amygdala was recorded in vivo from a bipolar electrode on a chart recorder (Graphtec Linearcorder WR 3310, Hugo Sachs) before and after stimulus delivery. Before and after kindling acquisition, the basal afterdischarge threshold (ADT) was determined. It reflects the electrical susceptibility of the stimulated region for triggering of paroxysmal neuronal activity and was recorded using an ascending stimulus procedure (Freeman and Jarvis, 1981). The initial current intensity (10 μA) was increased in steps of about 20% of the previous current at intervals of 1 min until an AD (≥3 s) was elicited. Determination of the threshold for induction of ADs was repeated at intervals of 2–3 days until all animals exhibited reproducible seizures thresholds.
Evaluation of drug effects in fully kindled rats
As previously described (Tober et al., 1996), the following seizure parameters were evaluated following stimulation with the ADT current: afterdischarge duration (ADD, total duration of afterdischarges), seizure severity scored according to the classification of Racine (Racine et al., 1972), behavioural seizure duration and total duration of behavioural changes. The effect of ICA-110381 was assessed in groups of 9–11 fully kindled rats by determination of the threshold for the induction of ADs. The control threshold for induction of ADs was determined 2 days prior to each drug treatment. Only animals with a stable control threshold were included in the following drug testing experiment. For control determinations, animals received an application of the vehicle with the same pre-treatment time used for drug testing. On the day of experiment, the same animals were administered orally with a certain dose of ICA-110381 (3, 10 and 30 mg kg−1). After 30 min, stimulation was initiated three 20% steps below the previously determined individual control threshold. For measuring the ADT, the current intensity was increased in steps of about 20% of the previous current at intervals of 1 min until an AD (≥3 s) was elicited. In parallel, the influence of the drug on seizure severity, seizure duration, total duration of behavioural changes and duration of ADs was measured during stimulation with the ADT current. To evaluate the side effect potential of the tested drug, behaviour, motor function and rectal body temperature were determined prior to experiment and 25 min after drug treatment.
For behavioural observation, animals were placed in an open arena (box of ellipsoidal shape, 70–100 cm) 5 min before electrical stimulation for 1 min. Motor deficit (ataxia, abnormal gait and stance, loss of placing response and muscle tone) and behavioural alterations (e.g. stereotyped behaviour, Straub tail, tremor) were visually determined. Afterwards, motor impairment was identified using a rotarod procedure. Inability of trained rats to maintain its equilibrium for 1 min in at least one of three trials on a rotating rod (8 rpm, 8 cm diameter) was used as an indication of the impairment. Rectal body temperature was measured by means of an electronic thermometer (ama-digit, Amarell Electronic, Kreutzwertheim, Germany) before and after treatment.
All kindling animals were used repeatedly for testing different doses of the drug. At least 7 days were interposed between two drug administrations in order to prevent or minimize changes in drug effects due to long-lasting alterations in receptor function or due to drug accumulation or tolerance. After termination of experiments, placement of stimulating electrode was examined histologically in all animals by toluidine blue staining of serial coronal slices of the right basolateral amygdala. Only animals with right placement of the bipolar electrode were included in final evaluation. Satellite non-kindled female Wistar rats were used to determine the plasma brain exposure at the time point of ADT determination. Two rats each were treated orally with 3, 10 and 30 mg kg−1 of ICA-110381 respectively. Plasma and brain were collected 30 min post application for bioanalytics.
Electrophysiological in vitro recordings
Whole-cell voltage-clamp recordings from fluorescent CHO cells were carried out at room temperature using a MultiClamp 700B amplifier (Molecular Devices). Data acquisition was performed using pClamp9 (Molecular Devices, Sunnyvale, CA, USA). Current records were filtered at 3 kHz and digitized at 10 kHz using a Digidata 1440A (Molecular Devices). Series resistance was compensated by 70%. Two-electrode voltage-clamp measurements in oocytes were performed three days after cRNA injection at room temperature in ND96 using a Turbo TEC amplifier (NPI Electronics, Tamm, Germany) and pClamp8 software (Molecular Devices).
In extracellular recordings, the viability of slices was verified by monitoring stratum radiatum stimulus induced field potential responses using a bipolar stimulation electrode. Hippocampal extracellular field potentials were recorded from stratum pyramidale of area CA1 and CA3. Microelectrodes with a tip opening of ∼1 μm diameter were pulled from borosilicate glass capillaries (Science Product GmbH, Hofheim, Germany) using a P-87 puller (Sutter Instruments, Novato, CA) and filled with ACSF (resistance 5–10 MΩ). Signals were pre-amplified using an in-house built amplifier and low-pass filtered at 1 kHz. Signals were stored on computer hard disc for off-line analysis using Spike2 3.21 software (CED 1401, Cambridge Electronic Design Ltd, Cambridge, UK) at a sampling frequency of 5 kHz. Patch pipettes were pulled using a PC-10 puller (Narishige, London, UK). Cells were visualized using an upright microscope equipped with a 40× water-immersion objective. Recordings were obtained from neurons located in the pyramidal layer CA1 of the hippocampus using a MultiClamp 700B (Axon Instruments Inc., Foster City, CA). Signals were filtered with 3 kHz, digitized at 10–100 kHz and acquired using the Clampex 9.0 software (Axon Instruments Inc.). Bridge balance was set using MultiClamp 700B and adjusted during the recording session. The liquid junction potential (∼−10 mV) was not compensated for.
Data analysis
Data analyses were performed using Clampfit 9.0 (Molecular Devices), MATLAB (Version R2006b, MathWorks Inc., Natick, MA), Origin 8.0 (OriginLab Corporation Inc., Northampton, MA) or Spike2 version 5 (Cambridge Electronic Design). Conductance were calculated by the equation 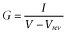 , where I is the tail current amplitude measured immediately after the transient capacitance current after stepping to −120 mV from potentials between −120 and +50 mV, V is the potential of the tail current and Vrev is the K+ reversal potential. For clearly demonstrating the effect of ICA-110381, the current at the end of the −120 mV tail current step after a hyperpolarizing pulse of −120 mV in the absence of a drug was considered as a CHO background leak and subtracted before conductance were calculated. As a consequence, the control current at −120 mV in the conductance–voltage relationship is nearly zero. The conductance–voltage relationship was fitted using the Boltzmann equation
, where I is the tail current amplitude measured immediately after the transient capacitance current after stepping to −120 mV from potentials between −120 and +50 mV, V is the potential of the tail current and Vrev is the K+ reversal potential. For clearly demonstrating the effect of ICA-110381, the current at the end of the −120 mV tail current step after a hyperpolarizing pulse of −120 mV in the absence of a drug was considered as a CHO background leak and subtracted before conductance were calculated. As a consequence, the control current at −120 mV in the conductance–voltage relationship is nearly zero. The conductance–voltage relationship was fitted using the Boltzmann equation 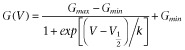 , where Gmax is the maximum conductance, Gmin is the minimum conductance, V1/2 is the voltage of half maximal activation and k is the slope factor. To avoid errors in calculating V1/2 and k, if Gmax is affected by the drug, G were normalized to Gmax in the presence of the drug.
, where Gmax is the maximum conductance, Gmin is the minimum conductance, V1/2 is the voltage of half maximal activation and k is the slope factor. To avoid errors in calculating V1/2 and k, if Gmax is affected by the drug, G were normalized to Gmax in the presence of the drug.
The deactivation time was estimated by fitting the trace using a simplex algorithm least-squares exponential fitting routine. Double exponential equations of the form 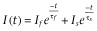 , where I(t) is the current at time t; If and Is are the amplitudes of the fast and slow deactivation components, respectively; and τf and τs are their respective deactivation time constants To compare decay times between different experimental conditions, we used a weighted mean decay time constant:
, where I(t) is the current at time t; If and Is are the amplitudes of the fast and slow deactivation components, respectively; and τf and τs are their respective deactivation time constants To compare decay times between different experimental conditions, we used a weighted mean decay time constant: 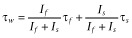 .
.
To determine the input resistance of a cell (Rinput), a hyperpolarizing voltage step of 10 mV from a holding potential of −60 mV was applied. The resonance frequency of a cell was determined from the voltage response to a sinusoidal current injection with a linear frequency increase over time (Puil et al., 1986; Hutcheon et al., 2000; Boehlen et al., 2010). The amplitude of the input was adjusted to avoid changes in the membrane potential over 10 mV during sinusoidal current injection. The average of five trials was used to determine the average resonance frequency. The input frequency corresponding to the highest impedance was noted as resonance frequency. The resonance strength (Q-value) was determined as the ratio of the peak impedance to the impedance at by analysis lowest detectable resonance frequency (0.36 Hz; Boehlen et al., 2010; 2011).
For extracellular field potential recordings, data analysis was performed using Spike 2 version 5 (Cambridge Electronic Design) and MATLAB (Version R2006b, MathWorks Inc.). In each experiment, peak power and peak frequency of field potential oscillations in the γ and θ frequency bands (30–40 Hz and 4–15 Hz, respectively) were determined from 100 s data sections immediately before drug application and immediately before washout, at 60 min after drug application. For spectral analysis, power spectra were calculated employing a fast Fourier transform algorithm with a FFT size of 1.221 Hz and a Hanning window (Spike2 software). The main oscillation frequency was taken as the peak frequency determined from the respective power spectra, and the power value corresponding to the peak frequency was taken as peak power. To facilitate comparison between experiments, peak power was normalized to control values before drug application.
All results were expressed as mean ± SEM. If not otherwise stated, the Student's t-tests were used to test for statistically significant differences (Origin 8.0, OriginLab Corporation Inc.). Differences were considered significant at P ≤ 0.05. In the figures and Tables significance levels are indicated as *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
Results
ICA-110381 activates KCNQ2- and KCNQ2/3-mediated potassium currents
To quantify the effect of ICA-110381 – its chemical structure is displayed as inset in Figure 1B – on KCNQ potassium currents, we performed whole-cell patch-clamp recordings from CHO cells expressing KCNQ2, KCNQ2/3, KCNQ3/5 and KCNQ 4 subunits. To elicit outward currents, cells were held at −70 mV, and voltage steps of 1 s to potentials between −90 and +30 mV were applied followed by a voltage step to a potential of −70 mV. The depolarizing voltage steps slowly activated a series of non-inactivating outward currents (Figure 1A). Under control conditions, repolarization to −70 mV led to a slow deactivation towards the initial current level.
Figure 1.
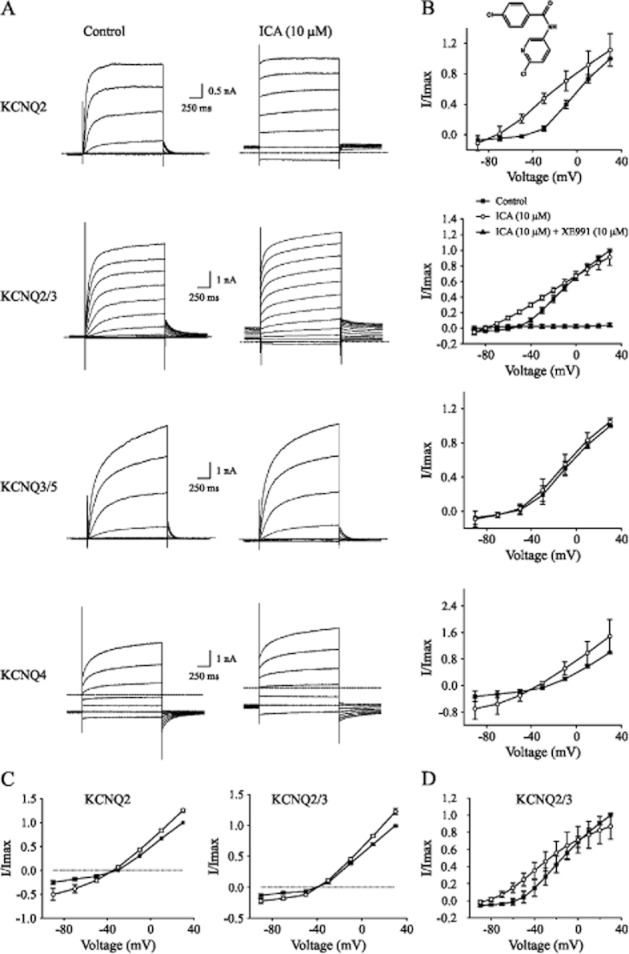
ICA-110381 shows an improved selectivity for KCNQ2/3- over KCNQ3/5-mediated currents. (A) Representative whole-cell currents recorded from individual CHO cells expressing KCNQ2, KCNQ2/3, KCNQ3/5 or KCNQ4 under control conditions and after bath application of ICA-110381 (10 μM, chemical structure is shown as inset in Figure 1B). The membrane potential was stepped from −90 to +30 mV for 1 s in 10 mV (KCNQ2/3) or 20 mV increments. (B) Current–voltage relationship under control conditions, after application of ICA-110381 (ICA) and in the presence of XE911 (20 μM) from currents at the end of the depolarizing step. Currents were normalized to the maximal current under control conditions. (C) Current–voltage relationship of currents from CHO cells expressing KCNQ2 or KCNQ2/3 recorded in high K+ (25 mM extracellular) under control conditions, after application of ICA (10 μM). (D) Current–voltage relationship of currents of currents from CHO cells expressing KCNQ2/3. Extracellular Na+ was replaced by NMG+.
After bath application of ICA-110381 (10 μM), outward current amplitudes in KCNQ2-, KCNQ2/3- and KCNQ4-transfected cells increased accompanied by acceleration of activation. The deactivation as indicated by the prolonged time of recovery to baseline current was markedly slowed. In some recordings, we observed an increase in holding current before the next voltage step, indicating that deactivation was still incomplete after 5 s. The effect of ICA-110381 on KCNQ2-, KCNQ2/3- and KCNQ4-mediated currents was fully reversible after 10 min of washout. Application of the KCNQ inhibitor XE991 (20 μM) in the presence of ICA-110381 caused a complete block of the current (Figures 1B and 2A). For current–voltage (IV) relationships (Figure 1B), the current at the end of the depolarizing voltage step was normalized to the maximal current obtained under control conditions and plotted as a function of the step potential. Application of ICA-110381 caused a voltage-dependent increase in I/Imax [KCNQ2: ΔI = 40 ± 1% of Imax at −30 mV and ΔI = 11.1 ± 0.6% of Imax at 30 mV, n = 7; KCNQ2/3: ΔI = 20 ± 1% of Imax at −40 mV and ΔI = 9 ± 0.05% of Imax at 30 mV, n = 9; KCNQ4 (recorded in 25 mM K+): ΔI = 50 ± 12% of Imax at 30 mV], resulting in a linearization of the IV curve in KCNQ2-containing channels. This suggests that ICA-110381 reduced the voltage dependence of the current and turned it to a ‘steady’ current. To directly test whether the effect of ICA-110381 is specific on KCNQ-expressing cells, we applied 10 μM ICA-110381 on non-transfected cells (n = 7) without any effect (data not shown). Furthermore, the steady current was blocked by XE991 (20 μM) as shown in Figure 1B for KCNQ2/3-mediated currents and in Figure 2A for KCNQ2-mediated currents. Together, our data strongly indicate that this ‘steady’ current is mediated by KCNQ channels.
Figure 2.
ICA-110381 slowed deactivation and shifted activation curve in KCNQ2-mediated currents. (A) Example whole-cell currents recorded from individual CHO cells expressing KCNQ2, under control conditions, after bath application of 1 μM ICA-11038 and after bath application of XE 991 (20 μM). Currents were activated from a holding potential of −70 mV by depolarizing pulses to +40 mV for 500 ms and deactivated by 1 s hyperpolarizing pulses to potentials between −120 and +40 mV, followed by a step to −120 mV. (B) Conductance–voltage activation curves under control conditions, after application of 1 μM ICA-110381 (ICA) and 1 μM retigabine (RTG). Data were obtained from the tail currents at −120 mV elicited by the protocol described in panel A. Conductance were normalized to the maximal conductance under control conditions after leak subtraction. (C) Deactivation times obtained from biexponential fits of tail currents at −120 mV after a hyperpolarizing pulse of −40 mV under control conditions in the presence of 1 μM ICA and in the presence of 1 μM RTG. (D) Concentration-dependent increase of tail current at −120 mV after a hyperpolarizing pulse of −40 mV and weighted deactivation time obtained by fitting the same trace.
To test whether ICA-110381 had an effect on the ion selectivity of KCNQ2/3 channels, extracellular Na+ was replaced by NMG+. Substitution did not cause significant differences in KCNQ2/3-mediated currents either under control conditions or in the presence of ICA-110381 (P > 0.05, n = 5) as shown for the IV curve of KCNQ2/3-mediated currents recorded in extracellular NMG+ (Figure 1D). We next increased the extracellular K+ concentration to 25 mM (high K+), leading to a theoretical reversal potential for the current of −42 mV. Figure 1C shows the IV relationship for KCNQ2- and KCNQ2/3-mediated currents recorded in high K+ under control conditions and in the presence of 1 μM ICA-110381. The experimental reversal potential is about −40 mV (without correction of the liquid junction potential) both under control conditions and in presence of ICA-110381, and it was hence not affected by the drug. This suggests that ICA-110381 did not change the ion selectivity of KCNQ2/3 channels.
Overall, our results are consistent with the view that ICA-110381 acts as a voltage-independent opener of KCNQ channels, with an efficacy for KCNQ2 > KCNQ4 > KCNQ3/5. Accordingly, ICA-110381-induced current augmentation also occurred at hyperpolarizing potentials at which KCNQ current are not activated under control conditions. Importantly, application of ICA-110381 up to concentrations of 10 μM had no significant effect on KCNQ3/5-mediated currents (Figure 1A).
Characterization of ICA-induced current modulation
To examine the effect of ICA-110381 more closely, we applied a voltage protocol, in which the current was first fully activated by stepping to +40 mV, subsequently stepped for 1.25 s to various potentials between +40 and −120 mV, followed by a final step to −120 mV for 0.5 s. (Tatulian et al., 2001). In some experiments, the duration of the final step was increased to 7 s. To increase the tail current, recordings were performed in high (25 mM) extracellular K+. The tail current at −120 mV correlates with the relative conductance activated at the end of the previous pulse and was used for the generation of voltage–conductance activation curves (Figures 2B and 3B) under control conditions, in the presence of ICA-110381 of the same cells as those used for control measurements and in the presence of 1 μM RTG from other cells. It must be noted that there were no significant differences between these cells under control conditions. For illustration, conductances were normalized to the maximal conductance under control conditions in Figures 2 and 3. The potentials for half maximal activation (V1/2) were obtained by fitting activation curves to the Boltzmann equation (see Methods); they are summarized in Table 1. In agreement with the literature, we observed a hyperpolarizing shift of V1/2 for all investigated subunits in the presence of 1 μM RTG (Main et al., 2000; Tatulian et al., 2001; Wickenden et al., 2008; Linley et al., 2012). In contrast, 10 μM ICA-110381 had nearly no effect on KCNQ3/5 channels but a clear effect on KCNQ4 channels, whereas only 1 μM ICA-110381 induced a nearly threefold larger hyperpolarizing shift of the activation curve than 1 μM RTG in KCNQ2-containing channels. The ICA-110381-induced shift of the activation curve is accompanied by a large increase in the slope factor k, which remained nearly unchanged in the presence of RTG (Table 1). The increase in slope factor reflects an increased relative conductance at lower membrane potentials and indicates a reduced voltage dependence of channel conductance in the presence of ICA-110381. Whereas we did not observe an increase in Gmax neither by ICA-110381 (1 μM) nor by RTG (1 μM) in KCNQ2-containing channels, we found an insignificant increase in Gmax by 17 ± 4% for KCNQ4-containing channels and RTG significantly increased Gmax in KCNQ3/5 heteromers by 37 ± 5% (n = 5, P < 0.05, Table 1).
Figure 3.

ICA-110381 slowed deactivation and shifted activation curve in KCNQ2/3-, KCNQ3/5- and KCNQ4-mediated currents. (A) Example whole-cell currents recorded from individual CHO cells expressing KCNQ2/3 KCNQ3/5 or KCNQ4 under control conditions and after bath application of 1 μM ICA-11038 (KCNQ2/3) or 10 μM ICA-11038 (KCNQ3/5, KCNQ4). Currents were activated from a holding potential of −70 mV by depolarizing pulses to +40 mV for 500 ms and deactivated by 1 s hyperpolarizing pulses to potentials between −120 and +40 mV, followed by a step to −120 mV. Please note, that the step to −120 mV is longer (5 s) for KCNQ4. (B) Conductance–voltage activation curves under control conditions, after application of 1 μM (KCNQ2/3), 10 μM (KCNQ3/5 and KCNQ4) ICA-110381 (ICA) and 1 μM retigabine (RTG). Data were obtained from the tail currents at −120 mV elicited by the protocol described in panel A. Conductance were normalized to the maximal conductance under control conditions after leak subtraction. (C) Deactivation times from tail currents at −120 mV after a hyperpolarizing pulse of −40 mV under control conditions in the presence of 1 μM ICA and in the presence of 1 μM RTG. For KCNQ2/3-mediated currents, deactivation times were obtained from biexponential for KCNQ3/5- and KCNQ4-mediated currents from monoexponetial fits.
Table 1.
Effect of ICA-110381 and RTG on conductance–voltage activation curve of Kv7-mediated currents
Δ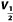 (mV) (RTG 1 μM) (mV) (RTG 1 μM) |
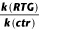 |
 |
n | Δ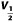 (mV) (ICA**) (mV) (ICA**) |
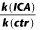 |
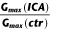 |
n | |
|---|---|---|---|---|---|---|---|---|
| KCNQ2 | −19.5 ± 2.3* | 1.0 | 1.06 ± 0.06 | 6 | −33.0 ± 1.9 | 1.5 | 1.04 ± 0.06 | 9 |
| KCNQ2/3 | −29.3 ± 3.8 | 1.9 | 1.2 ± 0.2 | 6 | −67.3 ± 5.8 | 3.3 | 1.11 ± 0.09 | 8 |
| KCNQ3/5 | −9.0 ± 1.2 | 1.2 | 1.37 ± 0.05 | 5 | −10.2 ± 1.3 | 1.1 | 0.97 ± 0.1 | 5 |
| KCNQ4 | − | − | − | −105.1 ± 19 | 5.1 | 1.17 | 3 |
For obtaining 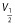 and k, the conductances in the presence of the drug were normalized to the maximal conductance in the presence of the drug.
and k, the conductances in the presence of the drug were normalized to the maximal conductance in the presence of the drug.
Concentration: 1 μM for KCNQ2 and KCNQ2/3 and 10 μM for KCNQ3/5 and KCNQ4.
To quantify the slowing of deactivation, we fitted the tail current mono- or biexponentially at −120 mV following a previous voltage step to −40 mV. The fitting results are summarized in Figures 2C and 3C. For KCNQ2 and KCNQ2/3, 1 μM ICA-110381 (n = 8, P = 0.01) and 1 μM RTG (n = 6, P = 0.01) significantly increased the weighted deactivation time constant. A significant slowing of deactivation by 10 μM ICA-110381 was also observed in KCNQ3/5 heteromers (n = 6, P = 0.05) and KCNQ4 homomers (n = 3) as well as a pronounced increase of the deactivation time constant in the presence of 1 μM RTG for KCNQ3/5 heteromers (n = 6, P = 0.05).
The concentration-dependent effect of ICA-110381 on conductance at −40 mV and on deactivation time of KCNQ2 homomers is illustrated in Figure 2D. Whereas application of 10 μM ICA-110381 led to a nearly twofold increase in conductance, there is a drastic increase in the deactivation time constant.
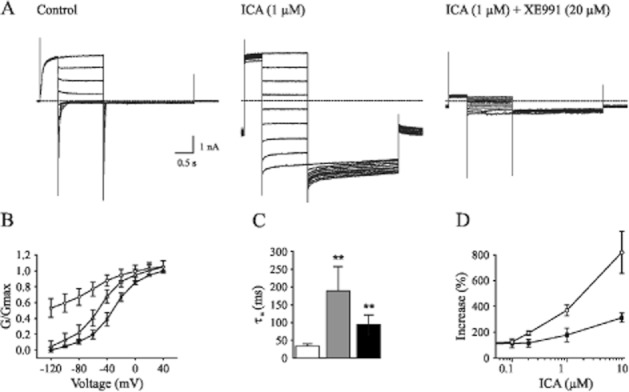
Combined action of ICA-110381 and RTG
We next examined the functional interactions between ICA-110381 and RTG on KCNQ2-mediated currents. To avoid saturating effects, we applied ICA-110381 and RTG at low concentrations between 0.1 and 0.5 μM in extracellular solutions containing high K+. Figure 4A illustrates the drug-dependent prolongation of tail currents obtained by the same voltage protocol as described in Figure 2. The effect of 0.5 μM RTG on deactivation of the tail current is illustrated in Figure 4B. Fitting of deactivation time constants of the tail current following a potential of −40 mV shows a synergistic effect of both drugs on deactivation (Figure 4C). In the presence of ICA-110381 (0.2 μM), voltage–conductance activation curves showed a hyperpolarizing shift with ΔV1/2 = 6.7 ± 0.2 mV (n = 6), which was increased to ΔV1/2 = 17.1 ± 0.8 mV (n = 6) after additional application of RTG (0.5 μM). When RTG (0.5 μM) is applied in the absence of ICA, V1/2 is shifted by 4.3 ± 0.2 mV. The slope factor k was increased by 30% after application of ICA-110381 (0.2 μM). Interestingly, addition of 0.5 μM RTG in the presence of 0.2 μM ICA-110381 led to a linearization of the voltage–conductance relationship and a further increase of the slope factor by 26%, although RTG itself did not cause linearization and had no effect on the slope factor. Our data suggest that simultaneous application of both drugs at low concentrations caused (i) a roughly additive effect on hyperpolarizing shift of the GV curve and on deactivation and (ii) increase in efficacy of ICA-11038, leading to a further reduction of voltage dependence of KCNQ2-mediated currents.
Figure 4.
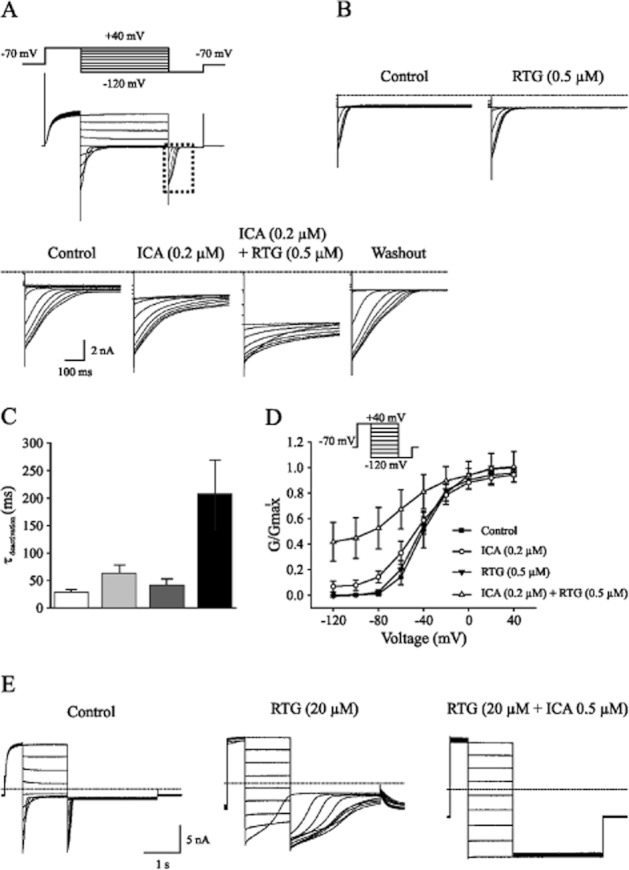
Co-application of ICA-110381 and RTG has a synergistic effect. (A) Tail currents recorded from individual CHO cells expressing KCNQ2 under control conditions, after bath application of 0.2 μM ICA-11038 (ICA), 0.2 μM ICA plus 0.5 μM retigabine (RTG) and after washout. (B) Tail currents recorded from another CHO cells expressing KCNQ2 under control conditions and in the presence of 0.5 μM RTG. (C) Deactivation times fitted monoexponentially from tail currents at −120 mV after a hyperpolarizing pulse of −40 mV under control conditions in the presence of 0.2 μM ICA, in the presence of 0.5 μM RTG and in the presence of 0.2 μM ICA and 0.5 μM RTG. (D) Conductance–voltage activation curves under control conditions, after application of 0.2 μM ICA, after application of 0.5 μM RTG and after co-application of 0.2 μM ICA and 0.5 μM RTG. (E) Example whole-cell currents recorded from individual CHO cells expressing KCNQ2 under control conditions and after bath application of 20 μM RTG and after additional application of 0.5 ICA-11038.
To test if ICA-110381 had an additional effect on KCNQ2-mediated currents in the presence of saturating concentrations of RTG, we applied RTG at concentrations between 20 and 50 μM. In the presence of these high concentrations, the kinetics of the KCNQ2-mediated current was lost due to a lack of deactivation. However, addition of ICA-110381 led to a complete absence of deactivation generating a steady current (Figure 4E).
KCNQ2 mutant W236L remained sensitive to ICA-110381
Additionally, we investigated the effect of ICA-110381 on KCNQ2 and the RTG-insensitive tryptophan mutant KCNQ2-W236L (Schenzer et al., 2005; Du et al., 2011) expressed in Xenopus oocytes. Figure 5A illustrates that ICA-110381 (10 μM) augmented the current amplitude at +40 mV by about 30%, accompanied by an acceleration of activation kinetics similar to the effect observed for KCNQ2/3 in CHO cells. In contrast to RTG, ICA-110381 also enhanced the current amplitude in KCNQ2-W236L (Figure 5B). The current–voltage relationship in Figure 5C shows that ICA-110381 produced a hyperpolarizing shift in the activation curve of KCNQ2-mediated currents, which is also present in KCNQ2-W236L-mediated currents (Figure 5D). In both constructs, this shift in the activation curves was clearly dependent on concentration of ICA-110381. Although the effect of 0.1 μM ICA-110381 was smaller in KCNQ2-W236L than in the wild type, the data suggest that the target site of ICA-110381 might be different from that of RTG.
Figure 5.
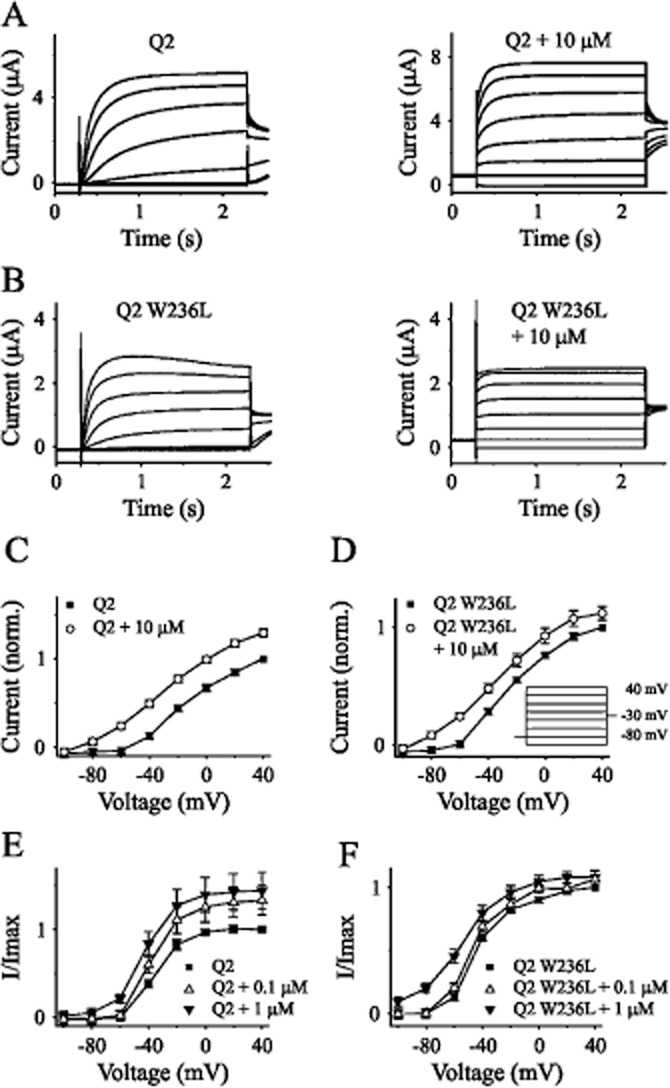
ICA-110381 activates KCNQ2-W236L. Typical current traces of oocytes expressing KCNQ2 (A) and KCNQ2-W236L (B) before and after the application of 10 μM ICA-110381 (ICA). The voltage protocol used for experiments is shown in panel D. From a holding potential of −80 mV, oocytes were clamped to values between −100 and +40 mV, followed by a constant tail pulse of −30 mV. Current–voltage (I/V) curves of KCNQ2 (n = 8) (C) and KCNQ2-W236L (n = 9) (D). Voltage dependence of I/Imax curves of KCNQ2 (n = 10) (E) and the mutant KCNQ2-W236L (n = 10) (F) measured in absence or presence of 0.1 and 1 μM ICA respectively.
ICA-110381 decreased neuronal excitability and altered resonance in CA1 hippocampal neurons
To investigate the physiological importance of ICA-110381, we examined its effects in adult rat hippocampal CA1 pyramidal cells. The voltage responses to a current ramp injection (150 pA s−1) under control conditions and 10 min after bath application of 10 μM ICA-110381 are shown in Figure 6A. As expected for an activation of a potassium conductance in the presence of ICA-110381, we measured a significant reduction of Rinput at −60 mV and a hyperpolarizing shift of the resting membrane potential (RMP), resulting in an increase of action potential threshold (Table 2).
Figure 6.
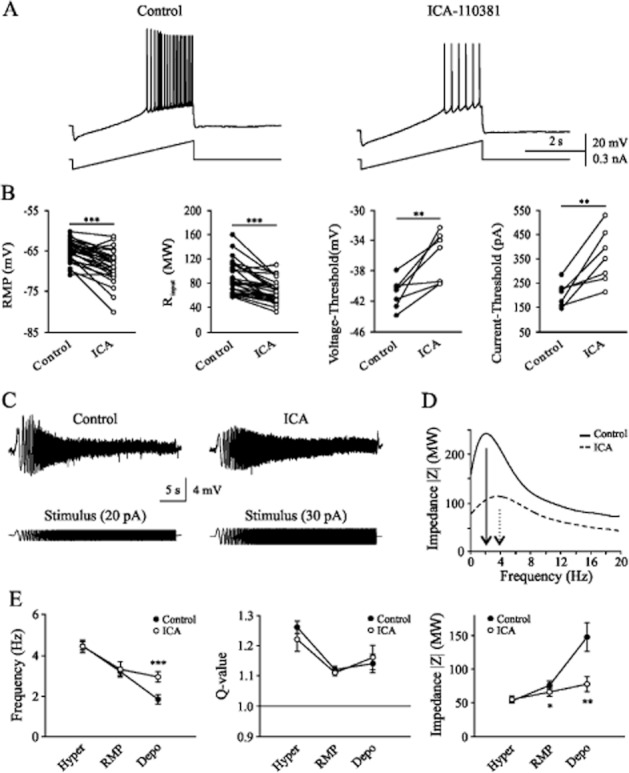
ICA-110381 alters intrinsic properties of hippocampal CA1 pyramidal neurons. (A) The voltage responses of a neuron to injection of a current ramp at the soma, under control conditions (left), after 10–15 min bath application of 10 μM ICA-110381 (right). (B) The resting membrane potential (RMP), the input resistant (Rinput), the voltage and current threshold in individual neurons under control conditions and after bath application of ICA-110381. (C) Representative recordings of the neuronal voltage response to sinusoidal current injection with constant peak to peak amplitude and linear increasing frequency (0–20 Hz). (D) Resulting impedance profile of the representative recordings. (E) Quantitative analysis of the mean resonance frequency, the corresponding Q-value and the impedance at hyperpolarized potential, at rest and at depolarized potential. Asterisks denote significance levels of P < 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
Table 2.
ICA-110381 affects cellular properties of hippocampal CA1 pyramidal cells
| Cellular properties | Control | ICA-110381 | P (n) |
|---|---|---|---|
| Rinput (MΩ) | 88.9 ± 5.1 | 66.4 ± 3.6 | <0.001 (28) |
| RMP (mV) | −65.0 ± 0.5 | −68.6 ± 0.8 | <0.001 (28) |
| Voltage threshold ramp (mV) | −41.0 ± 0.7 | −36.2 ± 1.2 | <0.01 (7) |
| Current threshold ramp (pA) | 205.7 ± 18.3 | 358.8 ± 42 | <0.01 (7) |
| Number of spikes at 400 pA | 10.0 ± 0.8 | 0.9 ± 0.5 | <0.001 (7) |
| AP-amplitude (mV) | 84.0 ± 1.8 | 72.7 ± 1.3 | <0.01 (7) |
| AP-duration (ms) | 2.8 ± 0.2 | 3.4 ± 0.3 | <0.05 (7) |
| AP-half duration (ms) | 1.12 ± 0.05 | 1.26 ± 0.02 | <0.05 (7) |
Since IM was suggested to contribute to neuronal resonance in some cell types (Hu et al., 2002; Schreiber et al., 2004), we investigated if ICA-110381 alters the intrinsic resonance behaviour of CA1 pyramidal cells. To this end, a sinusoidal current with constant peak-to-peak amplitude and linear increasing frequency (0–20 Hz) was injected (Figure 6C). The resulting voltage response was recorded before and 10 min after bath application of 10 μM ICA-110381. Under control conditions, the mean resonance frequency at RMP was 3.19 ± 0.21 Hz, and the mean Q-value was 1.12 ± 0.01 (n = 7, Figure 6E). At hyperpolarized membrane potentials (∼−78 mV), the resonance frequency was 4.5 ± 0.2 Hz (n = 7) with a Q-value of 1.26 ± 0.02 (n = 7). Near the threshold potential (∼−54 mV), the resonance frequency was lower (1.8 ± 0.2 Hz, n = 9), and only at these potentials did ICA-110381 increase the resonance frequency (to 2.9 ± 0.3 Hz, n = 9, P < 0.001). The resonance strength was not affected at all tested potentials, whereas the membrane impedance of the cell was reduced when depolarized from 147.2 ± 20.8 MΩ to 77.4 ± 11.3 MΩ (P < 0.01), at RMP (under control condition and under the new uncompensated RMP) from 75.5 ± 5.7 MΩ to 65.6 ± 6.1 MΩ (P < 0.05) but not when hyperpolarized (from 52.4 ± 2.8 MΩ to 55.2 ± 5.2 MΩ).
ICA-110381 does not affect postsynaptic currents
In order to investigate a potential impact on synaptic transmission, the effects of bath applied ICA-110381 on spontaneous and miniature postsynaptic currents were quantified. Spontaneous synaptic currents represent a mixture of action-potential-dependent and -independent synaptic release events. Action-potential-independent currents recorded in the presence of the sodium channel blocker TTX will be called miniature currents in the context of this study. Voltage-clamp whole-cell recordings were performed at −70 mV in the absence and presence of TTX (1 μM). Excitatory postsynaptic currents (EPSCs) were isolated by blocking GABAA receptors (5 μM bicuculline) and inhibitory postsynaptic currents (IPSCs) by blocking glutamate receptors (10 μM DNQX, 60 μM APV) using symmetric chloride concentrations. The amplitude and frequency were analysed under control conditions and 10–15 min after the onset of ICA-110381 application. Representative recordings of spontaneous EPSCs and in presence of TTX (mEPSCs) under control conditions and after application of 10 μM ICA-110381 are illustrated in Figure 7A and B. No significant differences were seen in the mean sEPSCs frequency of 3.4 ± 0.4 Hz (n = 14), in the mean mEPSCs frequency of 1.9 ± 0.4 Hz (n = 8) as well as in the amplitudes of either sEPSCs (−17.6 ± 0.9 pA, n = 14) or mEPSCs (−13.3 ± 1.2 pA, n = 8, Figure 7C). Sample recordings of sIPSCs under control conditions and after application of ICA-110381 are illustrated in Figure 7D. Typical mIPSCs recorded in presence of 1 μM TTX from a different cell before and after additional application of ICA-110381 are shown in Figure 6E. The mean sIPSCs frequency of 9.2 ± 0.7 Hz (n = 10) and the mean mIPSCs frequency of 5.3 ± 0.8 Hz (n = 5) were not affected by ICA-110381 application (Figure 6F). Similar to EPSCs, ICA-110381 had also no significant effect on amplitudes of sIPSCs (−29.1 ± 3.1 pA, n = 10) and mIPSCs (−25.7 ± 2.0 pA, n = 5) as illustrated in Figure 7G. In contrast, application of 50 μM RTG significantly reduced the mean of sIPSC frequency to 7.0 ± 0.9 Hz (Figure 7F) and the mean of sIPSC amplitudes to −22.7 ± 4.8 pA (n = 5, data not shown). Unlike RTG, ICA-110381 did not affect the decay of sIPSCs (Figure 7G and H).
Figure 7.
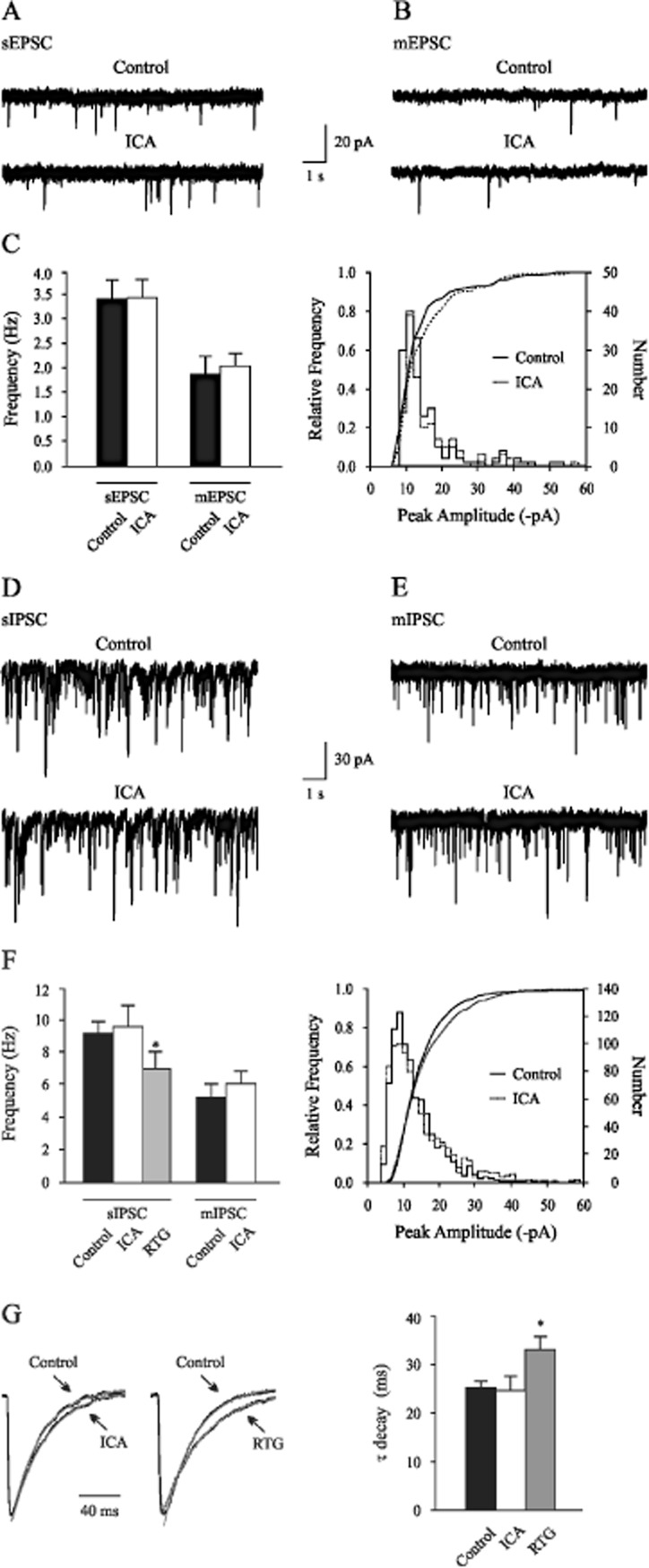
Properties of postsynaptic currents were not changed by ICA-110381. (A) Continuous recordings showing EPSCs from a rat hippocampal CA1 neuron in the absence and (B) in the presence of TTX (1 μM) under control conditions and 10 min after bath application of ICA-110381 (ICA). (C) Summary data showing mean frequency of sEPSCs and mEPSCs under control conditions and in the presence of ICA. Amplitude distributions of events recorded in under control conditions and in the presence of ICA. (D) Representative recordings of sIPSCs and (E) in the presence of TTX before and after application of ICA-110381. (F) Quantitative analyses of the mean sIPSC frequency under control conditions, in the presence of ICA-110381 (10 μM) and in the presence of RTG (50 μM) and mIPSC frequency under control conditions and in the presence of ICA-110381. Amplitude distributions of events recorded in under control conditions and in the presence of ICA-110381. Superimposed lines show cumulative amplitude histograms (P > 0.05, Kolmogorov–Smirnov test). (G) Average sIPSCs waveforms (right) obtained by aligning 50 individual events on their rising phase were normalized and superimposed (left) and the decay time was quantitative analysed (right). Whereas the decay of the waveform was unaffected by ICA-110381 (left), RTG (right) slowed the decay phase significantly. Asterisks denote significance levels of P < 0.05 (*).
Effects of ICA-110381 on kainate-induced network oscillations
IM has previously been implicated in hippocampal network oscillations since Retigabine suppressed γ and θ frequency network oscillations (Boehlen et al., 2009). In addition, network oscillations and neuronal excitability are tightly coupled as previously shown for IM (Leao et al., 2009). Since ICA-110381 profoundly affected IM and neuronal excitability, we investigated its influence on network oscillations. Bath application of 100 nM kainate induces γ frequency oscillations (30–40 Hz) in horizontal slices and θ-like oscillations (4–15 Hz) in coronal slices with mostly longitudinal connectivity (Gloveli et al., 2005; Boehlen et al., 2009). Kainate-induced network oscillations commenced 15–30 min and stabilized within 60 min (Boehlen et al., 2009).
Figure 7A shows typical recordings of γ and θ frequency oscillations from area CA3 under control conditions and after application of 5 μM ICA-110381, displaying a strong suppression of both γ and θ frequency oscillations in areas CA3. Similarly, ICA-110381 suppressed γ frequency oscillations recorded from area CA1 (data not shown). Figure 7B shows the corresponding power spectra of recordings in Figure 8A. Power spectrum analysis of γ frequency oscillations showed a major peak in area CA3 around 34 Hz (CA3: 33.4 ± 1.2 Hz, n = 12 and CA1: 29.7 ± 1.0 Hz, n = 7). The mean peak power was 2.3 ± 0.7 × 10−3 mV2 in area CA3 and 1.3 ± 0.8 × 10−3 mV2 in area CA1. The power spectrum analysis of θ frequency oscillations (n = 9) in area CA3 showed a mean peak frequency of 11.8 ± 0.5 Hz with an averaged peak power of 2.4 ± 0.5 × 10−5 mV2.
Figure 8.
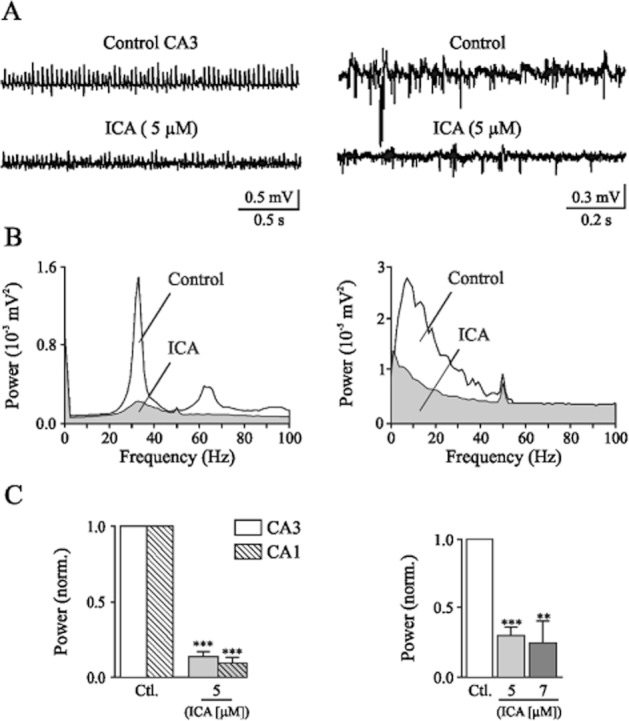
Kainate-induced network oscillations in rat hippocampal slices are reduced by ICA-110381. (A) Sample recordings in horizontal slices from stratum pyramidale in area CA3 (left) showing kainate-induced γ frequency oscillations under control conditions (top trace), following a 60 min bath application of 5 μM ICA-110381 (bottom trace) and in coronal slices from stratum pyramidale in area CA3 (right) showing kainate-induced θ frequency oscillations (top trace), following application of 5 μM ICA-110381 (bottom trace). (B) Corresponding power spectra for the recordings shown in panel A. Power spectra for γ oscillations (left) and θ oscillations (right) under control conditions, after 60 min of 5 μM ICA-110381 application. (C) Bar plots summarizing the results for γ oscillations in area CA3 and area CA1 for the effect of ICA-110381 and the results for theta oscillations for the effect of ICA-110381 on averaged normalized peak power. Asterisks denote significance levels of P < 0.01 (**) and P < 0.001 (***).
Figure 8C summarizes the modulation of ICA-110381 on γ and θ frequency network oscillations. The peak power of γ oscillations was reduced by 86.27 ± 0.04% of control in area CA3 (n = 12, P < 0.001) and by 92.2 ± 3.5% in area CA1 (n = 7, P < 0.001), while the peak frequency decreased insignificantly from 33.4 ± 1.2 Hz to 31.1 ± 2.7 Hz (P = 0.31) in area CA3 and in area CA1 significantly from 29.7 ± 1 Hz to 20.9 ± 3.5 Hz (P < 0.05). At a concentration of 5 μM, ICA-110381 diminished θ frequency oscillations by 69.6 ± 5.2% (n = 4, P < 0.001) and at 7 μM by 75.3 ± 15.7% (n = 5, P < 0.01). The averaged peak frequency was significantly reduced from 12.2 ± 0.5 Hz to 5.9 ± 0.3 Hz (P < 0.001) at 5 μM. Oscillations did not recover to control levels during prolonged wash-out (>90 min, data not shown).
ICA-110381 exhibits anticonvulsive properties
Considering that ICA-110381 acts as a potent activator of KCNQ2 and KCNQ2/3 potassium channels, we investigated its potential anticonvulsive properties using a rat amygdala kindling model, a widely used model for complex partial seizures. ICA-110381 was tested at 3, 10 and 30 mg kg−1 and exhibited a potent anticonvulsant effect (see Table 3). Typical seizure responses to a stimulation of 30 μA (control) and to a stimulation of 42 μA (ICA-110381 30 mg kg−1 p.o.) recorded from the same animal (Figure 9A). Figure 9B shows that significant elevation of ADT was already seen at 3 mg kg−1 p.o. (66.7 ± 32.3%, n = 9, P < 0.05). Increasing the dose to 10 mg kg−1 further reduced the seizure severity (n = 11, P < 0.05) and seizure duration (n = 11, P = 0.01). At the highest dose tested (30 mg kg−1), all measured parameters, afterdischarge duration (n = 10, P < 0.01), seizure severity (P < 0.01), seizure duration (P < 0.01) and total duration of behavioural changes were significantly reduced (P < 0.05).
Table 3.
Effects of ICA-110381 in fully amygdala-kindled rats. Control experiments were carried out 2 days prior to drug testing in the same group of rats
| Treatment | Dose Appl. route (mg kg−1) | Pre-treatment time (min) | No. of rats (n) | Threshold for induction of after discharges (μA) | Seizure recordings at afterdischarge threshold current | Plasma conc. (ng mL−1) | Brain conc. (ng g−1) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| After-discharge duration (s) | Seizure score (score) | Seizure duration (s) | Total duration of behavioural changes (s) | |||||||
| Control | 9 | 105.7 ± 29.8 | 76.0 ± 9.0 | 4.8 ± 0.2 | 50.2 ± 3.5 | 223.3 ± 12.7 | ||||
| ICA-110381 | 3 p.o. | 30 | 9 | 149.6 ± 30.1* | 64.4 ± 12.1 | 4.4 ± 0.4 | 38.3 ± 5.2 | 227.8 ± 36.8 | 91.9 ± 17.0 | 90.5 ± 6.4 |
| Control | 11 | 116.0 ± 20.3 | 58.0 ± 7.9 | 4.6 ± 0.3 | 44.6 ± 5.5 | 209.6 ± 27.2 | ||||
| ICA-110381 | 10 p.o. | 30 | 11 | 270.0 ± 53.0** | 37.4 ± 10.3 | 3.6 ± 0.4* | 26.0 ± 6.1** | 145.2 ± 47.9 | 447.2 ± 36.0 | 445.0 ± 125.4 |
| Control | 10 | 88.5 ± 16.0 | 61.2 ± 8.1 | 4.7 ± 0.3 | 43.4 ± 3.7 | 240.1 ± 24.2 | ||||
| ICA-110381 | 30 p.o. | 30 | 10 | 202.2 ± 38.1** | 15.3 ± 8.2** | 3.2 ± 0.4** | 11.1 ± 3.0** | 71.9 ± 40.4* | 468.7 ± 103.2 | 454.2 ± 65.3 |
Asterisks denote significance levels (Wilcoxon signed rank test for paired replicates) of P < 0.05 (*) and P < 0.01 (**).
Figure 9.
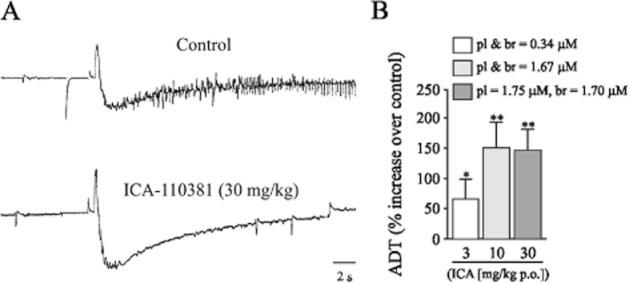
ICA-110381 exhibits anticonvulsive potential. (A) In vivo, seizure response to a stimulation of 30 μA (control, top trace) and to a stimulation of 42 μA (ICA-110381 30 mg kg−1 p.o., bottom trace) recorded from the same fully amygdala-kindled rat. (B) Quantitative analysis of the relative increase of the threshold for the induction of afterdischarges (ADT) in comparison with vehicle control at different concentrations of ICA-110381 in plasma (pl) and brain (br) (corresponding to orally applied doses, mg kg−1 p.o., n = 9, 11 and 10 respectively). Control experiments were carried out 2 days prior to drug testing in the same group of rats. Asterisks denote significance levels (Wilcoxon signed rank test for paired replicates) of P < 0.05 (*) and P < 0.01 (**).
ICA-110381 was well tolerated at all tested doses when the animals were observed in different tests during the last 5 min prior to electrical stimulation. There were no overt behavioural alterations noted in the open arena. All treated animals were able to perform the rotarod test and did not differ in their rectal body temperature measured before and after treatment.
Discussion
In this study, we described the effect of the benzanilide 4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid (ICA-110381) on KCNQ2, KCNQ2/3, KCNQ4 and KCNQ3/5 channels and investigated some implications on physiological and pathophysiological network activities. In currents mediated by KCNQ2 and KCNQ2/3, ICA-110381 produced a profound reduction of voltage dependence accompanied by a hyperpolarizing shift of the activation curve and a marked slowing in deactivation. This was also observed in KCNQ4 to a lesser extent. Because ICA-110381 had only minor effects on KCNQ3/5-mediated currents, ICA-110381 is much more potent for KCNQ2 containing channels comparable with the structural similar compound ICA-27243 (Wickenden et al., 2008). Despite the impaired voltage gating, ICA-110381 did not affect the potassium selectivity of KCNQ2- and KCNQ2/3-mediated currents or the antagonistic effect of XE991. Most of the effects produced by ICA-110381 were previously described for various compounds acting as KCNQ channel activators. So far, all activators of neuronal KCNQ channels produce a hyperpolarizing shift of V1/2 and also a slowing in deactivation was observed for a number of compounds like RTG (Main et al., 2000; Tatulian et al., 2001; Linley et al., 2012), zinc pyrithione (Xiong et al., 2007), meclophenamic acid (Peretz et al., 2005), NH6 (Peretz et al., 2007) and in compounds with a high structural similarity to ICA-110381 like ICA-27243 (Blom et al., 2010) and ztz240 (Gao et al., 2010). Furthermore, for KCNQ2 channels, a ‘voltage-independent current’ produced by ICA-27243 was described (Blom et al., 2010), an observation, that is in line with our results that ICA-110381 prolonged recovery from activation for many seconds resulting in a steady current in KCNQ2-expressing cells.
In our experiments in CHO cells, we have seen a maximal effect of RTG between 20 and 50 μM when KCNQ2 was expressed. In oocytes, RTG applied at a concentration of 100 μM did not augment currents mediated by KCNQ2 except for a prolonged deactivation (Blom et al., 2010). This discrepancy may be due to different expression systems used. Interestingly, Blom et al. (2010) have shown that ICA-27243 prolonged the tail currents of KCNQ2-mediated currents more than RTG alone, but this effect is much smaller than that one we observed with ICA-110381.
Our observation that ICA-110381 had similar effects in the RTG-insensitive mutant W236 suggests that the target site of ICA-110381 differs from that of RTG. RTG interacts with KCNQ channels by stabilizing the S5-6 domain hinge in the open state accompanied by a hyperpolarizing shift of the voltage activation curve and an increase in deactivation time (Schenzer et al., 2005; Wuttke et al., 2005; Brown and Passmore, 2009).
In hippocampal CA1 pyramidal cells, IM is predominantly mediated by KCNQ2/3 subunits (Wang et al., 1998), although KCNQ5 is also expressed in the hippocampus (Schroeder et al., 2000; Jensen et al., 2005). In mice, higher levels of KCNQ5 have been found in hippocampal CA3 pyramidal cells in contrast to lower levels in CA1 and CA2 pyramidal cells and mossy fibres (Tzingounis et al., 2010).
Our experiments indicate that the activation of the rat hippocampal IM by ICA-110381 led to a hyperpolarization of the cell and a reduction of membrane resistance at −60 mV. Similar effects in CA1 cells were previously observed after application of 10 μM RTG (Gu et al., 2005). Remarkably, in neurons of the entorhinal cortex, application of 100 μM RTG had no significant effect on Rinput at RMP but significantly reduced Rinput near the spiking threshold (Hetka et al., 1999).
Whereas ICA-110381 had no effect on postsynaptic currents in our experiments, RTG prolonged the decay phase of sIPSCs, suggesting modulation of postsynaptic GABAA receptor currents as well. A similar effect of RTG has been previously reported for mIPSC recorded from cultured cortical neurons (Otto et al., 2002). Effects on inhibitory and excitatory synaptic transmission in cultured hippocampal neurons have also been reported for the recently synthesized IM activators NH6 (Peretz et al., 2007).
The profound effect on neuronal excitability suggested that it might also affect network behaviour and particularly epileptiform activity. We found that ICA-110381 showed anticonvulsive properties in amygdala-kindled rats. The kindling model is predictive for complex partial seizures in man, and it is widely used for characterization of new potential antiepileptic drugs (McNamara et al., 1980; Loscher and Schmidt, 1988; Meldrum, 1997). Interestingly, the KCNQ2 subunit of potassium channels is up-regulated in the basolateral amygdala in amygdala-kindled rats, which is regarded to be an important compensatory mechanism to counteract hyperexcitability (Penschuck et al., 2005). ICA-110381 as potent opener of KCNQ2-containing channels was able to elevate the threshold for induction of ADs already at doses of 3 mg kg−1 p.o. corresponding to a plasma concentration of approximately 0.34 μM in rats. This effect on ADT was more pronounced at 10 mg kg−1 but did not further increase at 30 mg kg−1. This observation corresponds to the limitation in exposure at higher dose as shown in satellite animals (see Table 3). At 10 mg kg−1, the compound also reduced the seizure severity and duration; and at 30 mg kg−1, ADs were completely blocked. A similar broad spectrum of activity was observed in previous experiments with RTG, albeit RTG was already effective at lower doses, e.g. 0.01 mg kg−1 p.o. (Tober et al., 1996), and exhibited more potent effects compared by dose, which is potentially due to its broader mechanism of action. Furthermore, pharmacokinetic data of RTG point to approximately fourfold higher plasma levels at comparable doses in rats. Beside its role in epilepsy, the amygdala kindling model is also regarded to be predictive in terms of potential adverse effects of new AEDs (Loscher and Honack, 1991; Loscher and Schmidt, 1993). Since the amygdala is centrally involved in emotional behaviour as well as in the modulation of cognitive functions (LeDoux, 1992; Fanselow and Gale, 2003), the kindling process not only induces a greater susceptibility to seizures but also an altered susceptibility to psychotropic adverse effects. Therefore, new antiepileptic drugs are not only investigated regarding its anticonvulsant activity but also regarding behavioural alterations in fully kindled rats. ICA-110381 did not induce any behavioural alterations at tested doses up to 30 mg kg−1, indicating a good tolerability in female kindled rats. Drugs acting on partial epilepsies will often be employed in temporal lobe epilepsies involving also the hippocampal formation and the entorhinal cortex. These regions display pronounced network oscillation implicated in spatial orientation and formation of declarative memory. In the hippocampus, IM modulates network oscillations by modifying action potential firing in pyramidal neurons (Leao et al., 2009). The reduction of both θ and γ frequency network oscillations by ICA-110381 might predict side effects on cognitive functions and thereby limit the dose range in which these compounds could be therapeutically applied.
In the hippocampal formation, several types of neurons such as stellate cells in layer II of the medial entorhinal cortex and hippocampal pyramidal cells have been shown to resonate at θ frequencies (Hu et al., 2009; Boehlen et al., 2010). Modelling studies suggested IM to be crucial for intrinsic single-cell resonance (Hutcheon et al., 2000). Indeed, it has already been reported that the IM blocker XE991 suppresses θ resonance at depolarized subthreshold potentials in hippocampal pyramidal cells (Hu et al., 2002). In line with these observations and the voltage dependence of IM activation, our data indicate that an activation of IM by ICA-110381 significantly modifies single-cell resonance at near-threshold in CA1 pyramidal cells. Our recordings from CHO cells indicate that ICA-110381 diminished the voltage dependence of IM, thus leading to activation of IM also at resting or hyperpolarized membrane potentials. However, ICA-110381 had no effect on single-cell resonance at resting or hyperpolarized membrane potentials underlining the minor role of IM for single cell resonance at hyperpolarized potentials.
In summary, our results show that ICA-110381, which predominantly acts on KCNQ2-containing channels, shapes resonance and network oscillations in vitro and show anticonvulsant potential in vivo without affecting spontaneous synaptic transmission in the rat hippocampus in vitro.
Acknowledgments
We thank Elbion AG for synthesis of ICAGEN-110381, TJ Jentsch (Leibniz-Institut für Molekulare Pharmakologie and Max-Delbrück-Centrum für Molekulare Medizin, Berlin) for KCNQ cDNAs, IA Fleidervish, H Hu, K Vervaeke and C Henneberger for helpful discussions. This research was supported by DFG grants He1128/17-1 and Schw866/4-1, Hertie Foundation, Epicure and the Bernstein Center for Computational Neuroscience Berlin.
Glossary
- AED
antiepileptic drug
- ICA-110381
4-Chlor-N-(6-chlor-pyridin-3-yl)-benzamid
- IM
M-current
- NMG
N-methyl-glutamine
- RMP
resting membrane potential
- RTG
Retigabine
Conflict of interest
The funder had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors have declared that no competing interests exist.
References
- Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by 4-aminopyridine in rat entorhinal cortex hippocampal slices. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:33–39. doi: 10.1007/pl00005320. [DOI] [PubMed] [Google Scholar]
- Armand V, Rundfeldt C, Heinemann U. Effects of retigabine (D-23129) on different patterns of epileptiform activity induced by low magnesium in rat entorhinal cortex hippocampal slices. Epilepsia. 2000;41:28–33. doi: 10.1111/j.1528-1157.2000.tb01501.x. [DOI] [PubMed] [Google Scholar]
- Biervert C, Steinlein OK. Structural and mutational analysis of KCNQ2, the major gene locus for benign familial neonatal convulsions. Hum Genet. 1999;104:234–240. doi: 10.1007/pl00008713. [DOI] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460:109–116. doi: 10.1016/s0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- Blom SM, Schmitt N, Jensen HS. Differential effects of ICA-27243 on cloned K(V)7 channels. Pharmacology. 2010;86:174–181. doi: 10.1159/000317525. [DOI] [PubMed] [Google Scholar]
- Boehlen A, Kunert A, Heinemann U. Effects of XE991, retigabine, losigamone and ZD7288 on kainate-induced theta-like and gamma network oscillations in the rat hippocampus in vitro. Brain Res. 2009;1295:44–58. doi: 10.1016/j.brainres.2009.08.031. [DOI] [PubMed] [Google Scholar]
- Boehlen A, Heinemann U, Erchova I. The range of intrinsic frequencies represented by medial entorhinal cortex stellate cells extends with age. J Neurosci. 2010;30:4585–4589. doi: 10.1523/JNEUROSCI.4939-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehlen A, Heinemann U, Henneberger C. Heterogeneous voltage dependence of interneuron resonance in the hippocampal stratum radiatum of adult rats. Synapse. 2011;65:1378–1381. doi: 10.1002/syn.20971. [DOI] [PubMed] [Google Scholar]
- Brodie M, Lerche H, Gil-Nagel A, Elger C, Hall S, Shin P, et al. Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology. 2010;75:1817–1824. doi: 10.1212/WNL.0b013e3181fd6170. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neuron. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- Chen X, Johnston D. Properties of single voltage-dependent K+ channels in dendrites of CA1 pyramidal neurones of rat hippocampus. J Physiol. 2004;559:187–203. doi: 10.1113/jphysiol.2004.068114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuczwar P, Wojtak A, Cioczek-Czuczwar A, Parada-Turska J, Maciejewski R, Czuczwar SJ. Retigabine: the newer potential antiepileptic drug. Pharmacol Rep. 2010;62:211–219. doi: 10.1016/s1734-1140(10)70260-7. [DOI] [PubMed] [Google Scholar]
- Dalby-Brown W, Hansen HH, Korsgaard MPG, Mirza N, Olesen SP. K(v)7 channels: function, pharmacology and channel modulators. Curr Top Med Chem. 2006;6:999–1023. doi: 10.2174/156802606777323728. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Du XN, Zhang X, Qi JL, An HL, Li JW, Wan YM, et al. Characteristics and molecular basis of celecoxib modulation on K(v)7 potassium channels. Br J Pharmacol. 2011;164:1722–1737. doi: 10.1111/j.1476-5381.2011.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Gale GD. The amygdala, fear, and memory. Ann N Y Acad Sci. 2003;985:125–134. doi: 10.1111/j.1749-6632.2003.tb07077.x. [DOI] [PubMed] [Google Scholar]
- Forcelli PA, Soper C, Lakhkar A, Gale K, Kondratyev A. Anticonvulsant effect of retigabine during postnatal development in rats. Epilepsy Res. 2012;101:135–140. doi: 10.1016/j.eplepsyres.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Freeman FG, Jarvis MF. The effect of interstimulation interval on the assessment and stability of kindled seizure thresholds. Brain Res Bull. 1981;7:629–633. doi: 10.1016/0361-9230(81)90109-x. [DOI] [PubMed] [Google Scholar]
- French J, Abou-Khalil BW, Leroy R, Yacubian E, Shin P, Mansbach H, et al. Randomized, double-blind, placebo-controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology. 2011;76:1555–1563. doi: 10.1212/WNL.0b013e3182194bd3. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhang T, Wu M, Xiong Q, Sun H, Zhang Y, et al. Isoform-specific prolongation of Kv7 (KCNQ) potassium channel opening mediated by new molecular determinants for drug-channel interactions. J Biol Chem. 2010;285:28322–28332. doi: 10.1074/jbc.M110.116392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloveli T, Dugladze T, Saha S, Monyer H, Heinemann U, Traub RD, et al. Differential involvement of oriens/pyramidale interneurones in hippocampal network oscillations in vitro. J Physiol. 2005;562:131–147. doi: 10.1113/jphysiol.2004.073007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al Qatari M, Ye F, Wickenden AD, et al. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- Hetka R, Rundfeldt C, Heinemann U, Schmitz D. Retigabine strongly reduces repetitive firing in rat entorhinal cortex. Eur J Pharmacol. 1999;386:165–171. doi: 10.1016/s0014-2999(99)00786-4. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol. 2002;545:783–805. doi: 10.1113/jphysiol.2002.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Graham LJ, Storm JF. Complementary theta resonance filtering by two spatially segregated mechanisms in CA1 hippocampal pyramidal neurons. J Neurosci. 2009;29:14472–14483. doi: 10.1523/JNEUROSCI.0187-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheon B, Yarom Y. Resonance, oscillation and the intrinsic frequency preferences of neurons 3. Trends Neurosci. 2000;23:216–222. doi: 10.1016/s0166-2236(00)01547-2. [DOI] [PubMed] [Google Scholar]
- Jensen HS, Callo K, Jespersen T, Jensen BS, Olesen SP. The KCNQ5 potassium channel from mouse: a broadly expressed M-current like potassium channel modulated by zinc, pH, and volume changes. Brain Res Mol Brain Res. 2005;139:52–62. doi: 10.1016/j.molbrainres.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Schroeder BC, Kubisch C, Friedrich T, Stein V. Pathophysiology of KCNQ channels: neonatal epilepsy and progressive deafness. Epilepsia. 2000;41:1068–1069. doi: 10.1111/j.1528-1157.2000.tb00302.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen LV, Sandager-Nielsen K, Hansen HH. K(v) 7 (KCNQ) channel openers normalize central 2-deoxyglucose uptake in a mouse model of mania and increase prefrontal cortical and hippocampal serine-9 phosphorylation levels of GSK3β. J Neurochem. 2012;121:373–382. doi: 10.1111/j.1471-4159.2012.07704.x. [DOI] [PubMed] [Google Scholar]
- Lamas JA, Selyanko AA, Brown DA. Effects of a cognition-enhancer, linopirdine (DuP 996), on M-type potassium currents (IK(M)) and some other voltage- and ligand-gated membrane currents in rat sympathetic neurons. Eur J Neurosci. 1997;9:605–616. doi: 10.1111/j.1460-9568.1997.tb01637.x. [DOI] [PubMed] [Google Scholar]
- Leao RN, Tan HM, Fisahn A. Kv7/KCNQ channels control action potential phasing of pyramidal neurons during hippocampal gamma oscillations in vitro. J Neurosci. 2009;29:13353–13364. doi: 10.1523/JNEUROSCI.1463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. Brain mechanisms of emotion and emotional learning. Curr Opin Neurobiol. 1992;2:191–197. doi: 10.1016/0959-4388(92)90011-9. [DOI] [PubMed] [Google Scholar]
- Lerche H, Biervert C, Alekov AK, Schleithoff L, Lindner M, Klinger W, et al. A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann Neurol. 1999;46:305–312. doi: 10.1002/1531-8249(199909)46:3<305::aid-ana5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Linley JE, Pettinger L, Huang D, Gamper N. M channel enhancers and physiological M channel block. J Physiol. 2012;590:793–807. doi: 10.1113/jphysiol.2011.223404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, Honack D. Responses to NMDA receptor antagonists altered by epileptogenesis. Trends Pharmacol Sci. 1991;12:52. doi: 10.1016/0165-6147(91)90496-f. [DOI] [PubMed] [Google Scholar]
- Loscher W, Schmidt D. Which animal models should be used in the search for new antiepileptic drugs? A proposal based on experimental and clinical considerations. Epilepsy Res. 1988;2:145–181. doi: 10.1016/0920-1211(88)90054-x. [DOI] [PubMed] [Google Scholar]
- Loscher W, Schmidt D. New drugs for the treatment of epilepsy. Curr Opin Invest Drugs. 1993;2:1067–1095. [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Byrne MC, Dasheiff RM, Fitz JG. The kindling model of epilepsy: a review. Prog Neurobiol. 1980;15:139–159. doi: 10.1016/0301-0082(80)90006-4. [DOI] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Wuttke TV, Seebohm G, Lerche H. KV7 channelopathies. Pflugers Arch. 2010;460:277–288. doi: 10.1007/s00424-010-0831-3. [DOI] [PubMed] [Google Scholar]
- Marrion NV. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- Meldrum BS. Identification and preclinical testing of novel antiepileptic compounds. Epilepsia. 1997;38(Suppl. 9):S7–S15. doi: 10.1111/j.1528-1157.1997.tb05204.x. [DOI] [PubMed] [Google Scholar]
- Munro G, Erichsen HK, Mirza NR. Pharmacological comparison of anticonvulsant drugs in animal models of persistent pain and anxiety. Neuropharmacology. 2007;53:609–618. doi: 10.1016/j.neuropharm.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci. 2006;26:2053–2059. doi: 10.1523/JNEUROSCI.1575-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 2nd edn. London: Academic Press Inc; 1986. [Google Scholar]
- Penschuck S, Bastlund JF, Jensen HS, Stensbol TB, Egebjerg J, Watson WP. Changes in KCNQ2 immunoreactivity in the amygdala in two rat models of temporal lobe epilepsy. Brain Res Mol Brain Res. 2005;141:66–73. doi: 10.1016/j.molbrainres.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Peretz A, Degani N, Nachman R, Uziyel Y, Gibor G, Shabat D, et al. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Peretz A, Sheinin A, Yue C, Degani-Katzav N, Gibor G, Nachman R, et al. Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol. 2007;97:283–295. doi: 10.1152/jn.00634.2006. [DOI] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Porter RJ, Nohria V, Rundfeldt C. Retigabine. Neurother. 2007;4:149–154. doi: 10.1016/j.nurt.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puil E, Gimbarzevsky B, Miura RM. Quantification of membrane properties of trigeminal root ganglion neurons in guinea pigs. J Neurophysiol. 1986;55:995–1016. doi: 10.1152/jn.1986.55.5.995. [DOI] [PubMed] [Google Scholar]
- Racine R, Okujava V, Chipashvili S. Modification of seizure activity by electrical stimulation. 3. Mechanisms. Electroencephalogr Clin Neurophysiol. 1972;32:295–299. doi: 10.1016/0013-4694(72)90178-2. [DOI] [PubMed] [Google Scholar]
- Rogawski MA, Bazil CW. New molecular targets for Antiepileptic drugs: alpha 2 delta, SV2A, and K(v)7/KCNQ/M potassium channels. Curr Neurol Neurosci Rep. 2008;8:345–352. doi: 10.1007/s11910-008-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostock A, Tober C, Rundfeldt C, Bartsch R, Engel J, Polymeropoulos EE, et al. D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996;23:211–223. doi: 10.1016/0920-1211(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Rundfeldt C. The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur J Pharmacol. 1997;336:243–249. doi: 10.1016/s0014-2999(97)01249-1. [DOI] [PubMed] [Google Scholar]
- Rundfeldt C, Netzer R. The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells tranfected with human KCNQ2/3 subunits. Neurosci Lett. 2000;282:73–76. doi: 10.1016/s0304-3940(00)00866-1. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, et al. Molecular determinants of KCNQ (K(V)7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber S, Fellous JM, Tiesinga P, Sejnowski TJ. Influence of ionic conductances on spike timing reliability of cortical neurons for suprathreshold rhythmic inputs. J Neurophysiol. 2004;91:194–205. doi: 10.1152/jn.00556.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tober C, Rostock A, Rundfeldt C, Bartsch R. D-23129: a potent anticonvulsant in the amygdala kindling model of complex partial seizures. Eur J Pharmacol. 1996;303:163–169. doi: 10.1016/0014-2999(96)00073-8. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, et al. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci U S A. 2010;107:10232–10237. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wickenden AD McNaughton-Smith. Kv7 channels as targets for the treatment of pain. Curr Pharm Des. 2009;15:1773–1798. doi: 10.2174/138161209788186326. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Krajewski JL, London B, Wagoner PK, Wilson WA, Clark S, et al. N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol Pharmacol. 2008;73:977–986. doi: 10.1124/mol.107.043216. [DOI] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Sun H, Li M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol. 2007;3:287–296. doi: 10.1038/nchembio874. [DOI] [PubMed] [Google Scholar]
- Xiong QJ, Gao ZB, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci. 2008;29:99–107. doi: 10.1016/j.tips.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]