

Abstract

An efficient one-pot method for the synthesis of 2,3-disubstituted benzo[b]furans from commercially available 2-iodophenols, terminal acetylenes and aryl iodides has been developed utilizing Sonogashira reaction conditions. After an initial Sonogashira coupling of the 2-iodophenol with the terminal alkyne, cyclization involving the aryl iodide provides the 2,3-disubstituted benzo[b]furan in good to excellent yields. The use of microwave irradiation shortens the reaction times and minimizes the side products. This methodology is especially useful for the construction of libraries of highly substituted benzo[b]furans and their analogues.
Keywords: benzofuran, Sonogashira coupling, three-component reaction, microwave
1. Introduction
Benzo[b]furans have been studied extensively due to the high potential biological and pharmaceutical activity of this ring system.1 Thus, numerous synthetic methods to access this important scaffold have been developed.2 Among others, Pd-catalyzed reactions have proven to be highly efficient and convenient for the synthesis and functionalization of benzo[b]furans. Several review articles and books have been published that summarize previous and recent developments in this area.3 In 1996, Cacchi and co-workers reported that 2-(1-alkynyl)phenols in the presence of vinylic triflates and a palladium catalyst undergo cyclization to the corresponding 2,3-disubstituted benzo[b]furans.4 The authors proposed that this process most likely proceeds through a vinylic palladium intermediate, generated in situ from the vinylic triflate via oxidative addition to Pd(0). To access the starting 2-(1-alkynyl)phenols, a 3-step route has been employed in most cases. First, the OH group is protected with an appropriate protecting group (e.g. acetyl), then the Sonogashira coupling is conducted, and, following deprotection of the phenol, the desired 2-(1-alkynyl)phenol is obtained, generally in moderate yields. The major problem with the direct Sonogashira reaction between a 2-iodophenol and a terminal alkyne is that the coupling is often inefficient. In addition, if basic reaction conditions are employed, 3H-benzofurans are often formed as products, instead of the desired 2-(1-alkynyl)phenol.
In order to transform Cacchi's process into a three-component process, Flynn and coworkers employed MeMgCl as an additive to mask the phenol oxygen of the iodophenol (1). They were then able to conduct an efficient Sonogashira coupling and subsequent cyclization to a benzofuran (3) without isolating the corresponding 2-(1-alkynyl)phenol (2) (Scheme 1).5 A number of highly substituted benzofurans 3 have been obtained in moderate to good yields using this approach. The authors applied their methodology to the synthesis of (±)-frondosin B and its analogues, and benzo[b]furan-containing inhibitors of tubulin polymerization.6 However, this method has not been found suitable for the construction of libraries of 2,3-substituted benzofurans, due to the highly reactive nature of the MeMgCl reagent and its incompatibility with a large number of functional groups.
Scheme 1.
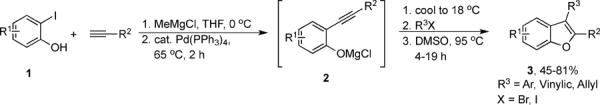
A one-pot approach to benzofurans using MeMgCl
Thus in 2004, Hu, Fathi, and Yang, in order to access a 210 member library of 2,3-disubstituted benzofurans, optimized the method developed by Cacchi starting from 2-(1-alkynyl)phenols, and, after their optimization studies, found more efficient conditions for the formation of 2,3-diarylsubstituted benzo[b]furans from 2-(1-alkynyl)phenols and aryl iodides. 7 However, considering the 3-step route required for synthesis of the 2-(1-alkynyl)phenols, the average yields of the final benzofurans (over 4 steps) were only moderate.
During the course of our own investigations into methodology for the synthesis of 2,3-disubstituted indoles under Sonogashira conditions,8 we have found that 2-iodophenols can also participate in an analogous process, providing an efficient and convenient new route to 2,3-disubstituted benzofurans. This finding encouraged us to proceed with our own optimization studies of this process.
2. Results and Discussion
2.1. Optimization
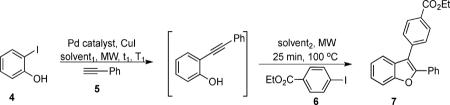
2-Iodophenol (4), phenyl acetylene (5), and ethyl 4-iodobenzoate (6) have been employed as starting materials under the reaction conditions we developed for the synthesis of indoles,8 providing the benzofuran 7 in a 51% yield (Table 1, entry 1). In order to improve the yield, optimization of the reaction conditions has been carried out.
Table 1.
Optimization of the reaction conditionsa
| entry | time (min) | temp.(°C) | solvent | ratio 4:5:6 | catalyst | yield 7b (%) | |
|---|---|---|---|---|---|---|---|
| step 1 | step 1 | step 1 | step 2 | ||||
| 1 | 15 | 60 | Et3N | MeCN | 1:1.05:1.1 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 51 |
| 2 | 15 | 60 | Et3N | MeCN | 1:1.05:1.1 | 2 mol % Pd(PPh3)4 1 mol % CuI | 23 |
| 3 | 15 | 60 | Et3N | MeCN | 1:1.05:1.1 | 2 mol % Pd(dppe)2 1 mol % CuI | 10 |
| 4 | 15 | 60 | Et3N | MeCN | 1:1.05:1.1 | 2 mol % Pd(OAc)2 4 mol % PPh3 1 mol % CuI | 6 |
| 5 | 15 | 60 | Et3N | MeCN | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 53 |
| 6 | 15 | 60 | iPr2NH | MeCN | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 50 |
| 7 | 15 | 60 | Et3N | DMF | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 34 |
| 8 | 15 | 60 | Et3N | THF | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 29 |
| 9 | 15 | 60 | Et3N | Toluene | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 15 |
| 10 | 25 | 80 | Et3N | MeCN | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 20 |
| 11c | 15 | 25 | Et3N | MeCN | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 53 |
| 12 | 30 | 25 | Et3N | MeCN | 1:1.05:1.0 | 2 mol % PdCl2(PPh3)2 1 mol % CuI | 73 |
| 13 | 30 | 25 | Et3N | MeCN | 1:1.05:1.0 | 3 mol % PdCl2(PPh3)2 2 mol % CuI | 86 |
| 14d | 30 | 25 | NMM/Et3N | MeCN | 1:1.2:1.0 | 3 mol % PdCl2(PPh3)2 2 mol % CuI | 89 |
| 15e | 30 | 25 | THF/Et3N | MeCN | 1:1.2:1.0 | 3 mol % PdCl2(PPh3)2 2 mol % CuI | 96 |
Unless otherwise noted, all of the reactions were carried out under microwave irradiation on a 1.0 mmol scale in microwave-resistant vials.
Isolated yields after column chromatography.
When the first step of the reaction was carried out at 25 °C, much cleaner reaction mixtures were obtained than at 60 °C.
0.5 mL of N-methylmorpholine (NMM)/1.5 mL of Et3N using anhydrous solvents under argon.
0.5 mL of THF/1.0 mL of Et3N, and CuI were added as a solution in 0.5 mL of Et3N using anhydrous solvents under argon.
Our initial examination of a number of palladium catalysts (Table 1, entries 1–4) indicated that bis(triphenylphosphine)palladium dichloride provided the best results. Changing the base from triethylamine to diisopropylamine (entries 5 vs 6) did not improve the yield of the desired benzofuran 7. A study of the effect of various solvents on the second cyclization step revealed that DMF afforded product 7 in a 34% yield (entry 7), whereas THF and toluene were less efficient, providing only 29% and 15% yields, respectively (entries 8 and 9). Thus, acetonitrile appeared to be the best solvent for this transformation. An increase in the temperature of the first step of the reaction to 80 °C decreased the yield of the desired product to 20% (entry 10), whereas conducting the first step at room temperature did not affect the yield of the product and the reaction was found to be cleaner based on TLC analysis when compared to the same reaction at 60 °C. Increasing the reaction time of the first step at room temperature to 30 minutes improved the yield of benzofuran 7 to 73% (entry 12). Slightly increasing the catalyst loading and finding the best Pd/Cu catalyst ratio (3 mol % and 2 mol % respectively) improved the yield of benzofuran 7 to 86% (entry 13).
After the initial evaluation of the scope of this process had been performed, the solubility of many iodophenols in triethylamine was found to be insufficient. The first step of the process was inefficient, leading to the exclusive formation of coupling products of the aryl iodides with the terminal alkyne and affording only low yields of the cyclized benzofurans. Thus, additional evaluation of the solvents for the first step was performed.
In this methodology, the choice of solvents plays a crucial role. For the first step, the ideal solvent needs to be suitable for an efficient Sonogashira reaction, but should not promote cyclization of the intermediate 2-(1-alkynyl)phenol, which will result in formation of an undesired 3H-benzofuran. The second step, on the other hand, requires a solvent, which will promote the cyclization. Based on our observations on the effect of different solvents on the cyclization step (Table 1; entries 5, 7–9), it can be concluded that a more polar solvent generally affords a more efficient cyclization process. The increased polarity of the solvent most likely produces less ion pairing between the phenolate and the Et3NH+ cation, which facilitates a more favorable attack of the phenolate on the alkyne triple bond. We, therefore, tried to add additional reagents to the triethylamine to improve the solubility of the iodophenols, but still retain reaction conditions favorable for the Sonogashira coupling. Solvents, like acetonitrile and DMF, solubilized the iodophenols well, but were not suitable for the first step, since they promote the cyclization subsequent. The best additives were found to be N-methylmorpholine (entry 14) and THF. Eventually, the ratio of 3/1 triethylamine/THF afforded the best results and was chosen as our optimized conditions. After we discovered that the reaction was sensitive to the palladium/copper ratio, we again looked at this factor. We subsequently found that only very low amounts of CuI were needed and it was better to add the exact amount of CuI desired as a 7.5M solution in Et3N. When running the reaction under an inert atmosphere and anhydrous conditions, this allowed us to increase the yield of the desired benzofuran 7 to 96% (entry 15).
2.2. Evaluation of the Scope
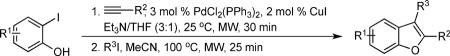
After “optimal” conditions for the formation of the benzo[b]furan 7 were found, an evaluation of the reaction scope was performed (Table 2).
Table 2.
Scope of the reaction
| entry | iodophenol | acetylene | aryl iodide | product | yielda (%) |
|---|---|---|---|---|---|
| 1 |
|
5 | 6 |
|
91 |
| 2 |
|
5 | 6 |
|
92 |
| 3 |
|
5 |
|
|
53 |
| 4 |
|
5 | 6 |
|
84 |
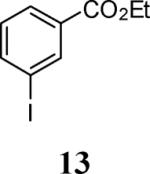
| 5 | 15 | 5 | 13 |
|
60b |
| 6 | 4 |
|
6 |
|
94 |
| 7 | 4 |
|
6 |
|
93 |
| 8 | 4 |
|
6 |
|
83 |
| 9 | 4 |

|
13 |
|
69c |
| 10 | 4 |
|
6 |
|
traced |
| 11 | 10 |
|
13 |
|
52 |
| 12 | 4 |
|
6 |
|
100 |
| 13 | 4 |
|
6 |
|
63e |
| 14 | 4 | 5 | PhI |
|
87 |
| 15 | 4 | 5 |
|
|
53f |
| 16 | 4 | 5 |
|
|
84 |
| 17 | 4 | 5 |
|
|
98 |
| 18 | 4 | 5 |
|
|
75 |
| 19 | 4 | 5 |
|
|
74 |
| 20 | 4 | 5 |
|
|
96 |
| 21 | 4 | 30 |
|
|
73 |
| 22 | 4 | 5 |
|
|
58 |
| 23 | 4 | 5 |
|
|
43 |
| 24 | 8 |
|
|
|
65 |
| 25 |
|
|
|
|
60 |
| 26 |
|
57 | 58 |
|
63 |
| 27 | 4 | 5 |
|
|
34 |
Isolated yields after column chromatography.
This compound was prepared on a large scale and recrystallized, what might have contributed to the lower yield.
1.0 Equiv of alkyne was employed.
The reaction afforded a complex mixture; for an alternative route to 27, see Scheme 2.
1.0 Equiv of alkyne was employed and the first step of the process was run at 60 °C.
The second step of the process was conducted at 80 °C with the addition of 10 mol % Pd(PPh3)4.
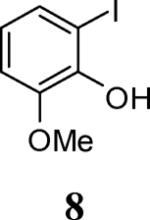
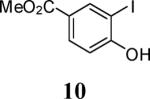
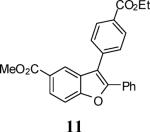
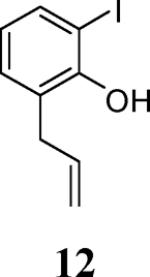
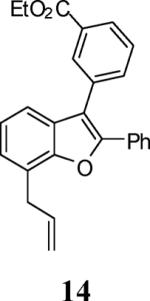
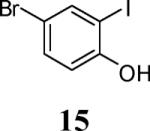
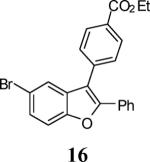
First, several different iodophenols were investigated under our optimized reaction conditions (Table 2, entries 1–5). The electron-rich methoxy-containing phenol 8, and the electron-poor ester-containing phenol 10 afforded the expected benzo[b]furans 9 and 11 in 91% and 92% yields, respectively (entries 1 and 2). When 6-allyl-2-iodophenol (12) was employed, benzofuran 14 was formed, albeit in only a 53% yield (entry 3). The presence of a bromine atom para to the hydroxy group was well tolerated and the bromo-containing benzo[b]furans 16 and 17 were isolated in 84% and 60% yields, respectively (entries 4 and 5). Even though the addition of THF greatly improved the solubility of many of the starting o-iodophenols, some substrates (e.g. 5-iodovanillin, 7-iodo-8-hydroxyquinoline-5-sulfonic acid, and 5-iodouracil) still exhibited insufficient solubility in the Et3N/THF mixture to afford good results. The poor solubility prevented the first coupling step from proceeding in acceptable yield and thus sharply reducing the yield of the three-component coupling product while increasing the number of side products.
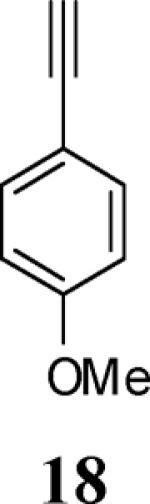
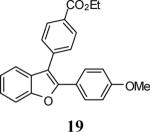
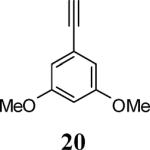
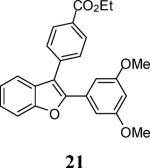
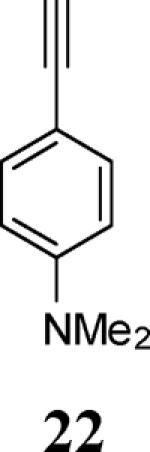
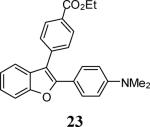
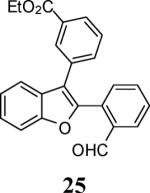
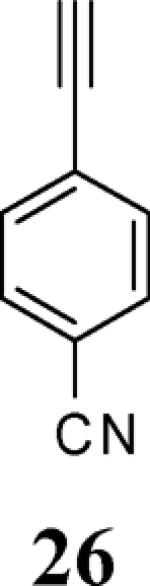
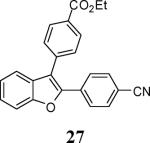
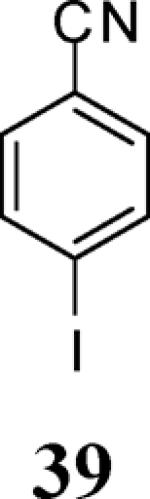
Various additional terminal alkynes have been studied (entries 6–12). Alkynes bearing electron-donating groups, such as alkynes 18, 20 and 22, were well tolerated, providing benzofurans 19, 21 and 23 in 94%, 93% and 83% yields, respectively (entries 6–8). The alkyne 24, containing an electron-withdrawing aldehyde group in the position ortho to the alkyne functionality was also tolerated, providing the benzofuran 25 in a 69% yield (entry 9). However, when a stronger electron-withdrawing cyano group (26) was present in the alkyne, no cyclization product was observed (entry 10). Instead a complex reaction mixture, containing the 3H-benzofuran 66, the coupling product of 26 with 6, as well as trace amounts of other products was obtained. This result can be rationalized by examining the nucleophilicity of the alkyne moiety. When an electron-withdrawing group is present, the electron density on the carbon-carbon triple bond is decreased, thus promoting cyclization of the OH group, without interception by the desired intermediate arylpalladium iodide. In the case of 4-ethynylbenzonitrile (26), using the corresponding TMS-protected phenol 65 and adding TBAF during the second step of the sequence, we were able to obtain the desired benzo[b]furan 27 in a 53% yield (Scheme 2).
Scheme 2.
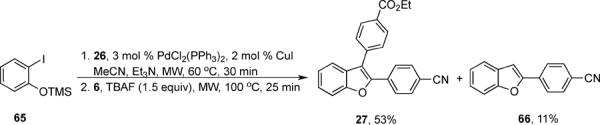
An alternative route employing 4-ethynylbenzonitrile
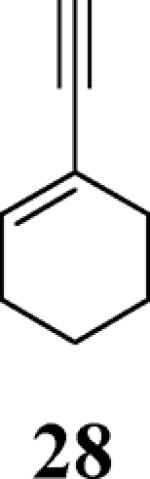
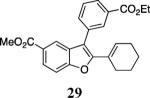
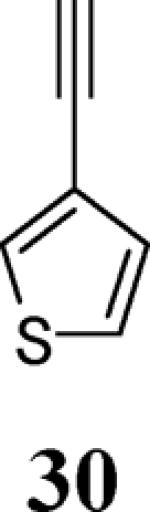
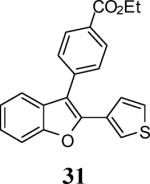
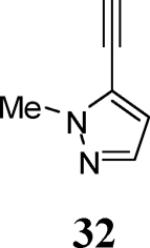
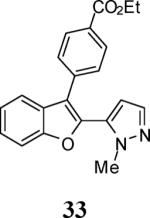
The use of 1-ethynylcyclohexene (28) afforded the desired benzo[b]furan 29 in a 52% yield (entry 11). The heterocycle-containing terminal alkynes 30 and 32 were also well tolerated under our reaction conditions, providing thiophenyl- and methylimidazolyl-substituted benzofurans 31 and 33 in 100% and 63% yields respectively (entries 12 and 13). Unfortunately, aliphatic acetylenes [e.g. 1-pentyne] led to formation of the desired benzo[b]furan in only trace amounts and afforded a complex reaction mixture. Various attempts to modify the reaction conditions failed to improve this reaction.
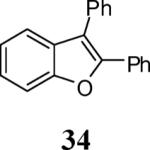
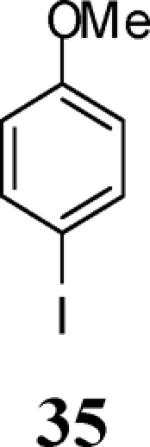
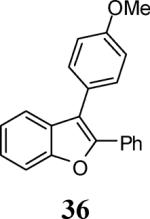
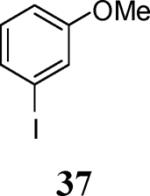
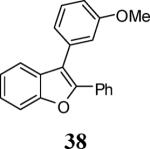
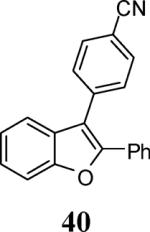
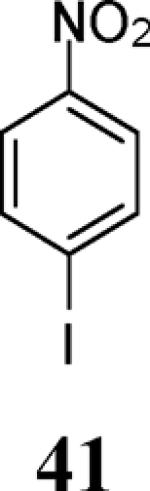
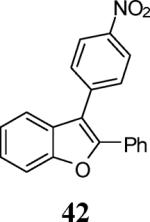
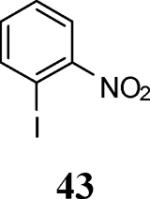
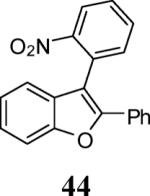
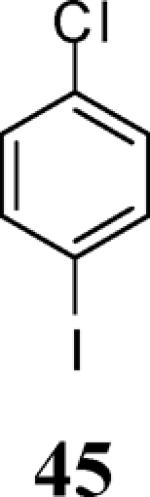
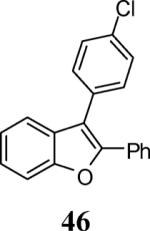
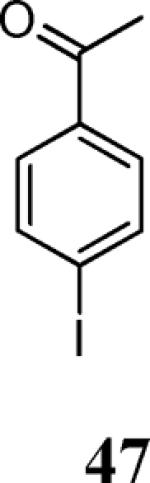
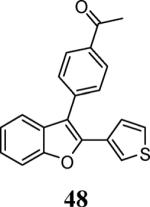
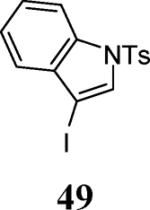
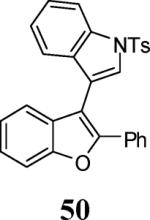
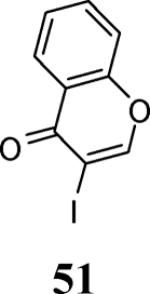
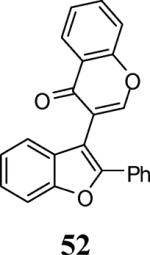
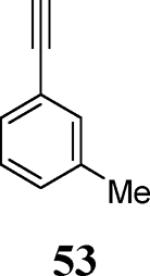
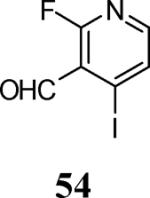
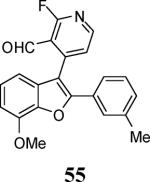
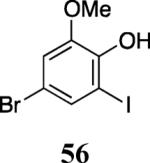
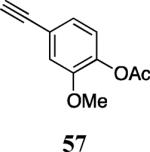
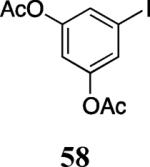
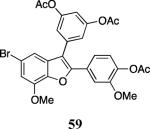
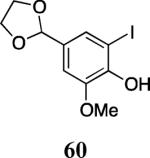
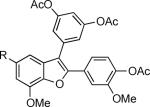
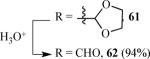
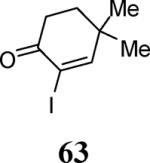
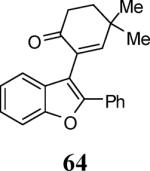
Finally, we examined the scope of the aryl iodides that can be used in this process under our optimized reaction conditions. Starting with iodobenzene, 2,3-diphenylbenzo[b]furan (34) was obtained in an 87% yield (entry 14). When 4-iodoanisole (35) was employed, only a 22% yield of benzofuran 36 was obtained under our optimized conditions. However, using a slightly lower temperature (80 °C) and an additional loading of the Pd catalyst in the second step improved the yield of the product 36 to 53% (entry 15). Surprisingly, the presence of an electron-donating methoxy group in the meta-position of the aryl iodide 37 was well tolerated and afforded the desired heterocycle 38 in an 84% yield (entry 16). Aryl iodides with electron-withdrawing groups, such as 4-iodobenzonitrile (39) and 4-iodonitrobenzene (41), also afforded the corresponding benzofurans 40 and 42 in 98% and 75% yields, respectively (entries 17 and 18). Placing the nitro group in the position ortho to the iodine did not affect the efficiency of the process, providing benzo[b]furan 44 in a 74% yield (entry 19). 4-Chloroiodobenzene (45) afforded the benzo[b]furan 46 in an excellent 96% yield (entry 20). The product 48 has been obtained in a 73% yield by employing 3-ethynylthiophene (30) and p-iodoacetophenone (47) (entry 21). Various heterocyclic aryl and vinylic iodides have been examined under our standard reaction conditions, providing the corresponding heterocyclic products in moderate to good yields (entries 22–24). N-Tosyl 3-iodoindole (49) afforded the desired benzofuran 50 in a 58% yield (entry 22). When employing 2-iodochromene (51), the bis-heterocyclic product 52 was obtained, albeit in only a 43% yield (entry 23). To our delight, this methodology proved to be useful for the synthesis of the highly substituted benzofurans 55, 59 and 61 (entries 24–26). 2-Fluoro-3-formyl-4-iodopyridine (54) coupled with 6-methoxy-2-iodophenol (8) and 3-tolyl acetylene (53) to afford the highly substituted benzofuran 55 in a 65% yield (entry 24). Bromo-substituted benzofuran 59 was obtained in a 60 % yield from the readily accessible starting materials 56, 57 and 58 (entry 25). Compound 61 was prepared in a 63% yield starting from 2-iodophenol 60 and was further converted into the corresponding aldehyde 62 in 94% yield (entry 26). Benzofurans 59 and 62 would appear to be an ideal intermediates for the synthesis of naturally-occurring Gnetuhainin F, a benzofuran from the class of oligostilbenes isolated from Gnetum hainanense species, a traditional Chinese medicine herb.9 Finally, the vinylic halide 2-iodo-4,4-dimethylcyclohex-2-enone (63) was allowed to react with o-iodophenol and phenyl acetylene to generate the corresponding heterocycle 64, albeit in only a 34% yield (entry 27).
2.3. Additional processes and further diversification
Recently, several processes describing Heck-type Pd(0) or Rh(I)-catalyzed reactions of 2-(1-alkynyl)phenols with alkenes have been reported.10 We were interested in knowing if our methodology could provide such a Heck-type transformation to afford 3-(1-alkenyl)benzofurans in a one-pot fashion from 2-iodophenol (4). Gratifyingly, by employing butyl acrylate and slightly modifying our reaction conditions for the second step of the process, we were able to obtain the olefinic product 67 in a 56% yield (Scheme 3). The alkyne 1-ethynylcyclohexene (28) also afforded the desired product 68 in a 70% yield.
Scheme 3.

Synthesis of benzofurans from iodophenols, alkynes and alkenes
We have also investigated the possibility of employing aryl boronic acids (analogous to a Suzuki-Miyaura coupling),11 plus aryl halides and terminal alkynes (analogous to a Sonogashira coupling)12 in a Sonogashira/cyclization/Suzuki-Miyaura sequential process. However, there was no evidence for formation of the expected 2,3-disubstituted benzo[b]furans in the two cases we examined using our standard reaction conditions. The major product in both cases was the corresponding 3H-benzofuran. This does not mean that there are no reaction conditions under which the desired Sonogashira/cyclization/Suzuki-Miyaura process won't occur.
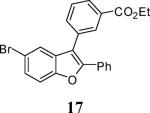
We have also attempted Pd-catalyzed couplings with the 5-bromobenzofurans 16 and 17 prepared by our benzofuran synthesis to illustrate how our benzofurans can be further diversified to provide a large variety of multisubstituted benzofurans for drug testing. Thus, Suzuki-Miyaura11 and Mizoroki-Heck12 couplings proceeded smoothly, affording the benzofurans 69 and 70 in 83% and 81% yields, respectively (Scheme 4).
Scheme 4.
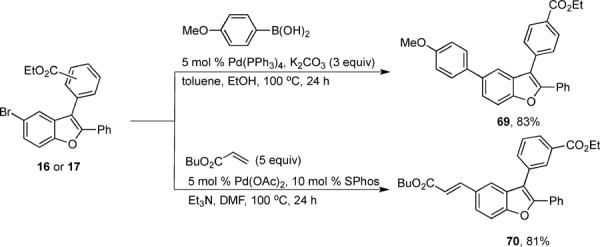
Pd-catalyzed diversification
3. Conclusions
A novel convenient multicomponent process for the synthesis of 2,3-disubstituted benzo[b]furans under Sonogashira conditions has been developed and the scope of this process studied. Microwave irradiation has been employed, providing higher yields, shorter reaction times and cleaner products. This methodology has proven quite general and should prove a valuable tool in the synthesis of combinatorial libraries of benzofurans.
4. Experimental
4.1. General remarks
All microwave reactions were carried out in sealed oven-dried microwave vials. A Biotage microwave reactor was used for all experiments run at or above 60 °C. A CEM microwave reactor was used for the room temperature microwave reactions. The 1H and 13C NMR spectra were recorded at 300 and 75.5 MHz or 400 and 100 MHz, respectively. The chemical shifts of the 1H NMR and 13C NMR spectra are reported relative to the residual signal of CDCl3 (δ 7.26 ppm for the 1H NMR and δ 77.23 ppm for the 13C NMR), acetone-d6 (2.05 ppm for the 1H NMR and δ 29.92 ppm for the 13C NMR) or DMSO-d6 (2.50 ppm for the 1H NMR and δ 39.51 ppm for the 13C NMR). All coupling constants (J) are reported in Hertz (Hz). All commercially obtained chemicals were used as received without further purification. Thin layer chromatography was performed using commercially prepared 60-mesh silica gel plates, and visualization was effected with short wavelength UV light (254 nm). All melting points were obtained using an EZ-Melt automated melting point apparatus and are uncorrected. High resolution mass spectra (HRMS) were obtained using an Agilent QTOF 6540 mass spectrometer (ESI at a voltage of 70 eV). All mass spectra (MS) were obtained using a GCT-Agilent 6890 gas chromatograph/mass spectrometer (EI at a voltage of 70 eV). All IR spectra were obtained using a Nicolet 380 FT-IR apparatus.
4.2. Preparation of the starting compounds for the three-component coupling
A majority of the starting materials were purchased from commercial sources and used as received. The following compounds were prepared following the procedure described in the literature: iodophenols 813 and 1214; 3-iodo-N-tosylindole (49),15 3-iodo-4H-chromen-4-one (51),16 2-iodo-4,4-dimethylcyclohexenone (63).17
4-Bromo-2-iodo-6-methoxyphenol (56)
Phenol 56 was prepared by a modification of a procedure described for the bromination of guaiacol.18 2-Iodo-6-methoxyphenol (8, 0.975 g, 3.9 mmol) was dissolved in dry DMF (0.8 mL), the reaction mixture was cooled to 0 °C, and then NBS (0.69 g, 3.9 mmol) in DMF (0.8 mL) was added dropwise. The reaction mixture was stirred for 30 min at 0 °C and then slowly quenched with an ice cold water/ethyl ether mixture at the same temperature. (The yield of the product dropped significantly when the reaction temperature was not kept at or below 0 °C). The organic fraction was separated, washed and dried over MgSO4. The reaction mixture was purified using column chromatography and the desired compound 56 was obtained as a brown solid (0.96 g, 75%): mp 77–79 °C; 1H NMR (300 MHz, CDCl3) δ 3.88 (s, 4H), 6.04 (s, 1H), 6.94 (d, J = 2.1 Hz, 1H), 7.43 (d, J = 2.1 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 56.7, 81.6, 112.5, 114.3, 132.4, 145.3, 146.4.
4-Ethynyl-2-methoxyphenyl acetate (57)
Commercially available 4-bromo-2-methoxyphenol (1.02 g, 5.0 mmol) and acetic anhydride (0.71 mL, 7.5 mmol) were dissolved in dichloromethane (10 mL). Then concentrated H2SO4 (25 mg) was added and the mixture was stirred for 30 min at rt. The reaction was then subjected to an aqueous work-up analogous to the one described in the literature,19 resulting in 4-bromo-2-methoxyphenyl acetate, obtained as a colorless solid, 1.21 g (99%): 1H NMR (300 MHz, CDCl3) δ 2.31 (s, 3H), 3.82 (s, 3H), 6.90 (d, J = 8.6 Hz, 1H), 7.03–7.12 (m, 2H). 4-Bromo-2-methoxyphenyl acetate (1.21 g, 4.9 mmol), Pd(OAc)2 (53.8 mg, 0.24 mmol), CuI (23 mg, 0.12 mmol), and tris(tert-butyl)phosphoniumtetrafluoroborate (69.6 mg, 0.24 mmol) were dissolved in diisopropylamine (10 mL) and purged with argon. Trimethylsilyl acetylene (1.38 mL, 9.8 mmol) was added and the reaction mixture was stirred at 40 °C for 2 h. The work-up was conducted analogous to a procedure described in the literature.20 The desired alkyne was obtained as a colorless solid (1.28 g, 100%). The resulting product was dissolved in THF (29 mL) and water (3.5 mL) and a 1M solution of TBAF in THF (5.7 mL) was added at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred for an additional 1 h. The volatile solvents were evaporated and the aqueous layer was extracted with ethyl acetate. The organic fractions were dried and evaporated, affording 0.74 g (80%) of the desired compound 57 as a colorless solid: mp 77–79 °C; 1H NMR (400 MHz, CDCl3) δ 2.31 (s, 3H), 3.07 (s, 1H), 3.82 (s, 3H), 6.99 (d, J = 8.0 Hz, 1H), 7.06–7.12 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 20.7, 56.0, 77.3, 83.2, 116.0, 120.8, 123.0, 125.1, 140.5, 150.9, 168.8.
3,5-Diacetoxyiodobenzene (58)
3,5-Dihydroxyiodobenzene (0.27 g, 1.16 mmol) was dissolved in dichloromethane (3 mL); Ac2O (0.33 mL, 3.49 mmol) and H2SO4 (1.2 mg) were added and the mixture was stirred at rt for 1 h. Then concentrated aq NaHCO3 solution was added at 0 °C and the mixture was allowed to warm up to room temperature. The organic phase was collected, dried (MgSO4) and evaporated. Compound 58 was obtained as a colorless solid (0.34 g, 91%) and used without further purification: mp 77–80 °C; 1H NMR (400 MHz, CDCl3) δ 2.28 (s, 3H), 6.92 (t, J = 2.0 Hz, 1H), 7.36 (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.3, 69.5, 92.7, 115.6, 128.4, 151.3, 168.8; HRMS calcd for C10H9IO4 [M+Na]+ 342.9438, found 342.9441.
4-(1,3-Dioxolan-2-yl)-2-iodo-6-methoxyphenol (60)
Compound 60 was prepared following a procedure described for an analogous reaction.21 5-Iodovanillin (1.0 mmol) and ethylene glycol (5.0 mmol) were dissolved in toluene. Then acidic aluminum oxide was added and the resulting mixture was refluxed for 24 h. After cooling, the mixture was filtered, washed with dichloromethane/water and the organic phase was dried (MgSO4) and evaporated. Column chromatography using ethyl acetate/hexanes (1:3) as the eluent afforded 196 mg (62%) of product 60 as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 3.91 (s, 3H), 3.97–4.06 (m, 2H), 4.08–4.16 (m, 2H), 5.69 (s, 1H), 6.16 (s, 1H), 6.96 (s, 1H), 7.42 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 56.5, 65.5, 80.9, 95.7, 103.0, 108.9, 129.1, 131.6, 146.2, 146.6.
4.3. General procedure for the one-pot, three-component Sonogashira/Cacchi type coupling to synthesize benzofurans
The 2-iodophenol (0.5 mmol) and dichlorobis(triphenylphosphine)palladium (10.5 mg, 3 mol %) were placed in a 5 mL microwave vial and purged with argon. Dry THF (0.5 mL) was added and the reaction mixture was stirred until the iodophenol completely dissolved. Then dry triethylamine (1.0 mL) and a 3.8M solution of CuI in dry triethylamine (0.5 mL) were added and the mixture allowed to stir for 10 min. Then 1.2 equiv of the corresponding alkyne was added; the vial was capped, purged with argon and placed in the microwave reactor for 30 min at 25 °C. The corresponding aryl iodide (0.5 mmol) and dry acetonitrile (2 mL) were added and the reaction mixture was heated in the microwave reactor at 100 °C for 25 min. After cooling, the solvents were evaporated and column chromatography using ethyl acetate/hexane as the eluent afforded the desired products.
Ethyl 4-(2-phenylbenzofuran-3-yl)benzoate (7)
Yellow solid, 164.2 mg (96%): mp 100–103 °C; 1H NMR (400 MHz, CDCl3) δ 1.42 (t, J = 7.1 Hz, 3H), 4.41 (q, J = 7.1 Hz, 2H), 7.21–7.26 (m, 1H), 7.26–7.36 (m, 4H), 7.49 (d, J = 7.3 Hz, 1H), 7.52–7.66 (m, 5H), 8.14 (d, J = 8.1 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 14.6, 61.3, 111.4, 116.7, 119.9, 123.3, 125.1, 127.4, 128.7, 128.9, 129.8, 129.8, 130.4, 131.6, 131.9, 137.9, 151.3, 154.2, 166.6; HRMS calcd for C23H18O3 [M+H]+ 342.1256, found 342.1260.
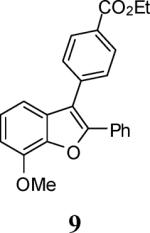
Ethyl 4-(7-methoxy-2-phenylbenzofuran-3-yl)benzoate (9)
Bright yellow solid, 169.6 mg (91%): mp 125–127 °C; 1H NMR (400 MHz, CDCl3) δ 1.44 (t, J = 7.1 Hz, 3H), 4.08 (s, 3H), 4.43 (q, J = 7.1 Hz, 2H), 6.87 (d, J = 7.9 Hz, 1H), 7.10 (d, J = 7.8 Hz, 1H), 7.19 (t, J = 7.9 Hz, 1H), 7.28–7.36 (m, 3H), 7.59 (d, J = 8.0 Hz, 2H), 7.61–7.70 (m, 2H), 8.14 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.6, 56.4, 61.3, 107.4, 112.3, 117.0, 124.0, 127.5, 128.7, 128.9, 129.8, 129.9, 130.3, 130.3, 131.4, 137.9, 143.6, 145.6, 151.6, 166.6; HRMS calcd for C24H20O4 [M+H]+ 372.1362, found 373.1438.
Methyl 3-[4-(ethoxycarbonyl)phenyl]-2-phenylbenzofuran-5-carboxylate (11)
Colorless solid, 162.3 mg (92%): mp 186–187 °C; 1H NMR (300 MHz, CDCl3) δ 1.44 (t, J = 7.1 Hz, 3H), 3.92 (s, 3H), 4.44 (q, J = 7.1 Hz, 2H), 7.29–7.47 (m, 3H), 7.46–7.73 (m, 5H), 8.09 (d, J = 8.6 Hz, 1H), 8.17 (d, J = 8.2 Hz, 2H), 8.20 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 14.6, 52.4, 61.4, 111.4, 117.1, 122.4, 125.8, 126.9, 127.4, 128.9, 129.4, 129.9, 129.9, 130.2, 130.6, 137.2, 152.7, 156.8, 166.5, 167.3; HRMS calcd for C25H20O5 [M+H]+ 401.1384, found 401.1395.
Ethyl 4-(7-allyl-2-phenylbenzofuran-3-yl)benzoate (14)
Colorless oil, 97.6 mg (53%): 1H NMR (300 MHz, CDCl3) δ 1.38 (t, J = 7.1 Hz, 3H), 3.79 (d, J = 6.7 Hz, 2H), 4.39 (q, J = 7.1 Hz, 2H), 5.11–5.33 (m, 2H), 5.06–5.38 (m, 2H), 6.16 (ddt, J = 16.8, 10.0, 6.6 Hz, 1H), 7.11–7.40 (m, 6H), 7.53 (t, J = 7.7 Hz, 1H), 7.59–7.73 (m, 3H), 8.10 (dt, J = 7.8, 1.5 Hz, 1H), 8.23 (t, J = 1.8 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 14.5, 29.9, 30.2, 34.2, 61.3, 116.5, 117.0, 118.1, 123.5, 123.9, 125.1, 127.2, 128.7, 129.0, 129.3, 130.0, 130.7, 131.0, 131.6, 133.7, 134.5, 136.1, 150.9, 152.6, 166.6; HRMS calcd for C26H22O3 [M+H]+ 382.1563, found 382.1558.
Ethyl 4-(5-bromo-2-phenylbenzofuran-3-yl)benzoate (16)
Colorless solid, 176.8 mg (84%): mp 135–137 °C; 1H NMR (400 MHz, CDCl3) δ 1.44 (t, J = 7.1 Hz, 3H), 4.43 (q, J = 7.1 Hz, 2H), 7.29–7.37 (m, 3H), 7.44 (d, J = 1.0 Hz, 2H), 7.56 (d, J = 8.3 Hz, 2H), 7.58–7.64 (m, 3H), 8.15 (d, J = 8.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 14.6, 61.4, 112.9, 116.3, 116.6, 122.7, 127.5, 128.0, 128.9, 129.4, 129.8, 129.9, 130.2, 130.6, 131.9, 137.2, 152.6, 153.0, 166.5; HRMS calcd for C23H17Br O3 [M+H]+ 421.0361, found 421.0434.
Ethyl 3-(5-bromo-2-phenylbenzofuran-3-yl)benzoate (17)
Colorless solid: mp 103–104 °C; 1H NMR (300 MHz, CDCl3) δ 1.39 (t, J = 7.1 Hz, 3H), 4.40 (q, J = 7.1 Hz, 2H), 7.28–7.35 (m, 3H), 7.44 (d, J = 1.2 Hz, 2H), 7.50–7.71 (m, 5H), 8.12 (dt, J = 7.6, 1.4 Hz, 1H), 8.17 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 14.5, 61.4, 112.9, 116.2, 116.5, 122.7, 126.4, 127.2, 127.9, 128.8, 128.9, 129.2, 129.4, 129.5, 130.0, 130.9, 131.8, 132.3, 132.8, 134.4, 152.4, 152.9, 166.5; HRMS calcd for C23H18BrO3 [M+H]+ 421.0434, found 421.0440.
Ethyl 4-[2-(4-methoxyphenyl)benzofuran-3-yl]benzoate (19)
Yellow solid, 175.0 mg (94%): mp 125–127 °C; 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H), 3.78 (s, 3H), 4.41 (q, J = 7.1 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 7.22 (t, J = 7.5 Hz, 1H), 7.29 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 7.7 Hz, 1H), 7.52–7.61 (m, 5H), 8.13 (d, J = 8.2 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 14.6, 55.4, 61.2, 111.2, 114.2, 115.2, 119.6, 122.9, 123.2, 124.6, 128.9, 129.5, 129.8, 130.3, 134.1, 138.2, 151.5, 154.1, 160.1, 166.6; HRMS calcd for C24H20O4 [M+H]+ 372.1362, found 372.1372.
Ethyl 4-[2-(3,5-dimethoxyphenyl)benzofuran-3-yl]benzoate (21)
Cream colored solid, 186.3 mg (93%): mp 102–104 °C; 1H NMR (400 MHz, CDCl3) δ 1.45 (t, J = 7.1 Hz, 3H), 3.70 (s, 6H), 4.44 (q, J = 7.1 Hz, 2H), 6.45 (s, 1H), 6.81 (s, 2H), 7.27 (t, J = 7.5 Hz, 1H), 7.37 (t, J = 7.7 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.63 (d, J = 8.0 Hz, 2H), 8.18 (dd, J = 8.2, 1.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.6, 55.5, 61.3, 101.6, 101.6, 105.2, 111.4, 117.1, 119.9, 123.3, 125.2, 129.8, 129.9, 130.3, 131.9, 137.9, 150.9, 154.0, 160.8, 166.5; HRMS calcd for C25H22O5 [M+H]+ 403.1540, found 403.1549.
Ethyl 4-[2-(4-(dimethylamino)phenyl)benzofuran-3-yl]benzoate (23)
Yellow-green oil, 160.1 mg (83%): 1H NMR (300 MHz, CDCl3) δ 1.47 (t, J = 7.2 Hz, 3H), 2.99 (s, 6H), 4.47 (q, J = 7.1 Hz, 2H), 7.29 (dt, J = 17.4, 7.0 Hz, 2H), 6.66 (d, J = 8.5 Hz, 2H), 7.29 (dt, J = 17.4, 7.0 Hz, 2H), 7.48–7.61 (m, 4H), 7.67 (d, J = 8.0 Hz, 2H), 8.19 (d, J = 8.0 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 14.5, 40.3, 61.1, 111.1, 111.9, 113.7, 117.9, 119.2, 123.0, 124.1, 128.5, 129.2, 129.8, 130.1, 130.2, 138.8, 150.6, 152.7, 153.9, 166.7; HRMS calcd for C25H23NO3 [M+H]+ 386.1751, found 386.1759.
Ethyl 4-[2-(2-formylphenyl)benzofuran-3-yl]benzoate (25)
Yellow oil, 128.6 mg (69%): 1H NMR (400 MHz, CDCl3) δ 1.36 (t, J = 7.2 Hz, 3H), 4.36 (q, J = 7.2 Hz, 2H), 7.36 (t, J = 7.5 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 7.47–7.64 (m, 5H), 7.73 (d, J = 7.8 Hz, 1H), 8.01 (t, J = 7.6 Hz, 2H), 8.14 (s, 1H), 10.07 (s, 1H); 13C NMR (101 MHz, CDCl3) δ 14.5, 61.3, 111.8, 117.6, 120.5, 120.7, 123.8, 125.8, 128.3, 128.3, 129.0, 129.3, 129.8, 130.5, 131.3, 131.6, 131.9, 132.9, 133.8, 134.3, 148.6, 155.1, 166.2, 191.0; HRMS calcd for C24H18O4 [M+H]+ 371.1278, found 371.1276.
Ethyl 4-[2-(4-cyanophenyl)benzofuran-3-yl]benzoate (27)
Colorless solid, 97.5 mg (53%): mp 135–137 °C; 1H NMR (400 MHz, CDCl3) δ 1.45 (t, J = 7.1 Hz, 3H), 4.45 (q, J = 7.1 Hz, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.57 (t, J = 8.1 Hz, 5H), 7.73 (d, J = 8.1 Hz, 2H), 8.19 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.6, 56.9, 61.4, 111.6, 111.9, 118.8, 119.7, 120.5, 123.8, 126.3, 127.3, 129.6, 129.8, 130.5, 130.7, 132.5, 134.7, 136.9, 148.7, 154.4, 166.4; HRMS calcd for C24H17NO3 [M+H]+ 367.1208, found 367.1212.
Ethyl 4-[2-(cyclohex-1-en-1-yl)benzofuran-3-yl]benzoate (29)
Yellow oil, 104.1 mg (52%): 1H NMR (400 MHz, CDCl3) δ 1.40 (t, J = 7.1 Hz, 3H), 1.62 (t, J = 3.2 Hz, 4H), 2.12 (br s, 2H), 2.18 (br s, 2H), 3.88 (s, 3H), 4.40 (q, J = 7.1 Hz, 2H), 6.41–6.48 (m, 1H), 7.47 (d, J = 8.5 Hz, 1H), 7.54 (t, J = 7.7 Hz, 1H), 7.61–7.66 (m, 1H), 8.01 (dd, J = 10.8, 1.5 Hz, 2H), 8.09 (d, J = 7.8 Hz, 1H), 8.13 (d, J = 1.9 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.5, 21.9, 22.6, 25.9, 26.6, 52.2, 61.3, 110.8, 115.5, 121.9, 125.2, 126.4, 127.9, 128.9, 128.9, 130.6, 131.0, 131.1, 131.2, 133.2, 134.5, 154.5, 156.0, 166.6, 167.4; HRMS calcd for C25H25O5 [M+H]+ 405.1697, found 405.1706.
Ethyl 4-[2-(thiophen-3-yl)benzofuran-3-yl]benzoate (31)
Yellow oil, 174.2 mg (100%): 1H NMR (300 MHz, CDCl3) δ 1.45 (t, J = 7.2 Hz, 3H), 4.44 (q, J = 7.1 Hz, 2H), 7.21 (t, J = 5.2 Hz, 1H), 7.24–7.30 (m, 2H), 7.35 (t, J = 7.6 Hz, 1H), 7.47 (d, J = 7.2 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.58–7.70 (m, 3H), 8.18 (d, J = 8.4 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 14.6, 61.3, 111.3, 115.9, 119.8, 123.4, 123.8, 125.0, 126.1, 126.2, 129.7, 130.0, 130.3, 131.6, 137.7, 148.3, 154.0, 166.6; HRMS calcd for C21H16O3S [M+H]+ 349.0893, found 349.0900.
Ethyl 4-[2-(1-methyl-1H-pyrazol-5-yl)benzofuran-3-yl]benzoate (33)
Green amorphous solid, 109.4 mg (63%): 1H NMR (400 MHz, CDCl3) δ 1.41 (t, J = 7.1 Hz, 3H), 3.63 (s, 3H), 4.40 (q, J = 7.1 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 7.38 (t, J = 7.3 Hz, 1H), 7.42–7.49 (m, 1H), 7.55 (t, J = 6.8 Hz, 3H), 7.60–7.72 (m, 2H), 8.11 (d, J = 7.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.6, 61.3, 111.6, 120.2, 123.7, 125.5, 128.1, 128.6, 128.7, 129.1, 129.9, 130.4, 132.1, 132.2, 132.3, 136.8, 154.8, 166.4 (N-CH3 does not show up); HRMS calcd for C21H18N2O3 [M+H]+ 347.1390, found 347.1398.
2,3-Diphenylbenzofuran (34).22
Yellow-green solid, 117.6 mg (87%): mp 116–119 °C [lit. mp 123 °C]23; 1H NMR (300 MHz, CDCl3) δ 7.20–7.36 (m, 5H), 7.46 (m, 6H), 7.56 (d, J = 8.2 Hz, 1H), 7.66 (d, J = 7.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 111.3, 117.7, 120.3, 123.1, 124.9, 127.2, 127.8, 128.6, 128.6, 129.2, 129.9, 130.5, 130.9, 133.1, 150.7, 154.2.
3-(4-Methoxyphenyl)-2-phenylbenzofuran (36).24
Yellow amorphous solid, 79.7 mg (53%): 1H NMR (400 MHz, CDCl3) δ 3.89 (s, 3H), 6.98–7.05 (m, 2H), 7.23–7.27 (m, 1H), 7.28–7.36 (m, 4H), 7.41–7.45 (m, 2H), 7.50 (d, J = 8.5 Hz, 1H), 7.56 (d, J = 8.2 Hz, 1H), 7.69 (dd, J = 8.2, 1.6 Hz, 2H).
3-(3-Methoxyphenyl)-2-phenylbenzofuran (38).25
Yellow oil, 125.7 mg (84%): 1H NMR (400 MHz, CDCl3) δ 3.83 (s, 3H), 6.98–7.03 (m, 1H), 7.13 (dd, J = 11.5, 4.9 Hz, 2H), 7.24–7.45 (m, 6H), 7.56–7.63 (m, 2H), 7.75 (dd, J = 8.1, 1.5 Hz, 2H).
3-(4-Cyanophenyl)-2-phenylbenzofuran (40)
Yellow solid, 140.1 mg (98%): mp 113–115 °C, 1H NMR (400 MHz, CDCl3) δ 7.27 (t, J = 7.5 Hz, 1H), 7.32–7.39 (m, 4H), 7.48 (d, J = 7.7 Hz, 1H), 7.59 (dt, J = 11.4, 4.6 Hz, 5H), 7.69–7.78 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 111.4, 111.6, 115.9, 118.9, 119.6, 123.6, 125.4, 127.5, 128.9, 129.2, 129.3, 130.0, 130.5, 132.9, 138.3, 151.8, 154.3; HRMS calcd for C21H13NO [M+H]+ 296.107, found 296.1073.
3-(4-Nitrophenyl)-2-phenylbenzofuran (42).25
Yellow solid, 114.5 mg (73%): mp 135–138 °C; 1H NMR (300 MHz, CDCl3) δ 7.26–7.28 (m, 1H), 7.32 (d, J = 7.6 Hz, 1H), 7.36–7.39 (m, 4H), 7.52 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 7.2 Hz, 3H), 7.70 (d, J = 8.2 Hz, 2H), 8.33 (dd, J = 8.7, 1.7 Hz, 2H).
3-(2-Nitrophenyl)-2-phenylbenzofuran (44)
Yellow crystals, 116.6 mg (74%): mp 119–122 °C; 1H NMR (400 MHz, CDCl3) δ 7.17–7.25 (m, 2H), 7.28–7.39 (m, 4H), 7.51 (dd, J = 7.5, 1.7 Hz, 1H), 7.52–7.60 (m, 3H), 7.62 (td, J = 7.7, 1.6 Hz, 1H), 7.68 (td, J = 7.5, 1.5 Hz, 1H), 8.13 (dd, J = 8.0, 1.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 111.6, 113.4, 119.5, 123.5, 125.2, 125.2, 126.9, 128.2, 128.9, 128.9, 129.4, 129.8, 130.1, 133.6, 133.6, 149.9, 151.4, 154.0; HRMS calcd for C20H13NO3 [M+H]+ 316.0968, found 316.0977.
3-(4-Chlorophenyl)-2-phenylbenzofuran (46)
Yellow solid, 145.6 mg (96%): mp 100–101 °C; 1H NMR (400 MHz, CDCl3) δ 7.24–7.28 (m, 1H), 7.35 (dd, J = 10.2, 5.7 Hz, 4H), 7.45 (s, 4H), 7.47 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 8.2 Hz, 1H), 7.64 (dd, J = 7.9, 1.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 111.4, 116.5, 119.9, 123.3, 125.1, 127.3, 128.7, 128.8, 129.5, 130.0, 130.5, 131.3, 131.5, 133.7, 143.7, 150.9; HRMS calcd for C20H13ClO [M+H]+ 305.0728, found 305.0731.
1-[4-(2-(Thiophen-3-yl)benzofuran-3-yl)phenyl]ethanone (48)
Cream colored solid, 116.1 mg (73%): mp 172–174 °C; 1H NMR (400 MHz, CDCl3) δ 2.68 (s, 3H), 7.20 (d, J = 5.1 Hz, 1H), 7.27 (dd, J = 7.3, 5.5 Hz, 2H), 7.35 (t, J = 7.7 Hz, 1H), 7.47 (d, J = 7.8 Hz, 1H), 7.55 (d, J = 8.1 Hz, 1H), 7.65 (dd, J = 8.6, 5.6 Hz, 3H), 8.09 (d, J = 8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 26.9, 111.4, 115.8, 119.8, 123.4, 123.9, 125.1, 126.1, 126.3, 129.1, 129.6, 130.2, 131.6, 136.5, 138.1, 148.3, 154.0, 197.9; HRMS calcd for C20H14O2S [M+H]+ 318.0715, found 318.0731.
3-(2-Phenylbenzofuran-3-yl)-1-tosyl-1H-indole (50)
Yellow amorphous solid, 134.1 mg (58%): 1H NMR (400 MHz, CDCl3) δ 2.40 (s, 3H), 7.14 (t, J = 7.5 Hz, 1H), 7.25 (ddd, J = 21.5, 15.1, 8.0 Hz, 7H), 7.37 (dd, J = 15.9, 8.5 Hz, 3H), 7.57–7.64 (m, 3H), 7.76 (s, 1H), 7.85 (d, J = 8.2 Hz, 2H), 8.13 (d, J = 8.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 21.9, 108.1, 111.5, 114.1, 114.9, 120.3, 121.4, 123.2, 123.7, 125.1, 125.3, 125.4, 126.9, 127.1, 128.6, 128.7, 130.0, 130.2, 130.5, 130.6, 135.4, 135.7, 145.3, 152.1, 154.3; HRMS calcd for C29H21NO3S [M+H]+ 463.1242, found 463.1315.
3-(2-Phenylbenzofuran-3-yl)-4H-chromen-4-one (52)
Cream colored solid, 73.4 mg (43%): mp 183–184 °C; 1H NMR (400 MHz, CDCl3) δ 7.22–7.28 (m, 1H), 7.29–7.40 (m, 4H), 7.40–7.45 (m, 1H), 7.50 (ddd, J = 8.2, 7.2, 1.1 Hz, 1H), 7.53–7.59 (m, 2H), 7.73–7.78 (m, 3H), 8.01 (s, 1H), 8.36 (dd, J = 8.0, 1.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 107.8, 111.5, 118.3, 118.5, 120.5, 123.3, 124.5, 125.0, 125.8, 126.7, 127.3, 128.9, 128.9, 130.4, 130.5, 134.1, 153.1, 154.3, 155.4, 156.7, 176.2; HRMS calcd for C23H14O3 [M+H]+ 339.1016, found 339.1015.
2-Fluoro-4-[7-methoxy-2-(m-tolyl)benzofuran-3-yl]nicotinaldehyde (55)
Yellow solid, 116.0 mg (65%): mp 177–180 °C; 1H NMR (300 MHz, CDCl3) δ 2.33 (s, 3H), 4.09 (s, 3H), 6.83 (d, J = 7.9 Hz, 1H), 6.90 (d, J = 8.0 Hz, 1H), 7.19 (t, J = 3.2 Hz, 4H), 7.36 (d, J = 5.1 Hz, 1H), 7.47 (s, 1H), 8.46 (d, J = 5.1 Hz, 1H), 10.05 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 21.7, 56.5, 108.0, 111.5, 124.6, 124.9, 125.2, 125.2, 127.9, 128.9, 129.0, 130.7, 131.0, 139.1, 140.5, 143.6, 145.8, 149.3, 152.3, 152.6, 153.6, 161.4, 164.7, 187.1, 187.1 (extra peaks due to 13C-19F coupling); HRMS calcd for C22H16FNO3 [M+H]+ 361.1114, found 362.1187.
2-(4-Acetoxy-3-methoxyphenyl)-5-bromo-3-(3,5-diacetoxyphenyl)-7-methoxybenzofuran (59)
Compound 59 was synthesized following the general procedure for a three-component Sonogashira/Cacchi type cyclization and was obtained as a brown amorphous solid (340.6 mg, 60%): 1H NMR (400 MHz, CDCl3) δ 2.29 (s, 6H), 2.31 (s, 3H), 3.67 (s, 3H), 4.03 (s, 3H), 6.95 (s, 1H), 7.00 (s, 1H), 7.03 (d, J = 8.3 Hz, 1H), 7.09 (d, J = 1.2 Hz, 2H), 7.15 (s, 1H), 7.21 (s, 1H), 7.35 (d, J = 8.3 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 20.9, 21.3, 55.9, 56.6, 110.9, 111.0, 111.1, 114.9, 115.6, 115.7, 116.6, 119.8, 120.6, 123.3, 128.2, 132.6, 134.2, 140.4, 142.2, 145.7, 151.2, 151.5, 151.8, 168.9; HRMS calcd for C28H24BrO9 [M+H]+ 583.0525, found 583.0598.
2-(4-Acetoxy-3-methoxyphenyl)-3-(3,5-diacetoxyphenyl)-5-(1,3-dioxolan-2-yl)-7-methoxybenzofuran (61)
Compound 61 was synthesized following the general procedure for the three-component Sonogashira/Cacchi type cyclization and was obtained as a bright yellow oil (340.6 mg, 63%): 1H NMR (400 MHz, CDCl3) δ 2.28 (s, 6H), 2.30 (s, 3H), 3.67 (s, 3H), 4.00–4.05 (m, 2H), 4.07 (s, 3H), 4.14–4.23 (m, 2H), 5.84 (s, 1H), 6.97–7.05 (m, 3H), 7.12 (d, J = 2.2 Hz, 2H), 7.19 (d, J = 13.7 Hz, 2H), 7.37 (dd, J = 8.3, 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 20.8, 21.3, 55.9, 56.4, 65.5, 104.2, 105.5, 110.9, 111.1, 115.4, 116.5, 117.6, 119.7, 120.7, 123.2, 128.6, 131.2, 134.1, 134.8, 140.2, 143.8, 145.4, 151.1, 151.7, 168.9, 168.9; HRMS calcd for C31H29O11 [M+H]+ 577.1704, found 577.1711.
2-(4-Acetoxy-3-methoxyphenyl)-3-(3,5-diacetoxyphenyl)-5-formyl-7-methoxybenzofuran (62)
Compound 61 (85.4 mg, 0.155 mmol) was dissolved in THF (0.3 mL), 10% aq HCl (62 μL) was added at 0 °C, and the reaction mixture was stirred for 20 min. An aqueous work-up was conducted according to a procedure described in the literature.26 Compound 62 was obtained as a yellow oil (75.8 mg, 92%): 1H NMR (400 MHz, CDCl3) δ 2.29 (s, 6H), 2.30 (s, 3H), 3.67 (s, 3H), 4.08 (s, 3H), 6.99–7.06 (m, 2H), 7.14 (d, J = 2.1 Hz, 2H), 7.18 (s, 1H), 7.37 (dd, J = 8.3, 1.8 Hz, 1H), 7.40 (s, 1H), 7.61 (s, 1H), 9.96 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 20.9, 21.3, 55.9, 56.4, 105.1, 111.1, 115.7, 116.7, 118.5, 119.8, 120.6, 123.4, 127.9, 131.5, 133.9, 133.9, 140.6, 146.2, 146.8, 151.3, 151.9, 152.2, 168.9, 191.8, 191.9; HRMS calcd for C29H25O10 [M+H]+ 533.1442, found 533.1443.
4,4-Dimethyl-2-(2-phenylbenzofuran-3-yl)cyclohex-2-enone (64)
Yellow solid, 54.3 mg (34%): mp 100–106 °C; 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 6H), 2.08 (t, J = 6.8 Hz, 2H), 2.73 (t, J = 6.8 Hz, 2H), 6.82 (s, 1H), 7.23 (t, J = 7.5 Hz, 1H), 7.31 (d, J = 8.8 Hz, 3H), 7.33–7.48 (m, 2H), 7.52 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 7.3 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 27.8, 33.9, 35.3, 36.3, 111.3, 112.5, 115.5, 120.1, 122.9, 124.7, 127.0, 128.6, 130.2, 130.3, 130.9, 151.9, 154.0, 160.7, 197.2; HRMS calcd for C22H20O2 [M+H]+ 339.1356, found 339.1348.
4.4. General procedure for the synthesis of benzofurans 67 and 68 by a three-component Sonogashira/Heck type coupling
2-Iodophenol (0.5 mmol) and bis(triphenylphosphine)palladium dichloride (10.5 mg, 3 mol %) were placed in a 5 mL microwave vial and purged with argon. Dry THF (0.5 mL) was added and the reaction mixture was stirred until the iodophenol completely dissolved. Then dry triethylamine (1.0 mL) and a 3.8M solution of CuI in dry triethylamine (0.5 mL) were added and the mixture allowed to stir for 10 min. Then 1.2 equiv of the corresponding alkyne was added; the vial was capped, purged with argon and placed in the microwave reactor for 30 min at 25 °C. The corresponding alkene (2.5 mmol), dry acetonitrile (2 mL), benzoquinone (0.5 mmol), and anhydrous KOAc (1.5 mmol) were added and the reaction mixture was heated in a microwave reactor at 60 °C for 25 min. After allowing the reaction to cool and a standard aqueous work-up, the reaction mixture was subject to column chromatography using ethyl acetate/hexane as the eluent to afford the desired products.
n-Butyl (E)-3-(2-phenylbenzofuran-3-yl)acrylate (67)
Orange amorphous solid, 88.9 mg (56%): 1H NMR (400 MHz, CDCl3) δ 1.00 (t, J = 7.4 Hz, 3H), 1.48 (h, J = 7.3 Hz, 2H), 1.67–1.76 (m, 2H), 4.26 (t, J = 6.6 Hz, 2H), 4.14–4.41 (m, 2H), 6.71 (s, 1H), 7.31–7.41 (m, 2H), 7.52 (dt, J = 13.4, 6.9 Hz, 4H), 7.78 (d, J = 8.0 Hz, 2H), 7.90 (d, J = 7.5 Hz, 1H), 8.05 (d, J = 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 14.0, 19.5, 31.0, 64.6, 111.8, 112.8, 115.5, 119.4, 121.2, 123.9, 125.5, 126.9, 128.7, 129.1, 129.9, 136.0, 154.7, 157.7, 167.6; HRMS calcd for C21H21O3 [M+H]+ 321.1485, found 321.1491.
n-Butyl (E)-3-[2-(cyclohex-1-en-1-yl)benzofuran-3-yl]acrylate (68)
Yellow amorphous solid, 113.6 mg (70%): 1H NMR (400 MHz, CDCl3) δ 0.99 (t, J = 7.4 Hz, 3H), 1.45 (dd, J = 14.8, 7.4 Hz, 2H), 1.68–1.82 (m, 6H), 2.32 (s, 2H), 2.52 (s, 2H), 4.24 (t, J = 6.6 Hz, 2H), 6.33 (s, 1H), 6.56 (d, J = 16.1 Hz, 1H), 7.26–7.31 (m, 2H), 7.45 (s, 1H), 7.81 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 16.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 19.4, 21.9, 22.5, 26.2, 26.6, 31.0, 64.4, 111.4, 111.8, 117.6, 120.9, 123.6, 124.9, 126.8, 128.4, 134.9, 136.7, 153.9, 160.6, 167.9; HRMS calcd for C21H24O3 [M+H]+ 324.1725, found 325.1799.
4.5. Elaboration of the bromo-containing benzofurans by Pd-catalyzed couplings
Ethyl 4-[5-(4-methoxyphenyl)-2-phenylbenzofuran-3-yl]benzoate (69)
In a 2 mL microwave vial, compound 16 (42.2 mg, 0.1 mmol), 4-methoxyphenylboronic acid (18.2 mg, 0.12 mmol), and tetrakis(triphenylphosphine)palladium (5.8 mg) were dissolved in a 1:1 mixture of EtOH/DMF (1.6 mL), then 1M aq Cs2CO3 (0.25 mL) was added and the mixture was heated in a microwave reactor at 120 °C for 20 min. The mixture was diluted with saturated aq Na2SO4 and extracted with ethyl acetate (3 × 15 mL), dried (MgSO4) and evaporated. Column chromatography using ethyl acetate/ hexanes (1:10) as the eluent afforded 35.6 mg (80%) of benzofuran 69 as a colorless solid: mp 127–129 °C; 1H NMR (400 MHz, CDCl3) δ 1.44 (dd, J = 7.4, 6.8 Hz, 3H), 3.85 (d, J = 0.5 Hz, 3H), 4.44 (q, J = 7.1 Hz, 2H), 6.97 (d, J = 8.2 Hz, 2H), 7.30–7.38 (m, 3H), 7.53 (t, J = 8.1 Hz, 3H), 7.63 (dt, J = 17.1, 5.9 Hz, 6H), 8.17 (d, J = 7.7 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 14.6, 55.6, 61.3, 111.5, 114.4, 116.9, 117.9, 124.5, 127.4, 128.6, 128.8, 128.9, 129.8, 129.9, 130.3, 130.4, 130.5, 134.2, 136.8, 137.9, 151.9, 153.6, 159.1, 166.6; HRMS calcd for C30H24O4 [M+H]+ 449.1747, found 449.1675.
Ethyl 3-[5-((E)-3-butoxy-3-oxoprop-1-en-1-yl)-2-phenylbenzofuran-3-yl]benzoate (70)
In a 2 mL vial, compound 17 (41.5 mg, 0.1 mmol), n-butyl acrylate (17.9 μL, 0.12 mmol), palladium acetate (0.6 mg) and SPhos (4.1 mg) were dissolved in DMF (0.5 mL). Then triethylamine (0.12 mL) was added and the mixture was heated at 100 °C for 24 h. The mixture was diluted with brine and extracted with ethyl ether (3 × 15 mL), dried (MgSO4) and evaporated. Column chromatography using ethyl acetate/ hexanes (1:10) as the eluent afforded 37.1 mg (81%) of benzofuran 70 as a cream colored solid: mp 77–80 °C; 1H NMR (400 MHz, CDCl3) δ 0.96 (t, J = 7.3 Hz, 3H), 1.42 (m, 5H), 1.69 (m, 2H), 4.20 (t, J = 6.7 Hz, 2H), 4.41 (q, J = 7.1 Hz, 2H), 6.41 (d, J = 15.9 Hz, 1H), 7.29–7.36 (m, 3H), 7.50–7.68 (m, 7H), 7.76 (d, J = 16.0 Hz, 1H), 8.14 (d, J = 7.7 Hz, 1H), 8.21 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 13.9, 14.5, 19.4, 31.0, 61.4, 64.6, 111.9, 116.7, 117.4, 120.1, 125.2, 127.2, 128.8, 129.1, 129.3, 129.5, 130.0, 130.2, 130.9, 131.8, 132.9, 134.4, 144.9, 152.2, 155.2, 166.4, 167.4; HRMS calcd for C30H28O5 [M+H]+ 468.1937, found 469.2010.
Supplementary Material
Acknowledgements
We gratefully acknowledge the National Science Foundation and the National Institutes of Health Kansas University Center of Excellence in Chemical Methodology and Library Development (P50 GM069663) for their generous financial support of this project. We would also like to thank the research groups of Professors Nicola Pohl and George Kraus for allowing us to use their CEM microwave reactors for the room temperature experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data Copies of 1H NMR and 13C NMR spectra of all novel compounds are available. Supplementary data related to this article can be found at http://dx.doi.org/xxx.
References
- 1.For recent selected examples, see: Boto A, Alvarez L. Heterocycles in Nat. Prod. Synthesis. 2011:99–152.. Ashwood VA, Field MJ, Horwell DC, Julien-Larose C, Lewthwaite RA, McCleary S, Pritchard MC, Raphy J, Singh L. J. Med. Chem. 2001;44:2276–2285. doi: 10.1021/jm010825z..
- 2.For some recent comprehensive reviews on the synthesis of benzofurans, see:Cacchi S, Fabrizi G, Goggiamani A. Org. Biomol. Chem. 2011;9:641–652. doi: 10.1039/c0ob00501k.. Yeung K-S, Peng S-X, Wu J, Hou X-L. Prog. Heterocycl. Chem. 2011;23:195–229.. Wu J. Five-membered Heterocycles: Benzofuran and Related Systems. Modern Heterocyclic Chemistry; 2011. pp. 593–633.. Mentwally MA, Abdel-Wahab BF, Al-Hiti GA. Curr. Org. Chem. 2010;14:48–64.. Miyata O, Takeda N, Naito T. Heterocycles. 2009;78:843–871.. De Luca L, Nieddu G, Porcheddu A, Giacomelli G. Curr. Med. Chem. 2009;16:1–20. doi: 10.2174/092986709787002826. and references therein..
- 3.a) Majumdar KC, Chattopadhyay B, Maji PK, Chattopadhyay SK, Samanta S. Heterocycles. 2010;81:517–584. [Google Scholar]; b) Cacchi S, Fabrizi G, Goggiamani A. Curr. Org. Chem. 2006;10:1423–1455. [Google Scholar]; c) Dudnik AS, Gevorgyan V. Catalyzed Carbon-Heteroatom Bond Formation. 2011:317–410. [Google Scholar]; d)For examples of Pd-catalyzed C-H activation leading to benzofuran formation, see:Ferreira EM, Zhang H, Stoltz BM. Tetrahedron. 2008;64:5987–6001. doi: 10.1016/j.tet.2008.01.052..
- 4.Arcadi A, Cacchi S, Del Rosario M, Fabrizi G, Marinelli F. J. Org. Chem. 1996;61:9280–9288. [Google Scholar]
- 5.Chaplin JH, Flynn BL. Chem. Commun. 2001:1594–1595. doi: 10.1039/b104624c. [DOI] [PubMed] [Google Scholar]
- 6.Flynn BL, Hamel E, Jung MK. J. Med. Chem. 2002;45:2670–2673. doi: 10.1021/jm020077t. [DOI] [PubMed] [Google Scholar]
- 7.Hu Y, Nawoschik KJ, Liao Y, Ma J, Fathi R, Yang Z. J. Org. Chem. 2004;69:2235–2239. doi: 10.1021/jo0303160. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y, Markina NA, Larock RC. Tetrahedron. 2009;65:8908–8915. doi: 10.1016/j.tet.2009.07.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Huang K-S, Wang Y-H, Li R-L, Lin M. Phytochemistry. 2000;54:875–881. doi: 10.1016/s0031-9422(00)00151-5. [DOI] [PubMed] [Google Scholar]; b) Li W-L, He K-K, Li Y, Hou Z-J. Acta Chimica Sinica. 2005;63:1607–1612. [Google Scholar]
- 10.a) Martinez C, Alvarez R, Aurrecoechea JM. Org. Lett. 2009;11:1083–1086. doi: 10.1021/ol8028687. [DOI] [PubMed] [Google Scholar]; b) Boyer A, Isono N, Lackner S, Lautens M. Tetrahedron. 2010;66:6468–6482. [Google Scholar]; c) Alvarez R, Martínez C, Madich Y, Denis JG, Aurrecoechea JM, de Lera AL. Chem. Eur. J. 2010;16:12746–12753. doi: 10.1002/chem.201001535. [DOI] [PubMed] [Google Scholar]
- 11.For selected recent reviews, see:aBubnov YN. Heterocycles. 2010;80:1–5.. McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447–2464. doi: 10.1039/b805701j..
- 12.For the most recent reviews, see:Chinchilla R, Najera C. Chem. Soc. Rev. 2011;40:5084–5121. doi: 10.1039/c1cs15071e.. Chinchilla R, Najera C. Chem. Rev. 2007;107:874–922. doi: 10.1021/cr050992x..
- 13.Weeratunga G, Jaworska-Sobiesiak A, Horne S, Rodrigo R. Can. J. Chem. 1987;65:2019–2023. [Google Scholar]
- 14.Larock RC, Lee NH. J. Org. Chem. 1991;56:6253–6254. [Google Scholar]
- 15.Amjad M, Knight DW. Tetrahedron Lett. 2004;45:539–541. [Google Scholar]
- 16.Gammill RB. Synthesis. 1979:901–903. [Google Scholar]
- 17.Jan N-W, Liu H-J. Org. Lett. 2006;8:151–153. doi: 10.1021/ol052638r. [DOI] [PubMed] [Google Scholar]
- 18.Fujikawa N, Ohta T, Yamaguchi T, Fukuda T, Ishibashi F, Iwao M. Tetrahedron. 2006;62:594–604. [Google Scholar]
- 19.Boudet N, Sase S, Sinha P, Liu C-Y, Krasovskiy A, Knochel P. J. Am. Chem. Soc. 2007;129:12358–12359. doi: 10.1021/ja074060h. [DOI] [PubMed] [Google Scholar]
- 20.Novák Z, Timári G, Kotschy A. Tetrahedron. 2003;59:7509–7513. [Google Scholar]
- 21.Kamitori Y, Hojo M, Masuda R, Yoshida T. Tetrahedron Lett. 1985;26:4767–4770. [Google Scholar]
- 22.Chiong HA, Daugulis O. Org. Lett. 2007;9:1449–1451. doi: 10.1021/ol0702324. [DOI] [PubMed] [Google Scholar]
- 23.Kamlet MJ, Dacons JC. J. Org. Chem. 1961;26:220–225. [Google Scholar]
- 24.Ohba H, Ikeda T, Kobayashi S, Taniguchi H. J. Chem. Soc., Chem. Commun. 1980:988–989. [Google Scholar]
- 25.Hu Y, Nawoschik K, Liao Y, Ma J, Fathi R, Yang Z. J. Org. Chem. 2004;69:2235–2239. doi: 10.1021/jo0303160. [DOI] [PubMed] [Google Scholar]
- 26.Fetter J, Bertha F, Poszávácz L, Simig G. J. Heterocycl. Chem. 2005;42:137–139. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.