

Abstract
Background and Purpose
SU4312, a potent and selective inhibitor of VEGF receptor-2 (VEGFR-2), has been designed to treat cancer. Recent studies have suggested that SU4312 can also be useful in treating neurodegenerative disorders. In this study, we assessed neuroprotection by SU4312 against 1-methyl-4-phenylpyridinium ion (MPP+)-induced neurotoxicity and further explored the underlying mechanisms.
Experimental Approach
MPP+-treated neurons and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated zebrafish were used to study neuroprotection by SU4312. NOS activity was assayed in vitro to examine direct interactions between SU4312 and NOS isoforms.
Key Results
SU4312 unexpectedly prevented MPP+-induced neuronal apoptosis in vitro and decreased MPTP-induced loss of dopaminergic neurons, reduced expression of mRNA for tyrosine hydroxylase and impaired swimming behaviour in zebrafish. In contrast, PTK787/ZK222584, a well-studied VEGFR-2 inhibitor, failed to prevent neurotoxicity, suggesting that the neuroprotective actions of SU4312 were independent of its anti-angiogenic action. Furthermore, SU4312 exhibited non-competitive inhibition of purified neuronal NOS (nNOS) with an IC50 value of 19.0 μM but showed little or no effects on inducible and endothelial NOS. Molecular docking simulations suggested an interaction between SU4312 and the haem group within the active centre of nNOS.
Conclusions and Implication
SU4312 exhibited neuroprotection against MPP+ at least partly via selective and direct inhibition of nNOS. Because SU4312 could reach the brain in rats, our study also offered a support for further development of SU4312 to treat neurodegenerative disorders, particularly those associated with NO-mediated neurotoxicity.
Keywords: SU4312, neuroprotection, Parkinson's disease, neuronal NOS, angiogenesis, MPP+
Introduction
SU4312 (3-[4-(dimethylamino)benzylidenyl]indolin-2-one) is a cell-permeable, potent and selective inhibitor of the VEGF receptor-2 (VEGFR-2) tyrosine kinase that has been designed as a candidate drug for cancer therapy (Sun et al., 1998). SU4312 competes with ATP for binding to VEGFR-2 and is able to completely block VEGF signalling in a non-competitive manner (Sun et al., 1998). SU4312 specifically inhibited VEGF-dependent angiogenesis without damaging normal cells (Tran et al., 2007; Miki et al., 2010) and also reduced the proliferation of multiple myeloma and leukaemia tumour cells in vitro (McMillin et al., 2010). It is suggested that the anticancer activity of SU4312 is achieved through direct inhibition of the proliferation of cancer cells and indirect suppression of angiogenesis. The recently discovered capabilities of SU4312 to block Aβ plaque-induced vessel formation in APP23 transgenic mice and to directly inhibit autophosphorylation of hte Parkinson's disease-associated leucine-rich repeat kinase 2 (LRRK2) highlight its potential to be developed for the treatment of neurodegenerative disorders (Schultheiss et al., 2006; Lee et al., 2010).
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a common neurotoxin widely used to produce Parkinson's disease models (Langston and Irwin, 1986). MPTP is converted into its active metabolite 1-methyl-4-phenylpyridinium ion (MPP+) by monoamine oxidase B (MAO-B) in the inner mitochondrial membrane (Tipton and Singer, 1993). MPP+ stimulates the production of superoxide radical and activates NOS to produce NO (Gonzalez-Polo et al., 2003; 2004b). Superoxide radical not only inhibits mitochondrial complex I of the electron transport chain but also reacts with NO to form peroxynitrite ion (ONOO-), the precursor of the tissue-damaging hydroxyl radical (Beckman et al., 1990). Thus, inhibition of NOS activity decreases the production of NO and further attenuates MPTP/MPP+-induced neurotoxicity (Przedborski et al., 1996).
In this study, we first examined the neuroprotective activity of SU4312 against MPP+-induced neuronal death in primary cerebellar granule neurons (CGNs) and dopaminergic cell lines, and MPTP-induced neurotoxicity in zebrafish. We demonstrated that the neuroprotective action of SU4312 was not closely correlated with its anti-angiogenic effect; instead, it might be mediated through the direct and selective inhibition of neuronal NOS (nNOS). Moreover, the substrate kinetics of NOS inhibition and molecular simulation indicated that SU4312 inhibited nNOS in a non-competitive manner.
Methods
Primary cerebellar granule neuron culture
All animal care and experimental procedures were conducted according to the ethical guidelines of ICMS, Macau University and the Animal Care Facility, The Hong Kong Polytechnic University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., ). A total of 15 animals were used in the experiments described here. Rat CGNs were prepared from 8-day-old Sprague–Dawley rats (The Animal Care Facility, The Hong Kong Polytechnic University) as described in our previous publication (Li et al., 2005). Briefly, neurons were seeded at a density of 2.7 × 105 cells·mL−1 in basal modified Eagle's medium (Life Technologies, Carlsbad, CA, USA) containing 10% FBS, 25 mM KCl, 2 mM glutamine and penicillin (100 units·mL−1) / streptomycin (100 μg·mL−1). Cytosine arabinoside (10 μM) was added to the culture medium 24 h after plating to limit the growth of non-neuronal cells. With the use of this protocol, more than 95% of the cultured cells were granule neurons.
Culture of cell lines
The human neuroblastoma SH-SY5Y cells were obtained from ATCC (Manassas, VA, USA). The cells were maintained in supplemented DMEM, 10% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin in a 37°C, 5% CO2 incubator. PC12 pheochromocytoma cells were also obtained from ATCC. The cells were cultured in medium that consisted of DMEM, 10% heat-inactivated horse serum, 5% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin in a 37°C, 5% CO2 incubator. All experiments were carried out 48 h after the cells were seeded.
Measurement of neurotoxicity
The survival of neurons in the presence of SU4312 and/or MPP+ was assayed by determining the activity of mitochondrial dehydrogenases with 3(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide (MTT) assay (Li et al., 2007). The assay was performed according to the specifications of the manufacturer (MTT kit I; Roche Applied Science, Penzberg, Germany). Briefly, the neurons were cultured in 96-well plates, 10 μL of 5 mg·mL−1 MTT labelling reagent was added to each well containing cells in 100 μL of medium, and the plates were incubated for 4 h in a humidified incubator at 37°C. After the incubation, 100 μL of the cytolytic solution (0.01 N HCl in 10% SDS solution) was added to each well for 16–20 h. Absorbance of the samples was measured at a wavelength of 570 nm with 655 nm as a reference wavelength. Unless otherwise indicated, the extent of MTT conversion in cells exposed to MPP+ is expressed as a percentage of the control.
Cytotoxicity was determined by measuring the release of LDH. Briefly, cells were precipitated by centrifugation at 500× g for 5 min at room temperature, 50 μL of the supernatants was transferred into new wells, and LDH was determined using the in vitro toxicology assay kit (Roche Applied Science). The absorbance of the samples was measured at a wavelength of 490 nm with 655 nm as a reference wavelength.
Fluorescein diacetate (FDA) / propidium iodide (PI) double staining assay
Viable granule neurons were stained with fluorescein formed from FDA, which is de-esterified only by living cells and PI can penetrate cell membranes of dead cells to intercalate into double-stranded nucleic acids. Briefly, after incubation with 10 μg·mL−1 FDA and 5 μg·mL−1 PI for 15 min, the neurons were examined and photographed using UV light microscopy; and the photographs compared with those taken with phase-contrast microscopy.
Hoechst staining assay
Chromatin condensation was detected by nuclear staining with Hoechst 33342, as described by Li et al., (2005). CGNs (2.7 × 106 cells) grown in a 35 mm dish were washed with ice-cold PBS and fixed with 4% formaldehyde in PBS. The cells were then stained with Hoechst 33342 (5 μg·mL−1) for 5 min at 4°C. The nuclei were visualized using a fluorescence microscope at ×400 magnification.
Measurement of levels of intracellular NO
Intracellular NO was monitored with 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM) diacetate, a pH-insensitive fluorescent dye that emits increased fluorescence after reaction with an active intermediate of NO formed during the spontaneous oxidation of NO to NO2 (Sheng et al., 2005). DAF-FM solution was added to the culture medium (final concentration: 5 μM). After 30 min in a CO2 incubator, cultures were washed twice with PBS and incubated for an additional 30 min to allow complete de-esterification of the intracellular diacetate for stronger fluorescence. The DAF-FM fluorescence in CGNs was quantified by a multi-detection microplate reader using excitation and emission wavelengths of 495 and 515 nm respectively. The measured fluorescence values were expressed as a percentage of the fluorescence in the control cells.
Maintenance of zebrafish and drug treatment
Wild-type zebrafish (AB strain) and Tg(fli-1 : EGFP) transgenic zebrafish were maintained as described in the Zebrafish Handbook (Westerfield, 1993). Zebrafish embryos were generated by natural pairwise mating (3–12 months old) and raised at 28.5°C in embryo medium (13.7 mM NaCl, 540 μM KCl, pH 7.4, 25 μM Na2HPO4, 44 μM KH2PO4, 300 μM CaCl2, 100 μM MgSO4, 420 μM NaHCO3, pH 7.4). Drugs were dissolved in DMSO and directly added into the zebrafish embryo medium to treat fish in 2–3 days (final concentration of DMSO was always less than 0.5% and showed no toxicity to zebrafish). An equal concentration of DMSO in embryo medium was used as vehicle control in each experiment.
Exposure to MPTP
Healthy zebrafish embryos were picked out and dechlorinated manually at 1 day post fertilization (dpf) and distributed into a 12-well plate with 20 fish embryos or a six-well microplate with 30 fish embryos in each well. In pilot experiments, several doses of MPTP were added to the embryo medium (final concentration from 50 to 800 μM), and treated 1 dpf fish embryo for 48 h, the optimal dose used (200 μM) induced significant decreases in brain diencephalic dopaminergic neurons and without any detectable systemic toxicities (data not shown). Thus, subsequent studies were done with 200 μM MPTP for whole-mount immunostaining and gene expression experiments.
As late as 3 dpf, zebrafish larvae showed very little spontaneous swimming, but by 5 dpf, they spontaneously swam longer distances and independently searched for food. Thus, the MPTP exposure needs to last 5 days from 1 dpf. In pilot locomotion behavioural tests, treatment for 3 days starting from 1 dpf with 200 μM MPTP in embryo medium killed all the fish larvae; however, after treatment for 2 days at 1 dpf with 200 μM MPTP and drug withdrawal for 3 days, the deficit behaviour was restored at 6 dpf. Finally, the optimal MPTP exposure was at 3 dpf, 2 days after treatment starting from 1 dpf with 200 μM MPTP. Zebrafish larvae were maintained in embryo medium containing 10 μM MPTP for another 3 days. The swimming distance decreased without any detectable systemic toxicities. Thus, subsequent locomotion behavioural studies were done with 200 μM MPTP for treatment for 2 days at 1 dpf then replacing with 10 μM MPTP for incubation for another 3 days.
Whole-mount immunostaining with antibody against tyrosine hydroxylase
Whole-mount immunostaining in zebrafish was performed as previously described by Zhang et al., (2011). Briefly, zebrafish were fixed in 4% paraformaldehyde in PBS for 5 h. Fixed samples were blocked (2% lamb serum and 0.1% BSA in PBST) for 1 h at room temperature. A mouse monoclonal anti-tyrosine hydroxylase (TH) antibody (Millipore, Billerica, MD, USA) was used as the primary antibody and incubated with samples overnight at 4°C. On the next day, samples were washed six times with PBST (each wash lasted 30 min), followed by incubation with secondary antibody according to the instruction provided by the Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). After staining, zebrafish were flat-mounted with 3.5% methylcellulose and photographed. Semi-quantification of the area of TH+ cells were assessed by an investigator, unaware of the drug treatment, using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, MD). Results are expressed as percentage of area of TH+ cells in untreated normal control groups.
Morphological observation of zebrafish
After drug treatment, zebrafish were removed from the microplate and observed for gross morphological changes of blood vessels under a fluorescence microscope (Olympus IX81 Motorized Inverted Microscope, Tokyo, Japan) equipped with a digital camera (DP controller, Soft Imaging System, Olympus). Images were analysed with Axiovision 4.2 (Carl Zeiss, Jena, Germany) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA, USA).
Locomotion behavioural test of zebrafish
After drug treatment, zebrafish larvae at 6 dpf were transferred into 96-well plates (1 fish per well and 12 larvae per group). Fishes that responded with excessive stress reaction (such as rapid and disorganized swimming or immobility for 2 min) due to the handling and monitoring of the behaviour were discarded. The experiments were performed in a calm, enclosed area. The larvae were allowed to habituate to the new environment for 30 min prior to experiments. Behaviour was monitored by an automated video tracking system (Viewpoint, ZebraLab, LifeSciences, Lyon, France). The 96-well plates and camera were housed inside a Zebrabox, and the swimming pattern of each fish was recorded for 10 min and repeated three times, once every other 10 min. The total distance moved was defined as the distance (in cm) that the fish had moved during one session (10 min).
Total RNA extraction, reverse transcription and real-time PCR
Total RNA was extracted from 30 zebrafish larvae of each treatment group using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RNA was reverse transcribed to single-strand cDNA using SuperScript™ III First-Strand Synthesis System for RT–PCR (Invitrogen™, Life Technologies), followed by real-time PCR using the TaqMan Universal PCR Master Mix and 2 μL TaqMan gene expression assay primers for the zebrafish th gene (assay ID:Dr03437803_g1, Life Technologies) in the ABI 7500 Real-Time PCR System (Life Technologies). The expression of th mRNA was normalized to the amount of bactin1 using the relative quantification method described by the manufacturer. The zebrafish bactin1 primers were 5′-CAACGGAAACGCTCATTGC-3′ (F) and 5′-CAAGATTCCATACCCAGGAAGGA-3′ (R) (Life Technologies).
NOS activity assays
For the in vitro NOS activity assay (Li et al., 2007), purified recombinant human nNOS, endothelial NOS (eNOS) and inducible NOS (iNOS) were bought from Alexis Biochemicals (Lausen, Switzerland). NOS activity was determined by monitoring the conversion of L-[3H]arginine to [3H]citrulline following the instructions provided in the kit (Merck, Darmstadt, Germany). The reaction mixture contained a final volume of 40 μL with 25 mM Tris–HCl at pH 7.4, 3 μM tetrahydrobiopterin, 1 μM FAD, 1 μM FMN, 1 mM NADPH, 0.6 mM CaCl2, 0.1 μM calmodulin, 2.5 μg of pure NOS enzyme, 5 μL L-[3H]arginine (Perkin Elmer, Waltham, MA) and different concentrations of the tested reagents. The reaction mixture was incubated at 22°C for 45 min. The reaction was quenched by adding 400 μL of stopping buffer (50 mM HEPES, pH 5.5, and 5 mM EDTA) for nNOS and eNOS reactions or by heating reaction tubes for iNOS. Unreacted L-[3H]arginine was then trapped by 100 μL of equilibrated resin provided in a spin cup followed by centrifugation for 30 s at 18 500× g. The [3H] in the filtrate was quantified by liquid scintillation counting.
Molecular docking
Molecular docking was performed using the ICM-Pro 3.6-1d program (Molsoft, San Diego, CA, USA) (Totrov and Abagyan, 1997). According to the ICM method, the molecular system was described using internal coordinates as variables. The biased probability Monte Carlo (BPMC) minimization procedure was used for global energy optimization. The BPMC global energy optimization method consists of the following steps: (1) a random conformation change of the free variables according to a predefined continuous probability distribution; (2) local energy minimization of analytical differentiable terms; (3) calculation of the complete energy including non-differentiable terms such us entropy and solvation energy; and (4) acceptance or rejection of the total energy based on the Metropolis criterion and return to step 1. A series of five grid potential representations of the receptor were automatically generated and superimposed that accounted for the hydrophobicity, carbon-based and hydrogen-based van der Waals boundaries, hydrogen bonding profile and electrostatic potential. The binding between SU4312 and NOS protein (PDB code for nNOS : 3NLV; PDB code for eNOS : 3NOS; and PDB code for iNOS : 1VAF) was evaluated by a binding score that reflected the quality of the complex. ICM docking was performed to find out the most favourable orientation. The resulting SU4312 and NOS protein complex trajectories were energy minimized, and the scores were computed.
Determination of SU4312 in the brain homogenate and plasma
To investigate whether SU4312 could reach the brain, an HPLC method was used to detect SU4312 in the brain homogenate and plasma of rats (Scott et al., 2004; Spitsin et al., 2008). Briefly, 15 min, 30 min and 1h after i.p. injection of SU4312, the animals were anaesthetised (40mg kg-1 sodium pentobarbital (Sigma-Aldrich) i.p.) and transcardially perfused with PBS containing heparin (1000 U·L−1) to remove any SU4312 in the cerebral vasculature. Then rats were decapitated, and brain tissue was homogenized (0.9% saline; 2 mL buffer per g brain tissue). SU4312 in the brain homogenate was extracted with 3 mL ethyl acetate. After evaporating the solvent, the residue was reconstituted in 50 μL methanol. SU4312 in the brain tissue was identified by an Agilent 1200 Series HPLC coupled with UV detector at a wavelength of 254 nm.
Plasma concentration of SU4312 was also assayed by an HPLC method. Briefly, SU4312 was injected i.p., and the right internal jugular vein cannulated, after anaesthesia (see above), to collect blood samples over 700 min after dosing. Blood was taken (0.25 mL per sample) and put into 1.5 mL heparinized microcentrifuge tube. After centrifugation at 1,500× g for 10 min, 0.1 mL plasma were transferred to a new 1.5 mL microcentrifuge tube. 0.1 mL of acetonitrile and 0.1 mL of methanol were added and mixed with vortex for 15 seconds. Then the mixtures were centrifuged at 12,000× g for 10 min. The supernatants were collected for HPLC analysis and 20 μL samples were injected into the HPLC system. The mobile phase consisted of a mixture of methanol and water (80:20, v/v) at a flow rate of 1 mL·min−1.
Data analysis and statistics
The data are expressed as the means ± SEM, and statistical significance was determined by one-way anova with Dunnett's test in the case of multiple comparisons with control or Tukey–Kramer means separation test for multiple comparisons among the treatment groups. Differences were accepted as significant at P < 0.05.
Materials
SU4312, MPP+ and MPTP were obtained from Sigma-Aldrich (St. Louis, MO, USA). PTK787/ZK222584 was purchased from LC Laboratories (Woburn, MA, USA). 7-nitroindazole (7-NI) and 1400W were from Merck.
Results
SU4312 but not PTK787/ZK222584 unexpectedly prevents MPP+-induced neuronal death in a concentration-dependent manner
At 8 days in vitro (DIV), CGNs were pre-treated with a range of concentrations of SU4312 (1, 3, 10, 20 or 30 μM) for 2 h and then treated with 35 μM MPP+ for 24 h. Cell viability was measured using the MTT and LDH assays. SU4312 prevented MPP+-induced cell death in a concentration-dependent manner (Figure 1A and B), without affecting cell proliferation or cell survival when given alone at the experimental concentrations (1–30 μM) for 26 h (data not shown). For comparison, PTK787/ZK222584 (vatalanib®), another specific VEGFR-2 inhibitor, was also tested in this model. PTK787/ZK222584 at 3–10 μM failed to block neuronal loss in vitro (Figure 1A and B). Moreover, SU4312 and PTK787/ZK222584 co-application did not affect neuroprotection by SU4312 against MPP+ in CGNs, suggesting that the neuroprotective effects of SU4312 might be independent of VEFGR-2 inhibition (Figure 1A and B).
Figure 1.

SU4312 prevents MPP+-induced apoptosis in a concentration-dependent manner. CGNs were treated with SU4312 and/or PTK787/ZK222584 at the indicated concentrations for 2 h and then exposed to 35 μM MPP+. Cell viability (A) and cytotoxicity (B) were measured at 24 h after MPP+ challenge by MTT and LDH assays respectively. (C) CGNs were co-treated with SU4312 or PTK787/ZK222584 and 35 μM MPP+. Cell viability was measured at 24 h after MPP+ challenge by MTT assay. (D) SU4312 blocks neuronal loss and reverses the morphological alterations induced by MPP+. CGNs were pre-incubated with or without 20 μM SU4312 and exposed to 35 μM MPP+ 2 h later. At 24 h after the MPP+ challenge, CGNs were assayed with FDA/PI double staining. (E) SU4312 blocks MPP+-induced neuronal apoptosis. CGNs were exposed to 35 μM MPP+ for 24 h with or without pre-treatment of 20 μM SU4312 for 2 h. The cultures were then assessed by Hoechst 33342 staining assay. The number of pyknotic nuclei with condensed chromatin was counted from representative photomicrographs and represented as a percentage of the total number of nuclei counted. Data, expressed as a percentage of the control, were the mean ± SEM of three separate experiments; *P < 0.05 and **P < 0.01 versus MPP+ group in panels A, B and C, or versus control in panel E; ##P < 0.01 versus MPP+ group in panel E (Tukey's test).
We also performed co-administration experiments and found that SU4312 (10–20 μM) still had neuroprotective effects against MPP+-induced neurotoxicity when it was given together with 35 μM MPP+ (Figure 1C). However, under the same conditions, PTK787/ZK222584 (10 μM) did not exert any neuroprotective effects (Figure 1C).
For analysis of apoptosis by microscopy, CGNs were pretreated with 20 μM SU4312 for 2 h and then exposed to 35 μM MPP+. Phase-contrast microscopy and FDA/PI double staining assays showed that SU4312 blocked MPP+-induced loss of neurons and reversed MPP+-induced morphological alterations, including abnormal cell bodies and extensive breaks in the neural network (Figure 1D). In addition, staining of pyknotic nuclei by Hoechst 33342 showed that SU4312 reversed nuclear condensation induced by MPP+ (Figure 1E).
To further investigate the neuroprotective effects of SU4312 in dopaminergic neurons, we have used SH-SY5Y cells and PC12 cells, two commonly used in vitro models of dopaminergic neurons. SH-SY5Y or PC12 cells were pre-treated with a range of concentrations of SU4312 for 2 h and then treated with 1 mM MPP+ for 24 h. Cell viability was measured using the MTT assay. SU4312 prevented MPP+-induced dopaminergic neuronal death at 3–10 μM in SH-SY5Y cells and at 10 μM in PC12 cells (Figure 2). PTK787/ZK222584 was also tested in the same models. PTK787/ZK222584 at 3 μM failed to block neuronal loss induced by MPP+ in both SH-SY5Y cells and PC12 cells (Figure 2). Treatment with 10 μM SU4312 or 3 μM PTK787/ZK222584 alone for 26 h did not show cell proliferative or cytotoxic effects (data not shown). However, SU4312 and PTK787/ZK222584 at higher concentrations were toxic to both SH-SY5Y cells and PC12 cells (data not shown).
Figure 2.
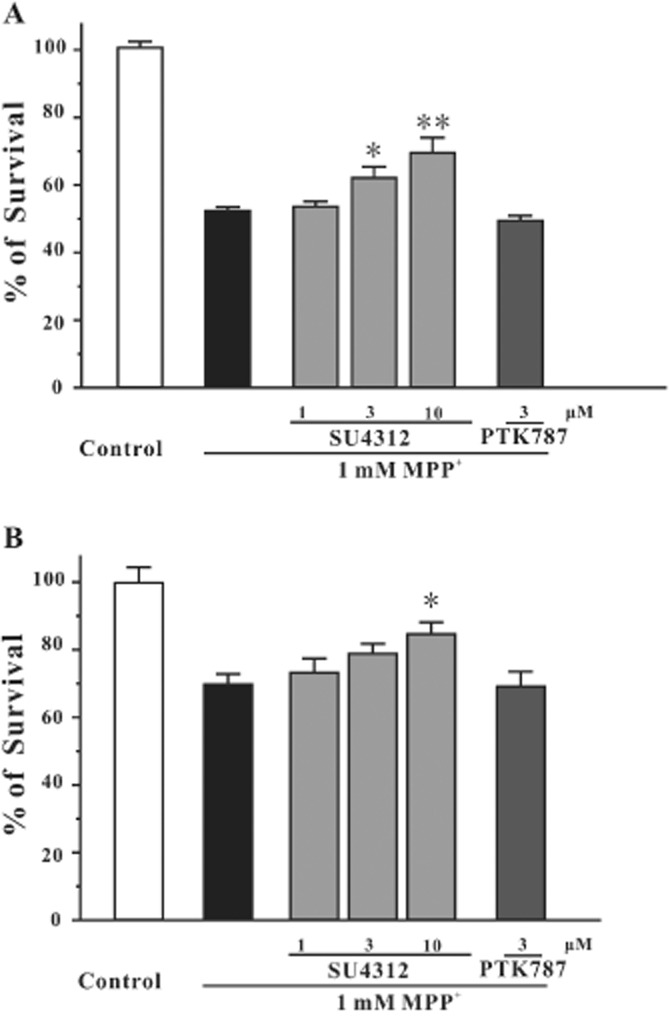
SU4312, but not PTK787/ZK222584, prevents MPP+-induced neurotoxicity in dopaminergic neurons. (A) SU4312, but not PTK787/ZK222584, prevents MPP+-induced neurotoxicity in SH-SY5Y cells. SH-SY5Y cells were treated with SU4312 or PTK787/ZK222584 at the indicated concentrations for 2 h and then exposed to 1 mM MPP+. Cell viability was measured by the MTT assay at 24 h after MPP+ challenge. (B) SU4312, but not PTK787/ZK222584, prevents MPP+-induced neurotoxicity in PC12 cells. PC12 cells were treated with SU4312 or PTK787/ZK222584 at the indicated concentrations for 2 h and then exposed to 1 mM MPP+. Cell viability was measured by the MTT assay at 24 h after MPP+ challenge. Data, expressed as percentage of control, were the mean ± SEM of three separate experiments; *P < 0.05 and **P < 0.01 versus MPP+ group (anova and Dunnett's test).
SU4312 but not PTK787/ZK222584 prevents MPTP-induced neurotoxicity in zebrafish
To test the neuroprotective effect of SU4312 and PTK787/ZK222584 in vivo, zebrafish embryos at 1 dpf were exposed to 200 μM MPTP for 2 days; the dopaminergic system in the brain of the zebrafish was determined by immunostaining of TH with a specific antibody and analysis of th gene expression by quantitative PCR. After MPTP treatment, ventral diencephalic TH populations, which are highly sensitive to MPTP exposure, were included in the analysis (Wen et al., 2008). The number of dopaminergic neurons in the diencephalons of zebrafish (indicated by red bracket) decreased dramatically (Figure 3A), and the level of th gene expression was down-regulated (Figure 3C). SU4312 alleviated the loss of dopaminergic neurons and decrease of th gene expression in a concentration-dependent manner. In contrast, PTK787/ZK222584 (0.3–3 μM) could not prevent MPTP-induced loss of dopaminergic neurons in the zebrafish (Figure 3).
Figure 3.
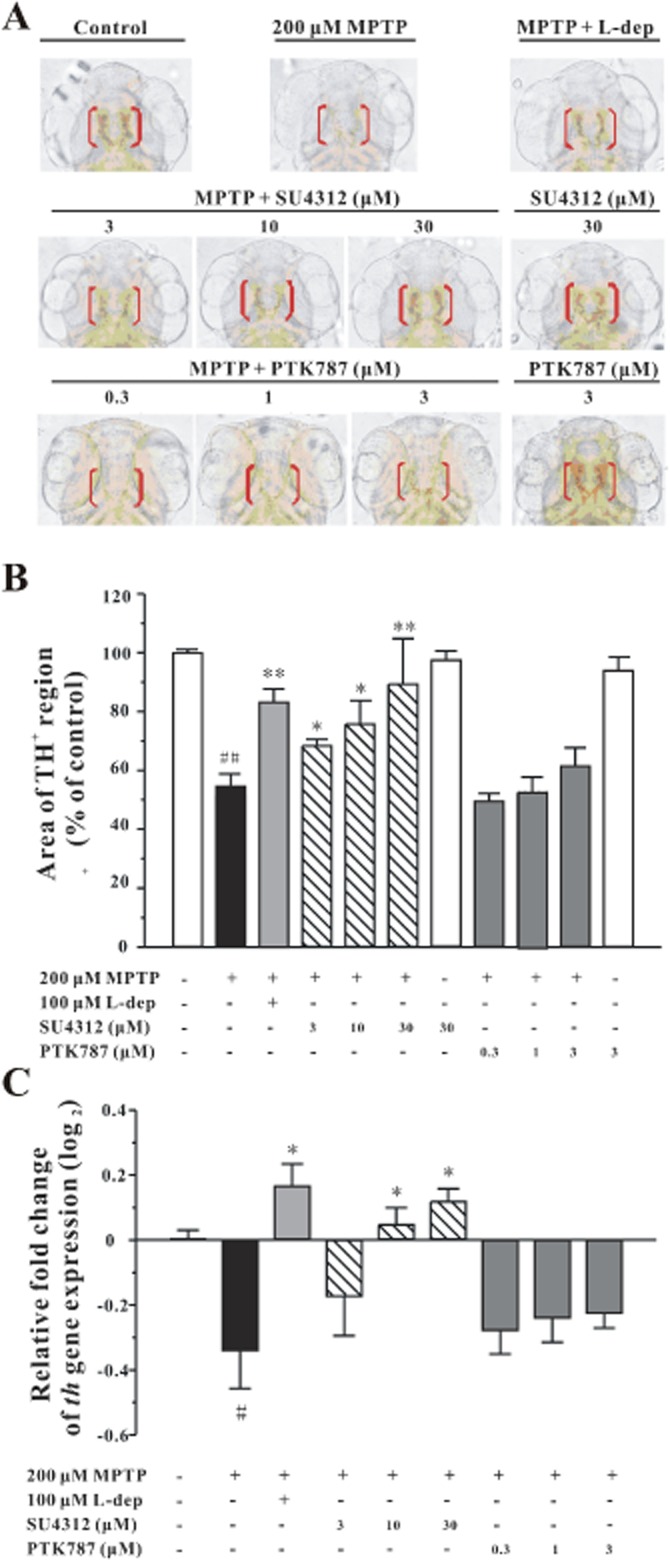
SU4312 protects against MPTP-induced neurotoxicity in zebrafish. Zebrafish embryos (1 dpf) were co-incubated with 200 μM MPTP and SU4312 or PTK787/ZK222584 at the indicated concentrations for 48 h, and zebrafish embryos that had been co-treated with MPTP and 100 μM L-deprenyl (L-dep, aMAO-B inhibitor) were used as the positive control. After treatment, zebrafish were collected to carry out immunohistochemistry, or total RNA extraction and real-time quantitative PCR. (A, B) SU4312, but not PTK787/ZK222584, prevents MPTP-induced TH+ neuronal loss in the brain of zebrafish in a concentration-dependent manner. (A) Representative pictures of dopaminergic neurons in the zebrafish brain from different treatment groups. Immunohistochemistry was performed with anti-TH primary antibody, and TH+ neurons in the diencephalic area of the zebrafish brain were considered as DA neurons. (B) Analysis of TH+ neurons in each treatment group, 20 fish embryos per group from three time-independent experiments. Values are expressed as a percentage of the control. (C) SU4312, but not PTK787/ZK222584, reverses th gene expression down-regulated by MPTP. Data were expressed as relative fold change of control (log2), *P < 0.05 and **P < 0.01 versus MPTP group; #P < 0.05 and ##P < 0.01 versus control (Tukey's test).
MPTP markedly altered the swimming behaviour of the zebrafish as a consequence of injury to dopaminergic neurons (McKinley et al., 2005). As shown in Figure 4A, the total distance travelled by the zebrafish larvae decreased significantly after exposure to MPTP. SU4312 but not PTK787/ZK222584 ameliorated MPTP-induced deficit of swimming behaviour. Under the same conditions, MPTP-induced deficit of swimming behaviour were also rescued by two positive control treatments, l-deprenyl ( an inhibitor of MAO-B) and l-DOPA (Figure 4A–C). Neither SU4312 nor PTK787/ZK222584 treatment alone notably altered the swimming behaviour of normal zebrafish larvae (Figure 4D).
Figure 4.
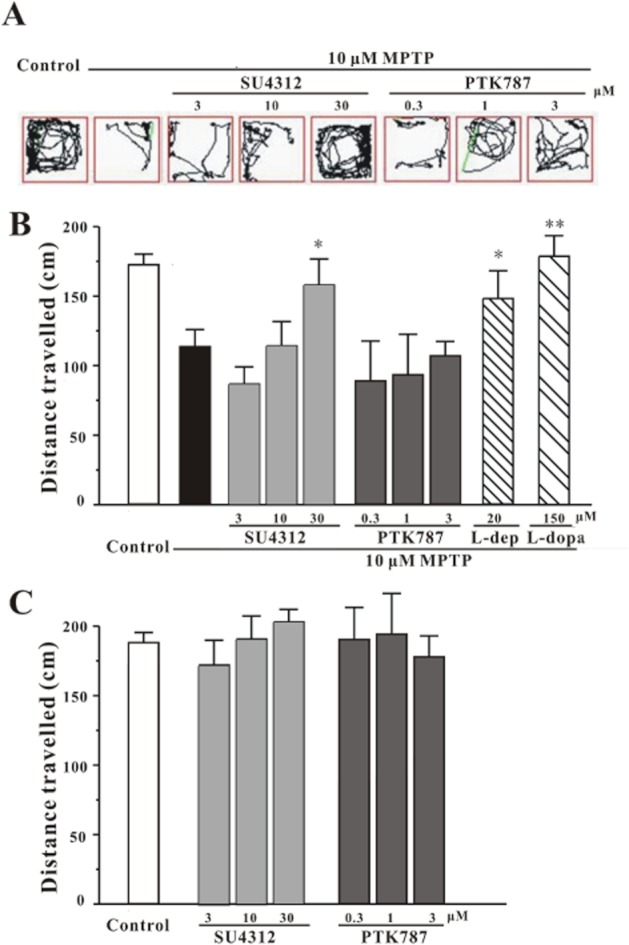
SU4312 attenuates the deficit of locomotion behaviour in zebrafish larvae induced by MPTP. (A–C) zebrafish embryos (1 dpf) were treated with 200 μM MPTP for 2 days, then co-incubated with 10 μM MPTP and SU4312 or PTK787/ZK222584 at the indicated concentrations for 3 days, and zebrafish larvae co-treated with MPTP and 20 μM L-deprenyl or 150 μM L-DOPA were used as the positive controls. After treatment, zebrafish were collected to carry out locomotion behaviour tests, using the Viewpoint Zebrabox system, and the total distance moved in 10 min was calculated. (A) Representative patterns of zebrafish locomotion traced from different treatment groups. (B) Statistical analysis of total distance travelled by each zebrafish larva in different treatment groups, 12 fish larvae per group from three independent experiments. (C) Zebrafish larvae (3 dpf) were treated with SU4312 or PTK787/ZK222584 but without MPTP at the indicated concentrations for 3 days, then the locomotion behaviour test was performed. The results represented the mean distance travelled by 36 larvae and are expressed in as cm travelled in 10 min. Values are mean ± SEM. *P < 0.05 and **P < 0.01 versus MPTP group (anova and Dunnett's test).
Neuroprotective effects of SU4312 are not directly correlated with its anti-angiogenic activity
We further determined if SU4312 and PTK787/ZK222584 within the particular concentration ranges exhibited any anti-angiogenic activities in Tg(fli1 : EGFP) transgenic zebrafish embryos. This transgenic model harbours EGFP gene under the control of the fli-1 promoter and thereby allows direct monitoring of endothelial cells under a fluorescence microscopy. Figure 5 shows the inhibitory effects of SU4312 and PTK787/ZK222584 at different concentrations on inter-segmental vessel (ISV) formation in zebrafish larvae. Compared with the vehicle control, SU4312 (3–30 μM) and PTK787/ZK222584 (0.3–3 μM) showed differential concentration-dependent inhibition of ISV formation in zebrafish larvae at 3 dpf (Figure 5). Accordingly, PTK787/ZK222584 exhibited more potent and effective anti-angiogenic activity than SU4312. These results suggested that SU4312 (3–30 μM) but not PTK787/ZK222584 (0.3–3 μM) could prevent MPTP-induced neuronal loss and locomotion deficit in zebrafish. However, the anti-angiogenic activities of these compounds were not correlated with their neuroprotective activities in vivo.
Figure 5.
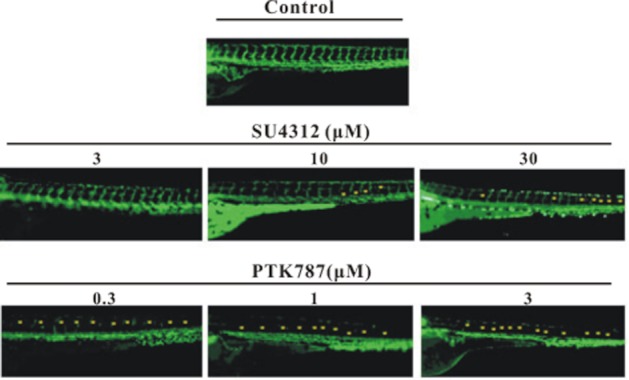
Comparison of anti-angiogenesis effects induced by SU4312 or PTK787/ZK222584 in zebrafish. Tg(Fli-1 : EGFP) transgenic zebrafish embryos (1 dpf) were treated with SU4312 or PTK787/ZK222584 at the indicated concentrations for 48 h. After treatment, inter-segmental vessels (ISV) of zebrafish were observed under fluorescent microscopy. The deficit of blood vessels was indicated by yellow asterisks.
SU4312 prevents MPP+-induced increase of intracellular NO
NO has been associated with MPP+-/MPTP-induced neurotoxicity (Hantraye et al., 1996; Przedborski et al., 1996). To clarify whether NO was involved in neuronal loss in CGNs caused by MPP+, DAF-FM diacetate was used to evaluate the intracellular NO level. MPP+ induced neuronal death and increased intracellular NO levels in a time-dependent manner (Figure 6A). For comparison, two NOS inhibitors were also selected to pre-treat neurons for 2 h before the addition of MPP+. A specific nNOS inhibitor, 7-NI, (3–10 μM), inhibited neuronal death and elevated level of intracellular NO induced by MPP+ (Figure 6B and C). In contrast, a specific inhibitor of iNOS, 1400W, failed to block neuronal death (Figure 6B).
Figure 6.

SU4312 reverses the elevated intracellular NO induced by MPP+ in CGNs. (A) MPP+ induces neuronal death and increases the level of intracellular NO in a time-dependent manner. CGNs were exposed to 35 μM MPP+ for different durations as indicated. Cell viability was measured by the MTT assay and intracellular NO level was measured using DAF-FM diacetate after the MPP+ challenge. (B) Selective nNOS inhibitor prevents MPP+-induced neurotoxicity. CGNs were treated with 7-NI or 1400W at the indicated concentrations for 2 h and then exposed to 35 μM MPP+. Cell viability was measured by the MTT assay at 24 h after the MPP+ challenge. (C) SU4312 reverses the elevated intracellular NO induced by MPP+. CGNs were pre-incubated with or without SU4312 or 7-NI at the indicated concentrations for 2 h and exposed to 35 μM MPP+. Intracellular NO level was measured using DAF-FM diacetate as a probe at 24 h after the MPP+ challenge. Data, expressed as percentage of control, were the mean ± SEM of three separate experiments; *P < 0.05 and **P < 0.01 versus MPP+ group in panels B and C (anova and Dunnett's test).
CGNs were pre-treated with SU4312 (3–30 μM) for 2 h and then exposed to MPP+ for another 24 h. This procedure attenuated the MPP+-triggered elevation of intracellular NO levels, indicating that SU4312 prevented MPP+-induced neuronal loss possibly through inhibiting NO overproduction (Figure 6C).
SU4312 selectively inhibits nNOS in a non-competitive manner
To investigate whether SU4312 directly inhibited NOS, activity of NOS was assayed in vitro. SU4312 directly and selectively inhibited recombinant human nNOS relative to iNOS, but there was no activity against eNOS. In contrast, PTK787/ZK222584 did not inhibit any of the NOS isoforms even at 1 mM (Table 1).
Table 1.
Selective inhibition of nNOS by SU4312 in vitro
| Drug | IC50 (μM) | ||
|---|---|---|---|
| nNOS | iNOS | eNOS | |
| SU4312 | 19.0 ± 1.0 | 389.9 ± 9.9 | N.D. |
| 7-NI | 1.5 ± 0.2 | 42.5 ± 1.0 | 3.5 ± 0.3 |
| PTK787 | ND | ND | ND |
The activity of recombinant human nNOS, iNOS and eNOS and their inhibition by chemicals were measured by the NOS assay kit. The IC50 value of each compound to inhibit nNOS, iNOS and eNOS was obtained from seven-point titration using Sigma Plot 9.0, where each individual point was an average of duplicate determinations at the same concentration from three independent experiments.
ND (not detectable) indicates that the corresponding compound did not inhibit the activity of NOS at 1 mM.
To investigate the mode of nNOS inhibition by SU4312, two concentrations (10 or 20 μM) of SU4312 were added to the nNOS reaction system containing a range of concentrations (5–40 μM) of l-[3H]arginine. Lineweaver–Burk plots in Figure 7A show that SU4312 inhibited nNOS in a non-competitive manner, and the Ki value of nNOS inhibition by SU4312 was 12.7 μM (Figure 7B).
Figure 7.
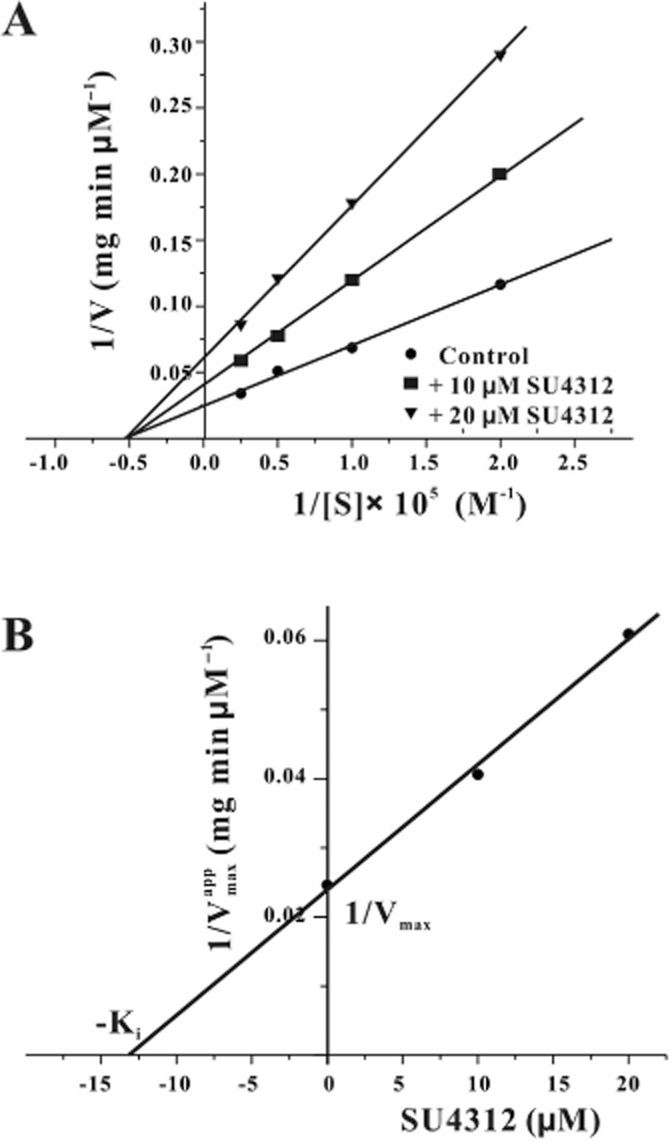
SU4312 inhibits nNOS in a non-competitive manner. (A) Kinetic analysis of nNOS inhibition with l-arginine by SU4312. Recombinant nNOS (2.5 μg) was assayed in either the presence (10 or 20 μM) or absence of SU4312 over a range of concentrations of l-[3H]arginine (5 to 40 μM). The plots of 1/V versus 1/[S] were fitted by a Lineweaver–Burk straight line with an intercept of 1/Vmax and a slope of Km/Vmax. The data were expressed as the means of three independent experiments. (B) The Ki value of SU4312 in the inhibition of nNOS. A graph of the apparent 1/Vmax from (A) versus concentration of SU4312 yielded a straight line.
Molecular docking simulation reveals the interaction between SU4312 and nNOS
To gain further insight into the mechanisms of interaction between SU4312 and nNOS, computational docking was performed. Both the cis- and trans-isomers of SU4312 (Figure 8C and D) showed favourable interaction with the haem group within the nNOS protein (PDB code: 3NLV), with a binding score of −31.25 and −38.98 respectively. As a reference, a binding score of −36.44 was obtained for a compound known to bind to nNOS 6-(-4-[2-({2,2-difluoro-2-[(2R)-piperidin-2-yl]ethyl}amino)ethoxy)-4-methylpyridin-2-amine (Xue et al., 2010). In both cis- and trans- forms, the N(CH3)2 group of SU4312 might interact with NH2+ of Pro565, and the NH of SU4312 might interact with the COO– of the haem group. The N,N-dimethyl aniline group is located in the binding pocket of nNOS, which is formed by the haem, Pro565 and Arg596 (Figure 8C and D). On the other hand, neither of the two SU4312 forms showed relatively favourable interactions with iNOS protein as reflected by their binding scores of −11.47 and −12.66, respectively; while no ‘druggable’ binding pocket could be found near the haem for eNOS.
Figure 8.
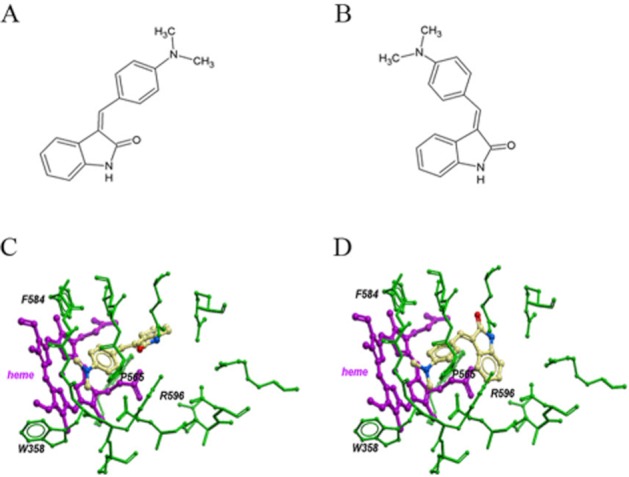
Molecular docking simulation of interactions between SU4312 and nNOS. The structures of cis- and trans-SU4312 were shown in panels A and B respectively. Molecular dockings show the binding of cis- and trans-SU4312 with nNOS (PDB code: 3NLV) in panels C and D respectively.
SU4312 can enter the brain
To investigate whether SU4312 could reach the brain, an HPLC method was used to detect SU4312 in brain tissue after i.p. injection. The plasma concentration–time profile of SU4312 after i.p. administration is shown in Figure 9A. Most importantly, SU4312 rapidly entered the brain and was detected in brain tissue 15 min after injection (Figure 9D). However, the brain concentration of SU4312 also rapidly decreased as almost no SU4312 was detected in the brain at 60 min after i.p. administration (Figure 9F).
Figure 9.

SU412 can reach the brain after i.p. administration. (A) Plasma concentration–time profile of SU4312 in rats after i.p. administration. After i.p.injection of 12 mg·kg−1 SU4312, serial blood samples were collected and analysed. Data are the mean ± SD (n = 3). (B–F) HPLC Chromatograms of (B) standard SU4312 solution (150 μg·mL−1); (C) brain homogenate extract from control rat; (D–F) brain homogenate extract from rat at 15 min (D), 30 min (E) and 1 h (F) after i.p. administration of SU4312 (12 mg·kg−1). Peak 1: cis-SU4312; Peak 2: trans- SU4312.
Discussion
SU4312 was originally designed as an anti-cancer drug candidate, targeting VEGFR-2. Under light illumination, SU4312 interchanged freely between the cis- and trans- forms in solution. These forms of SU4312 selectively inhibit VEGFR-2 with IC50 values of 0.8 (cis- form) and 5.2 (trans- form) μM respectively (Sun et al., 1998). Our results showed that SU4312, even at a concentration as high as 30 μM, did not induce any neurotoxicity in primary neuron cultures or in zebrafish. These results, together with those of a previous study that showed that prolonged SU4312 treatment (3 mg of SU4312 every 5 days for 12 weeks) did not damage retinal photoreceptors or ganglion cells in rodents (Miki et al., 2010), indicate the lack of toxicity of SU4312 towards neurons.
We have previously demonstrated neuroprotective effects of VEGF (10–300 ng·mL−1) against MPP+-induced neuronal death in CGNs, the same in vitro model that we used in the current study, by activating the VEGFR-2/Akt signalling pathway (Cui et al., 2011). Our unpublished results have also shown that VEGF did not affect production of NO at the concentrations exerting neuroprotective activity. If SU4312 acted only as a VEGFR-2 blocker to inhibit the VEGFR-2/Akt pathway, it would promote, not decrease, MPP+-induced neurotoxicity. Therefore, the neuroprotection of SU4312 must be independent of its anti-VEGFR-2 action. To further rule out the possibility that SU4312 protected neurotoxicity via inhibiting angiogenesis, PTK787/ZK222584 was applied to the same models. PTK787/ZK222584 inhibits VEGFR-2 with an IC50 value of 37 nM in vitro, which is about 20 times lower than that of cis-SU4312 (Sun et al., 1998). As expected, PTK787/ZK222584 at 1 μM inhibited angiogenesis as effectively as SU4312 at 30 μM, in zebrafish (Figure 5). Interestingly, PTK787/ZK222584 at the same concentration failed to inhibit MPP+-induced neurotoxicity in CGNs, SH-SY5Y cells, PC12 cells or MPTP-induced neurotoxicity in zebrafish. These results suggested that the neuroprotective effects of SU4312 were not closely correlated with its anti-angiogenic property.
How does SU4312 inhibit MPP+/MPTP-induced neurotoxicity? It is known that NO mediates MPP+- and MPTP-induced neurotoxicity both in vitro and in vivo (Przedborski et al., 1996; Gonzalez-Polo et al., 2004a). As shown in Figure 6, SU4312 inhibited the MPP+-induced increase of intracellular NO level, indicating that SU4312 may affect the formation or degradation of endogenous NO. Endogenous NO is produced only by NOS converting l-arginine to l-citrulline (Fedorov et al., 2004). Three isotypes of NOS, namely, nNOS, iNOS and eNOS, have been identified (Alderton et al., 2001),with nNOS is the predominant form in neurons in the CNS, The eNOS isoform is mainly present in cerebral vascular endothelial cells, whereas iNOS is expressed in astrocytes and microglia (Estevez et al., 1998). It is noteworthy that ablation of eNOS had no effects on MPP+-induced neurotoxicity (Gonzalez-Polo et al., 2004a). According to our results, MPP+-induced neuronal death was inhibited by the selective nNOS inhibitor 7-NI, but not by the selective iNOS inhibitor 1400W, and the increase of intracellular NO was also similarly reduced. Consequently, SU4312 may prevent MPP+-induced neurotoxicity by inhibiting nNOS. By assaying NOS activity in vitro, we found that SU4312 directly inhibited the activity of purified NOS, showing high selectivity toward nNOS. Furthermore, SU4312 did not alter Km but decreased the apparent Vmax, with a linear relationship between the apparent Vmax and SU4312 concentrations. All these results suggest that SU4312 prevented neurotoxicity at least partially by directly inhibiting nNOS in a non-competitive manner.
Docking simulation revealed a possible molecular interaction between isomers of SU4312 and nNOS. In cis-SU4312-nNOS and trans-SU4312-nNOS complexes, the NH group of SU4312 has close contact with the haem domain of nNOS. The haem domain is required for nNOS dimerization, a process necessary to convert inactive nNOS monomer into active dimers (Roman and Masters, 2006). The haem domain is also the final electron acceptor in the electron flow, which is required for NO production (Zhou and Zhu, 2009). The interaction between SU4312 and the haem domain of nNOS may disrupt nNOS dimerization and/or impair the electron transfer process, and consequently induce a non-competitive inhibition event. Furthermore, the binding pocket involving the haem, Pro565 and Arg596 of nNOS may facilitate the interaction between SU4312 and nNOS and support a prolonged inhibition.
Besides nNOS inhibition, other molecular mechanisms, such as inhibition of MAO-B inhibition and antagonism of kinases, may also contribute to the neuroprotective effects of SU4312. For example, some NOS inhibitors were found also to be MAO-B inhibitors that prevented MPTP neurotoxicity (Herraiz et al., 2009). Moreover, recent studies have shown that chemicals with the indolineone structure can inhibit LRRK2, a kinase associated with an increased risk of Parkinson's disease, implying that the neuroprotective effects of SU4312 might be due to LRRK-2 inhibition (Lee et al., 2010). To rule out the possibility that SU4312 protected against neurotoxicity via directly inhibiting MAO-B, we have assessed inhibition of MAO-B by SU4312, using the MAO-Glo™ assay kit (Promega Inc., Fitchburg, WI, USA). Our results have shown that SU4312 did not affect the activity of MAO-B (data not shown), suggesting the neuroprotective effects of SU4312 are not due to a direct inhibition of MAO-B. Other targets that SU4312 may act on for its neuroprotective effects, further experiments are currently being investigated in our laboratory.
In conclusion, our findings demonstrated that SU4312 exhibited neuroprotection against MPP+ at least partly via a selective and direct inhibition of nNOS. Although further studies are needed to confirm the neuroprotective effects of SU4312 in other neurodegenerative models or in patients, in view of the ability of SU4312 to reach the brain in rats, our results offer support for further development of SU4312 in the treatment of neurodegenerative disorders, particularly those associated with NO-mediated neurotoxicity.
Acknowledgments
This work was supported by grants from the Research Grants Council of Hong Kong (PolyU5609/09M and 5610/11M), The Hong Kong Polytechnic University (G-U952), the Science and Technology Development Fund (FDCT) of Macao SAR (078/2011/A3 and 045/2007/A3), the Research Committee of the University of Macau (MYRG139(Y1-L4)-ICMS12-LMY and UL017/09-Y1), and the National Natural Science Foundation of China (NSFC) (81202510). We sincerely thank Dr Peng Cho Tang (HEC Pharma, Shenzhen, China) and Dr Hua Yu (School of Chinese Medicine, The Hong Kong Baptist University, Hong Kong, China) for kindly providing suggestions during this study and Ms Josephine Leung for proofreading our manuscript.
Glossary
- CGNs
cerebellar granule neurons
- DAF-FM
4-amino-5-methylamino-2′,7′-difluorofluorescein
- DIV
days in vitro
- dpf
days post fertilization
- eNOS
endothelial NOS
- FDA
fluorescein diacetate
- iNOS
inducible NOS
- ISV
inter-segmental vessel
- MPP+
1-methyl-4-phenylpyridinium ion
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTT
3(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide
- nNOS
neuronal NOS
- PI
propidium iodide
- TH
tyrosine hydroxylase
- VEGFR-2
VEGF receptor-2, 7-NI, 7- nitroindazole
Conflict of interest
None.
References
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357(Pt 3):593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Li W, Han R, Mak S, Zhang H, Hu S, et al. PI3-K/Akt and ERK pathways activated by VEGF play opposite roles in MPP+-induced neuronal apoptosis. Neurochem Int. 2011;59:945–953. doi: 10.1016/j.neuint.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Estevez AG, Spear N, Thompson JA, Cornwell TL, Radi R, Barbeito L, et al. Nitric oxide-dependent production of cGMP supports the survival of rat embryonic motor neurons cultured with brain-derived neurotrophic factor. J Neurosci. 1998;18:3708–3714. doi: 10.1523/JNEUROSCI.18-10-03708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov R, Vasan R, Ghosh DK, Schlichting I. Structures of nitric oxide synthase isoforms complexed with the inhibitor AR-R17477 suggest a rational basis for specificity and inhibitor design. Proc Natl Acad Sci U S A. 2004;101:5892–5897. doi: 10.1073/pnas.0306588101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Soler G, Alvarez A, Fabregat I, Fuentes JM. Vitamin E blocks early events induced by 1-methyl-4-phenylpyridinium (MPP+) in cerebellar granule cells. J Neurochem. 2003;84:305–315. doi: 10.1046/j.1471-4159.2003.01520.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Soler G, Fuentes JM. MPP+: mechanism for its toxicity in cerebellar granule cells. Mol Neurobiol. 2004a;30:253–264. doi: 10.1385/MN:30:3:253. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Soler G, Rodriguezmartin A, Moran JM, Fuentes JM. Protection against MPP+ neurotoxicity in cerebellar granule cells by antioxidants. Cell Biol Int. 2004b;28:373–380. doi: 10.1016/j.cellbi.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, et al. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- Herraiz T, Aran VJ, Guillen H. Nitroindazole compounds inhibit the oxidative activation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxin to neurotoxic pyridinium cations by human monoamine oxidase (MAO) Free Radic Res. 2009;43:975–984. doi: 10.1080/10715760903159170. [DOI] [PubMed] [Google Scholar]
- Langston JW, Irwin I. MPTP: current concepts and controversies. Clin Neuropharmacol. 1986;9:485–507. [PubMed] [Google Scholar]
- Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, et al. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson's disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Pi R, Chan HH, Fu H, Lee NT, Tsang HW, et al. Novel dimeric acetylcholinesterase inhibitor bis7-tacrine, but not donepezil, prevents glutamate-induced neuronal apoptosis by blocking N-methyl-D-aspartate receptors. J Biol Chem. 2005;280:18179–18188. doi: 10.1074/jbc.M411085200. [DOI] [PubMed] [Google Scholar]
- Li W, Xue J, Niu C, Fu H, Lam CS, Luo J, et al. Synergistic neuroprotection by bis(7)-tacrine via concurrent blockade of N-methyl-D-aspartate receptors and neuronal nitric-oxide synthase. Mol Pharmacol. 2007;71:1258–1267. doi: 10.1124/mol.106.029108. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinley ET, Baranowski TC, Blavo DO, Cato C, Doan TN, Rubinstein AL. Neuroprotection of MPTP-induced toxicity in zebrafish dopaminergic neurons. Brain Res Mol Brain Res. 2005;141:128–137. doi: 10.1016/j.molbrainres.2005.08.014. [DOI] [PubMed] [Google Scholar]
- McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med. 2010;16:483–489. doi: 10.1038/nm.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki A, Miki K, Ueno S, Wersinger DM, Berlinicke C, Shaw GC, et al. Prolonged blockade of VEGF receptors does not damage retinal photoreceptors or ganglion cells. J Cell Physiol. 2010;224:262–272. doi: 10.1002/jcp.22129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci U S A. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman LJ, Masters BS. Electron transfer by neuronal nitric-oxide synthase is regulated by concerted interaction of calmodulin and two intrinsic regulatory elements. J Biol Chem. 2006;281:23111–23118. doi: 10.1074/jbc.M603671200. [DOI] [PubMed] [Google Scholar]
- Schultheiss C, Blechert B, Gaertner FC, Drecoll E, Mueller J, Weber GF, et al. In vivo characterization of endothelial cell activation in a transgenic mouse model of Alzheimer's disease. Angiogenesis. 2006;9:59–65. doi: 10.1007/s10456-006-9030-4. [DOI] [PubMed] [Google Scholar]
- Scott GS, Kean RB, Mikheeva T, Fabis MJ, Mabley JG, Szabo C, et al. The therapeutic effects of PJ34 [N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-N,N-dimethylacetamide.HCl], a selective inhibitor of poly(ADP-ribose) polymerase, in experimental allergic encephalomyelitis are associated with immunomodulation. J Pharmacol Exp Ther. 2004;310:1053–1061. doi: 10.1124/jpet.103.063214. [DOI] [PubMed] [Google Scholar]
- Sheng JZ, Wang D, Braun AP. DAF-FM (4-amino-5-methylamino-2′,7′-difluorofluorescein) diacetate detects impairment of agonist-stimulated nitric oxide synthesis by elevated glucose in human vascular endothelial cells: reversal by vitamin C and L-sepiapterin. J Pharmacol Exp Ther. 2005;315:931–940. doi: 10.1124/jpet.105.087932. [DOI] [PubMed] [Google Scholar]
- Spitsin S, Portocarrero C, Phares TW, Kean RB, Brimer CM, Koprowski H, et al. Early blood-brain barrier permeability in cerebella of PLSJL mice immunized with myelin basic protein. J Neuroimmunol. 2008;196:8–15. doi: 10.1016/j.jneuroim.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Tran N, Tang F, App H, Hirth P, McMahon G, et al. Synthesis and biological evaluations of 3-substituted indolin-2-ones: a novel class of tyrosine kinase inhibitors that exhibit selectivity toward particular receptor tyrosine kinases. J Med Chem. 1998;41:2588–2603. doi: 10.1021/jm980123i. [DOI] [PubMed] [Google Scholar]
- Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem. 1993;61:1191–1206. doi: 10.1111/j.1471-4159.1993.tb13610.x. [DOI] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Flexible protein-ligand docking by global energy optimization in internal coordinates. Proteins. 1997;(Suppl 1):215–220. doi: 10.1002/(sici)1097-0134(1997)1+<215::aid-prot29>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- Tran TC, Sneed B, Haider J, Blavo D, White A, Aiyejorun T, et al. Automated, quantitative screening assay for antiangiogenic compounds using transgenic zebrafish. Cancer Res. 2007;67:11386–11392. doi: 10.1158/0008-5472.CAN-07-3126. [DOI] [PubMed] [Google Scholar]
- Wen L, Wei W, Gu W, Huang P, Ren X, Zhang Z, et al. Visualization of monoaminergic neurons and neurotoxicity of MPTP in live transgenic zebrafish. Dev Biol. 2008;314:84–92. doi: 10.1016/j.ydbio.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Westerfield M. Eugene, OR: University of Oregon Press; 1993. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Brachydanio Rerio). edn. [Google Scholar]
- Xue F, Li H, Delker SL, Fang J, Martasek P, Roman LJ, et al. Potent, highly selective, and orally bioavailable gem-difluorinated monocationic inhibitors of neuronal nitric oxide synthase. J Am Chem Soc. 2010;132:14229–14238. doi: 10.1021/ja106175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Cheang LC, Wang MW, Lee SM. Quercetin exerts a neuroprotective effect through inhibition of the iNOS/NO system and pro-inflammation gene expression in PC12 cells and in zebrafish. Int J Mol Med. 2011;27:195–203. doi: 10.3892/ijmm.2010.571. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]