

Abstract
An 8-plex version of an isobaric reagent for the quantitation of proteins using shotgun methods is presented. The 8-plex version of the reagent relies on amine-labeling chemistry of peptides similar to 4-plex reagents. MS/MS reporter ions at 113, 114, 115, 116, 117, 118, 119, and 121 m/z are used to quantify protein expression. This technology which was first applied to a test mixture consisting of 8 proteins and resulted in accurate quantitation, has the potential to increase throughput of analysis for quantitative shotgun proteomics experiments when compared to 2-plex and 4-plex methods. The technology was subsequently applied to a longitudinal study of cerebrospinal fluid proteins from subjects undergoing intravenous immunoglobulin treatment for Alzheimer’s disease. Results from this study identify a number of protein expression changes that occur in cerebrospinal fluid after 3 and 6 months of treatment compared to a baseline and compared to a drug washout period. A visualization tool was developed for this dataset and is presented. The tool can aid in the identification of key peptides and measurements. One conclusion aided by the visualization tool is that there are differences in considering peptide-based observations versus protein-based observations from quantitative shotgun proteomics studies.
Keywords: iTRAQ, shotgun proteomics, Alzheimer’s disease, immunotherapy, cerebrospinal fluid
1 Introduction
There are an increasing number of approaches to quantify expression profiles from complex protein mixtures, such as those present in cell lysates and tissue samples including cerebrospinal fluid. Each of the many available approaches has distinct advantages and drawbacks that should be considered carefully before being applied. The two most commonly used methods rely either on gel-based or solution-based separation of proteins and/or peptides followed by mass spectrometry. In the case of gel-based methods, the quantitation of protein expression can be performed by image analysis of proteins stained either before [1] (e.g. DIGE) or after [2] the gel-run (e.g. Sypro Ruby, silver, or Coomassie blue). Among the drawbacks of the gel approach is concern over the qualitative and quantitative reproducibility of this method among different laboratories as well as limits on the ability to study hydrophobic proteins and low abundance proteins [3]. Another important consideration is the need for post-separation analysis of bands or spots of interest using mass spectrometry. Despite these concerns, the approach of using gels to monitor changes in protein expression has some advantages compared to shotgun approaches; for example, there is a relatively low cost for most investigators to acquire and analyze a dataset and studies that rely on gels can be scaled to a relatively large number of samples.
On the other hand, quantitative shotgun approaches that rely on multi-dimensional liquid phase separations followed by mass spectrometry can rely on quantitation using the mass spectrometer [4]. The use of shotgun methods addresses important issues related to the ability to study a broad class of proteins and improved quantitative reproducibility when compared to gel-based studies. Among the challenges associated with shotgun methods are issues about the identification of the same proteins from experiment to experiment, the high cost of the instruments, and the need to analyze very large datasets which results in lower throughput. A further challenge in shotgun proteomics is the need to consider peptide-based observations against protein-based observations. Because shotgun experiments do not provide data on intact proteins, it can be difficult to accurately monitor changes in protein expression because of post-translational modifications, alternative splicing, and homologous gene sequences.
Many of the most effective quantitative shotgun techniques measure changes in expression between two samples and there has been a desire to increase the multiplexing capability of these types of studies in general. One key development in this regard has been the advent of multiplexing technologies such as the isobaric tags for relative and absolute quantitation that are commercially available. In one version of this technology [5], each of four isobaric reagents labels peptides derived from four different samples. In MS/MS, a reporter ion series is observed at 114, 115, 116, and 117 m/z with no adverse effect on the sequencing ion series and these four peaks permit quantitation of peptides from the original samples.
In this study we report on the application of an 8-plex reagent series that performs similarly and increases throughput of analyses by a factor of two when compared to 4-plex technologies. These reagents were used to study human cerebrospinal fluid (CSF) collected from two individuals at four different timepoints during a clinical trial of intravenous immunoglobulin (IVIg) treatment. IVIg passive immunotherapy [6] has emerged as one of the most promising methods for altering the amyloid-related Alzheimer’s disease (AD) neuropathology. Although clinical trials relying on active immunization [7] have not continued, the initial observations from those studies suggested the possibility of altering the molecular pathology of the disease and suggested the possibility of improving clinical outcomes. More recently, intense interest has been directed at passive immunization strategies typically relying on monoclonal antibody based treatments. As a complement to monoclonal antibody treatments, we have considered the use of IVIg as a treatment strategy. IVIg is a collection of the antibodies available from healthy blood donors and is known to contain auto-antibodies against beta-amyloid (Aβ) protein. Although the presence of anti-Aβ antibodies [8] was the initial rationale for the use of this treatment strategy for AD, an equally compelling motivation is the potential anti-inflammatory effects that may be exerted on the Aβ cascade in subjects.
In a clinical trial, we observed positive clinical outcomes in AD subjects treated with IVIg. The subjects underwent a 6-month treatment of IVIg followed by a 3-month washout period. We collected CSF samples at the outset of the trial, after 3mo and 6mo of treatment, and then after a subsequent 3-month drug washout, resulting in four longitudinal CSF samples from each subject enrolled in the trial. With the goal of better understanding changes in CSF protein expression that occur, we pursued a quantitative shotgun proteomics study of eight samples (four longitudinal samples from each of two subjects) using new 8-plex isobaric reagents for quantitative shotgun proteomics.
2 Materials and Methods
2.1 Eight-protein test mixture
Eight tubes containing 52μg of an 8-protein mix were prepared as shown in Table 1. Two of the eight proteins, carbonic anhydrase and ovotransferrin, were distributed in varying ratios across eight tubes, while the remaining six proteins were distributed in equimolar concentrations. The protein content of each tube was reduced, alkylated, digested and differentially labeled with 8-plex iTRAQ reagents per manufacturer’s instructions (Applied Biosystems).
Table 1.
8-Protein test mixture and their distribution in the final mixture
| Protein | Distribution in-g | Expected ratios |
|---|---|---|
|
| ||
| carbonic anhydrase | 10:15:5:2:18:1:19:10 | 1:1.5:0.5:0.2:1.8:0.1:1.9:1 |
| ovotransferrin | 10:5:15:18:2:19:1:10 | 1:0.5:1.5:1.8:0.2:1.9:0.1:1 |
| β-galactosidase | 10:10:10:10:10:10:10:10 | 1:1:1:1:1:1:1:1 |
| apo-transferrin | 6:6:6:6:6:6:6:6 | 1:1:1:1:1:1:1:1 |
| bovine serum albumin | 6:6:6:6:6:6:6:6 | 1:1:1:1:1:1:1:1 |
| β-lactoglobulin | 6:6:6:6:6:6:6:6 | 1:1:1:1:1:1:1:1 |
| lysozyme | 3:3:3:3:3:3:3:3 | 1:1:1:1:1:1:1:1 |
| α-lactalbumin | 3:3:3:3:3:3:3:3 | 1:1:1:1:1:1:1:1 |
One μg of pooled, labeled sample was dried down and resuspended in Buffer A (2% ACN, 0.1% TFA) and injected onto an LC-Packings/Dionex Ultimate™ system for reverse phase chromatography. Peptides were trapped onto a guard column (C18, 3μm, Dionex), washed in Buffer A for 10min, and subsequently eluted onto an analytical column (PepMap C18, 3μm, 150mm × 0.1mm, Dionex). Peptides were eluted over a 45min gradient from 2% to 35% Buffer B (85% ACN, 5% isopropanol, 0.1% TFA) at a flow rate of 500nl/min. Column eluent was mixed with 6mg/ml alpha-hydroxycinnamic acid at 1.2ml/min and spotted in 13sec intervals on a MALDI target plate using a Probot (LC-Packings/Dionex). MS and MS/MS spectra were acquired on a 4800 TOF/TOF Analyzer (Applied Biosystems). A maximum of 10 unique precursors with a minimum signal/noise of 60 were selected per spot for MS/MS with 1kV acceleration.
2.2 CSF samples
The human subjects portion of this study was carried out with Institutional Review Board approval and oversight at Cornell University. IVIg (Baxter Healthcare) was administered at one of two doses, 0.4g/kg/wk or 0.4g/kg/2wk, to subjects diagnosed with probable Alzheimer’s disease according to clinically accepted criteria. CSF was collected by lumbar puncture at the beginning of the treatment regimen. CSF was also collected after 3mo of treatment and after 6mo of treatment. After 6mo, IVIg treatment was stopped and a further CSF sample was collected after 3mo of drug washout (i.e. 9mo from the beginning of the study). Clinical data including mini mental status examination (MMSE) scores were recorded at each timepoint. CSF was collected into four tubes (automated cell counts were performed on the first tube of fresh CSF to ensure minimal blood contamination) prior to being frozen at −80 °C. The second tube of CSF was thawed. Approximately 45μg of protein from each of the eight CSF samples were denatured, reduced with TCEP (tris (2-carboxyethylphosphine)), alkylated with MMTS (methyl methanethiosulfonate) in 500mM triethylammonium bicarbonate (TEAB, pH8.5), trypsin digested and subsequently frozen until labeling [9].
2.3 Labeling and separation
All steps (Figure 1) were performed using buffers and components for maximum compatibility with iTRAQ reagent chemistry per manufacturer’s instructions. In preparation for labeling, 22μg of each digest was diluted in 1M TEAB pH 8.5. The 8-plex iTRAQ reagents (113–119, 121) were prepared by adding 70μl of ethanol to each vial and peptides from each sample were differentially labeled as follows: Subject A, dose 0.4g/kg/wk, timepoint 0-label 113, timepoint 3mo-label 115, timepoint 6mo-label 117, and timepoint 9mo-label 119; Subject B, dose 0.4g/kg/2wk, timepoint 0-label 114, timepoint 3mo-label 116, timepoint 6mo-label 118, and timepoint 9mo-label 121. The peptide-label mixtures were incubated for 2hr at room temperature, at which point 0.5M taurine was added to quench any un-reacted iTRAQ reagent (30min). The 8 iTRAQ reagent-labeled samples were then pooled into a single vial for cation exchange chromatography. The reagent added an isobaric 305.1Da increase in mass to the peptides assuming each peptide was labeled once.
Figure 1.

A schematic depiction of the workflow for using 8-plex isobaric reagents in shotgun proteomics studies. The basic workflow is the same as used for 4-plex isobaric reagents.
2.4 Strong Cation Exchange (SCX) Chromatography
The pooled, labeled sample was diluted to 8mL with 10mM potassium phosphate, 25% acetonitrile (pH 2.1). The sample was then injected onto an Agilent 1100 with a PolySulfoethylA column (C18, 5μm, 200Å, 4.6mm × 100mm, Nest Group) for separation. Peptides were eluted from the column using an 11min gradient from 0–500mM KCl in 10mM potassium phosphate, 25% acetonitrile, pH 3 at a flow rate of 1ml/min into 19 distinct 1min size fractions. The fractions were collected and dried down in a vacuum centrifuge.
2.5 Reverse Phase Chromatography
SCX fractions were reconstituted in 2% ACN, 1% TFA in dI water. Seven less abundant fractions, as determined by inspection of SCX chromatograms, were pooled resulting in a total of 13 fractions for reverse phase chromatography. In contrast to the 8 protein test system, these samples were injected into a Tempo LC MALDI Spotting System, a single-stage HPLC and spotter (Applied Biosystems), equipped with a ProteCol (C18, 3μm, 120Å, 300μm × 10mm) trap column (SGE) and an Onyx Monolithic (C18, 150mm × 0.1mm) analytical column (Phenomenex). Samples eluting at 3μl/min during a 98% Buffer A (2% ACN, 0.1% TFA) to 45% Buffer B (98% ACN, 0.1% TFA) gradient over 30min were mixed 1:1 with 7mg/ml alpha-hydroxycinnamic acid spiked with 10mM ammonium phosphate and 10fmol/ml Glu1-Fibrinopeptide B (internal calibrant) and spotted every 3sec onto Opti-TOF LC/MALDI 123×81mm plates (Applied Biosystems). 599 300nl spots were collected per fraction with a maximum of two SCX fractions on each plate.
2.6 Mass spectrometry and database search
MALDI plates were analyzed on a 4800 MALDI TOF/TOF Analyzer. MS spectra were acquired in positive ion reflector mode over a mass range of 800 to 4000 m/z with 1200 laser shots per spot. MS data were processed with internal calibration and filtered through the following criteria to determine a maximum of 15 precursors for MS/MS per spot: minimum signal/noise of 25 and fraction-to-fraction mass tolerance of 200ppm. Tandem MS spectra were acquired with 2500 laser shots and a 200 resolution (FWHM) mass window per precursor. Fragmentation was induced with 2kV collision energy and air at 1.2×10−6 torr for the collision gas. MS/MS data were processed with default calibration.
The resulting peak lists were submitted for database sequence searches using GPS Explorer v3.6 software, relying on Mascot v2.1.03 software, and searched against the NCBInr database (08/15/2006 download date) using the following parameters: taxonomy human, trypsin cleavage, 50ppm precursor mass tolerance, 0.3Da MS/MS mass tolerance, 1 maximum missed cleavage, and 5 variable modifications (8-plex K, 8-plex Nterm, 8-plex Y, oxidation (M), MMTS (C)). Only the top 1000 hits per plate were reported. To measure false positive identification rates, a copy of the reversed NCBInr sequences was generated and searched with the same parameters.
Results from peptides with MS/MS GPS scores of >95% and >99% confidence are reported.
2.7 Data Analysis
The calculation for isotopic carryover was carried out in a method to similar to that previously reported [9] with correction, normalization, and scaling performed in R. The quality control data, available from the manufacturer of the reagents, were processed using Microsoft Excel. The resulting quantitative values were then normalized using the median.
2.8 Western blot analysis
Western blot analysis against clusterin/apolipoprotein J (ApoJ) was performed with 25μl of the following samples: Subject A, timepoint 0 (8-plex 113), timepoint 3mo (8-plex 115); Subject B, timepoint 0 (8-plex 114), timepoint 3mo (8-plex 116). An N-terminal antibody (Abcam, cat# ab35076) specific for amino acids 46–60 of human ApoJ was used at a 1:400 dilution and probed with an anti-rabbit IgG secondary antibody (Sigma, cat# A2556) at 1:30000. A duplicate blot was probed with a C-terminal antibody (Abcam, cat# ab39991) specific for the last 14 amino acids at a 1:1600 dilution and an anti-goat IgG secondary antibody (Sigma, cat# A4062) was used at a 1:30000 dilution. Enhanced chemifluorescence was detected on a FujiFilm FLA-3000 and bands were quantified using Multi-Analyst software version 1.1 (Bio-Rad). The mean count parameter was used to quantify band intensity.
3 Results and Discussion
3.1 Eight-protein test mixture
By studying the reporter ions in the low mass region of the MS/MS spectrum, one can quantify the amount of protein available in a given sample. Reporter ions are expected at 113, 114, 115, 116, 117, 118, 119, and 121 m/z (Figure 2). 112 and 120 m/z are excluded as reporter ions because these are the expected locations of the arginine and phenylalanine immonium ions. We tested the ability of the reagents to quantify a range of relative protein ratios in the presence of a 1:1:1:1:1:1:1:1 background.
Figure 2.

MS/MS of 2708.26 m/z from carbonic anhydrase from the test mixture of proteins quantified using 8-plex isobaric reagents. A zoom of the reporter ion region demonstrates the use of the 113, 114, 115, 116, 117, 118, 119, and 121 m/z ions to quantify relative protein abundance. Ions at 112 and 120 m/z are immonium ions for arginine and phenylalanine, respectively.
Figure 2 shows the MS/MS spectrum for a precursor ion (2708.26 m/z) from carbonic anhydrase. The expected ratios for this protein are 1:1.5:0.5:0.2:1.8:0.1:1.9:1, the calculated ratios (averaged over 12 peptides) are 1:1.45:0.50:0.19:1.92:0.13:1.75:0.94 with a standard deviation of 12.4%. The ovotransferrin expected ratios are 1:0.5:1.5:1.8:0.2:1.9:0.1:1, the calculated ratios over 24 peptides are 1:0.55:1.28:1.61:0.26:1.70:0.22:0.87 with a standard deviation of 14.1%. Average ratios for 203 peptides from remaining six proteins distributed in equimolar concentrations were measured and calculated at 1:1.04:0.93:0.90:1.17:0.92:1.16:1.06 with a standard deviation of 17.0%.
3.2 CSF
During the course of the IVIg treatment, we observed an increase in MMSE score of +4 points over the first 6mo which then declined by 3 points during the drug washout period (for a net change of +1 during the 9mo of the study). During this time, the expected change in MMSE would be −1.5 points over the first 6mo or a net of −2.5 points over 9mo. To identify and study changes in protein expression in cerebrospinal fluid, we used changes in expression quantified by the 8-plex iTRAQ reagents.
After a search against the NCBInr database, we observed 1187 peptides that matched with 95% confidence or greater (as defined by the GPS Explorer software) and 926 peptides that matched with 99% confidence or greater out of a total of 17,765 MS/MS (see Supplementary Information). These 1187 and 926 peptides correspond to 167 and 106 proteins, respectively, based on unique accession numbers in the NCBInr database. A peptide based search against the reverse NCBInr database yielded 8 matches at 95% confidence and 1 hit at 99% confidence suggesting false positive discovery rates of 0.8% and 0.1% for the 95% and 99% confidence levels in terms of peptide matches [10]. There were 7 protein-based matches at 95% in the reverse database and 1 match at the 99% confidence level in the reverse database for a 4% and a 0.9% false discovery rate at the protein level. Based on the MS/MS selection criteria and the resolution obtained by chromatography, we observed that additional precursor selection and subsequent MS/MS collection would not add significantly to the number of matches because MS/MS acquisition was not limited by the number precursor ions meeting the criteria.
3.3 Possible disease-modifying effects
We first studied the expression of all CSF peptides for each of the eight samples to identify changes in CSF protein expression that correlated with the clinical outcomes. Such changes are possible indicators of any disease-modifying effects that result from IVIg in these two Alzheimer’s disease subjects. To identify relevant peptides, we looked for changes between the 0-, 3- and 6-month timepoints that were consistent: either monotonically increasing or monotonically decreasing and without regard to the amount of the increase or decrease. We then considered only the subset of those peptide hits that had a trend reversal at the 9-month timepoint corresponding to the time at which drug washout occurred and at which we observed a reversal in the clinical outcomes. In subject A, we observed 69 peptides that met the criteria of decreasing expression during treatment and increasing expression upon washout. In subject B, 66 peptides met these criteria. Only 10 of these peptides are common to both subjects and the proteins and accession numbers corresponding to these peptides are shown in Table 2. Similarly, we observed 200 peptides in subject A that demonstrated increasing expression during treatment and decreasing expression upon washout, and 117 in subject B. Only 32 of these two sets of peptides were common to both subjects and the protein names and accession numbers corresponding to these peptides are shown in Table 3. We highlight that the protein names presented in Tables 2 and 3 reflect aggregate observations over multiple peptides. We discuss in the next section the benefits of considering peptide-based observations over protein observations, but present the data in Tables 2 and 3 as protein information to simplify the explanation of possible disease modifying effects from IVIg treatment.
Table 2.
Proteins observed to decrease in expression with IVIg treatment
| Protein Name | NCBI accession number |
|---|---|
|
| |
| albumin | gi|4502027 |
| α-2-macroglobulin | gi|66932947 |
| chain B, Mhc class I H-2db | gi|4699634 |
| clusterin / ApoJ | gi|32891795 |
| hemopexin | gi|11321561 |
| HP protein | gi|78174390 |
| plasma glutamate carboxypeptidase | gi|18088384 |
Table 3.
Proteins observed to increase in expression with IVIg treatment
| Protein Name | NCBI accession number |
|---|---|
|
| |
| α-1-antitrypsin | gi|1942629 |
| albumin | gi|4502027 |
| α-1-β-glycoprotein | gi|69990 |
| α-2-macroglobulin | gi|66932947 |
| apolipoprotein E | gi|178853 |
| beta-trace | gi|410564 |
| cystatin C | gi|73535286 |
| α-1-Pi-Pittsburgh & S195a trypsin complex | gi|34810822 |
| antibody-antigen complex | gi|15825713 |
| antibody | gi|42543654 |
| antibody | gi|42543650 |
| chromogranin B | gi|62089004 |
| clusterin / ApoJ | gi|32891795 |
| complement component 2 | gi|55961814 |
| insulin-like growth factor binding protein 7 | gi|42542649 |
| ITI heavy chain H4 | gi|13432192 |
| neuronal cell adhesion molecule isoform A | gi|81158226 |
| neurotrimin | gi|7705413 |
| secretogranin-2 / chromogranin C | gi|134464 |
| transferrin | gi|4557871 |
| unnamed protein product | gi|16554039 |
Many of the proteins with changes in expression are consistent with our expectations from IVIg treatment of AD subjects based on the improvement observed in the subjects. For example, with IVIg treatment a decrease in the expression of hemopexin was observed that was mitigated during drug washout. Others [11] used a 2DE proteomics approach to observe a decrease in CSF hemopexin in AD subjects relative to non-demented controls. This observation that lower levels are associated with an individual being more like a control rather than AD is consistent with our observations on hemopexin levels decreasing with IVIg treatment resulting in improved MMSE scores in AD patients. We also observed a decrease in α-2-macroglobulin, a protein genetically associated with AD [12], which has been implicated based on its ability to mediate the clearance and degradation of Aβ. A decrease in expression in CSF with IVIg treatment is consistent with decreasing amounts of Aβ deposits in treated subjects because lower levels of Aβ may be present which require less α-2-macroglobulin for clearance.
We observed an increase in the expression of a number of proteins upon IVIg treatment that was mitigated upon washout. As one might expect from IVIg treatment, a number of proteins that were increased are antibody-based molecules. Alpha-1β-glycoprotein is a protein that was elevated in this study during IVIg treatment and has been observed by others [13] as a CSF protein that is decreased in AD relative to controls. Similarly, Eder and colleagues [14] have observed an increase in chromogranin B in control subjects relative to AD. Other proteins with a known association to Alzheimer’s disease that changed upon IVIg treatment include apolipoprotein E, α-1-antitrypsin, complement component 2, and transferrin [15].
Using a linear-mixed effects model, we calculated the significance value for the quantitation of all peptides that satisfy a P<0.05 for each patient individually and that are consistent among both subjects. We observe 51 peptides corresponding to 29 proteins that meet this criteria as listed in Table 4 (see also Supplementary Information). We can further limit our observations by requiring that the changes observed be significant in each subject individually at P<0.05 and also that the changes are not significantly different (P<0.05) across both subjects. There are only 4 peptides that fit in this category, two of which are fragments of antibody molecules (as might be expected from IVIg treatment) and two of which are derived from one unknown protein product (accession number gi|16554039). Note that this last group of peptides requires that protein expression be quantitatively similar between both subjects at all timepoints, perhaps not a reasonable assertion given individual variability. The study to two subjects that respond to IVIg treatment does not provide enough statistical power for conclusions without validation using other methods; however, such time-course studies do offer the possibility of reducing the challenges associated with subject variability because each subject can serve as her/her own control.
Table 4.
Statistically-significant changes (linear-mixed effects model)
| Protein Name | NCBI accession number |
|---|---|
|
| |
| α1-antitrypsin | gi|4502027 |
| albumin | gi|4557871 |
| α-1-antichymotrypsin | gi|55958974 |
| amyloid precursor-like protein 1 | gi|229585 |
| angiotensinogen | gi|15825713 |
| antibody | gi|32891795 |
| apolipoprotein A-I | gi|4557287 |
| apolipoprotein E | gi|15930186 |
| calsyntenin 1 | gi|178853 |
| albumin complex | gi|34810822 |
| cystatin C | gi|73535286 |
| α-1-Pi-Pittsburgh & S195a trypsin complex | gi|1709301 |
| antibody-antigen complex | gi|55962140 |
| antibody | gi|177933 |
| antibody | gi|229526 |
| clusterin / ApoJ | gi|15029894 |
| hemopexin | gi|22219206 |
| antibody | gi|1942629 |
| antibody | gi|229528 |
| kallikrein 6 | gi|11321561 |
| N-cadherin | gi|253483 |
| orosomucoid 1 | gi|4557321 |
| prostaglandin D2 synthase | gi|456784 |
| protein Len, Bence-Jones | gi|55669910 |
| protein Rei, Bence-Jones | gi|55962673 |
| serpin peptidase inhibitor | gi|21669465 |
| albumin | gi|99031801 |
| transferrin | gi|16554039 |
| unnamed protein product | gi|16974102 |
3.4 Visualization tool
A key challenge that has been mentioned previously [16] is the need to balance peptide-based observations with protein-based observations in shotgun experiments. In the case of microbes, one may infer the expression level change of the intact protein based on an appropriate average of the changes for observed peptides. Such an inference assumes that all of the peptide-based observations accurately reflect the intact protein. Given the possibility of homologous genes resulting in two or more proteins that share identical and non-identical peptides as well as the possibility of certain post-translational modifications which may be present only on specific peptides, the averaging of observations from peptides may not accurately reflect expression changes at the protein level. In the case of higher organisms which have an even higher incidence of post-transcriptional control, such as alternative splicing events, as well as post-translational controls, such as protein/proteolytic processing (e.g. preproinsulin, proinsulin, and insulin), the assumption that all peptide observations reflect a given intact protein may be further flawed. In the case of cerebrospinal fluid, there are known to be a wide variety of possible polymorphisms, specific proteolysis, alternate splicing, and other post-translational modifications [17] that can make the “averaging” of the data for a given protein over multiple peptides inappropriate. One example is the amyloid precursor protein that is differentially processed resulting in a toxic peptide that accumulates in neuritic plaques in AD. Measurements from the differentially expressed (in AD) Aβ1-40 and Aβ1-42 fragments will both contribute to protein based measurements of Aβ. As another example, it has been found that proteolytic fragments of albumin can be useful markers of neurologic disease [15]. Protein fragments and their differential expression may be more discernable in the context of 2DE analyses where the measurement of different forms of a given protein are inherent in the technique resulting in separate quantification because protein identification is decoupled from quantitation; whereas, in shotgun experiments, the trend has been to allow protein identification to drive the quantitative analysis.
We prepared a visualization tool using the R programming environment to facilitate the visualization of peptide level measurements in the context of protein based observations. Data on peak areas, accession numbers, peptide start and end sites was exported to Microsoft Excel. These data were converted into a visual format for each protein (based on accession number) as shown in Figure 3 for albumin. All data displayed in Figure 3 is on a log base 2 scale.
Figure 3.

Visualization tool example for albumin developed in the R environment. As discussed in detail in the text, 3A are average peptide expression changes for both subjects, 3B and 3D are protein average expression changes for subjects A and B, 3C and 3E are peptide expression changes observed for subjects A and B, 3F is graphical representation of peptide expression changes using a heatmap to indicate increases or decreases in expression where the horizontal axis of the figure shows the amino acid position along the length of the protein.
The bottom panel of Figure 3(3F) displays a graphical representation of observed expression changes for all observed peptides associated with a given protein. The protein name and accession number are listed in Figure 3F, as is a heatmap. The heatmap intensity from red to yellow to green reflects the largest observed changes in expression across all peptides for the protein. Also in Figure 3F is a graphical representation of all of the peptide changes observed across the length of the protein. These changes are represented by rectangles consisting of eight boxes that are positioned according to the sequence position of that peptide from N-terminus to C-terminus running from left to right. Thus, a peptide hit that begins at residue 25 and continues for 20 residues, will have a rectangle consisting of eight boxes beginning at 25 and ending at 45. In this study, the 8 labels were used to study two subjects at four timepoints. The eight boxes provide information about each of the four timepoints across the two subjects (upper row of four boxes is Subject A and the lower row of boxes is Subject B). All data are presented as ratios against the 113 and 114 label (the 0 timepoint for each subject).
Figures 3C and 3E show the change in expression ratio for subjects A and B (respectively) for every peptide observed for a given protein. The initial peak area ratio is always 0 for the first timepoint because the data are plotted log base 2. Plot colors in Figures 3C and 3E indicate peptide expression similarity as determined by two-dimensional self-organizing maps clustering performed using the “som” package available in R. Figures 3B and 3D are the log average change for all observed peptides for subjects A and B, respectively. Figure 3A is the average of the observed expression change for every peptide within each subject with the black and red curves corresponding to subjects A and B, respectively.
Figure 3 is useful as a tool for visualizing specific observations about peptide expression changes, relative to other peptides of the same protein, observed in the same shotgun experiment. Furthermore, in cases involving two longitudinal sample sets of four timepoints each, such as the one studied here, the visualization of the data using this approach can aid in the identification of outlier data points or changes unique to one individual. In principle, the visualization tool can be extended to single subjects over eight timepoints, four subjects at two timepoints and other combinations. An additional benefit of viewing data as two sets of four timepoints is that the visualization tool can be more easily applied to studies that have relied on 4-plex reagents.
To better illustrate the issue of peptide versus protein-based observations and the benefits of data visualization tools, consider ApoJ, a protein that changed expression in this study that has been linked to AD [18]. Apolipoprotein J (also known as clusterin) appears to be both increasing and decreasing upon treatment (Tables 2 and 3). Data for clusterin using the visualization tool is shown in Figure 4. Some of the observed peptides (position 69–78) clearly show a decreased expression upon treatment, whereas other peptides (positions 199–214 and positions 326–336 to a lesser extent) show an increased expression upon treatment, at least in subject A. There are several possible reasons for the observed differences in expression at different positions along clusterin. Some possible explanations are individual specific responses to IVIg treatment, differential proteolytic processing of fragments during IVIg treatment, technical error, and others. The observation to note is that the N-terminal peptides (69–78) indicate a slight decrease in ApoJ expression for both subjects after 3mo of IVIg treatment whereas the C-terminal peptides (326–336, 386–443) indicate a general increase in ApoJ expression for subject A and no expression change for subject B after 3mo of treatment.
Figure 4.

Visualization tool applied to observed expression changes for clusterin / apolipoprotein J. As discussed in detail in the text, 4A are average peptide expression changes for both subjects, 4B and 4D are protein average expression changes for subjects A and B, 4C and 4E are peptide expression changes observed for subjects A and B, 4F is graphical representation of peptide expression changes using a heatmap to indicate increases or decreases in expression where the horizontal axis of the figure shows the amino acid position along the length of the protein.
To make a qualitative comparison between the shotgun approach and another analytical method, we performed a Western analysis on clusterin using commercially available antibodies to the N-terminal and C-terminal regions of the protein (Figure 5). Using an antibody against an N-terminal epitope, we measured no significant change in ApoJ expression for subject A and a decrease in ApoJ expression for subject B after 3mo of IVIg treatment. With the C-terminal antibody, we observed no expression change in subjects A and B after 3mo of IVIg treatment. The observations of expression changes using Western are consistent with the 8-plex iTRAQ measurements, although they are not quantitatively the same. Such an observation reinforces the importance of considering both peptide as well as protein-level expression changes in shotgun experiments as well as the importance of applying multiple analytical measurements to confirm protein expression changes.
Figure 5.
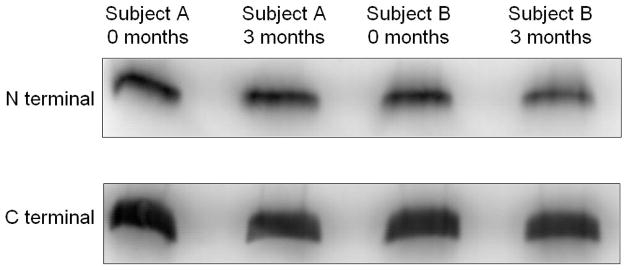
Western analysis of clusterin / apolipoprotein J using primary antibodies against N-terminal and C-terminal epitopes.
4.0 Concluding Remarks
We report on the use of an isobaric 8-plex reagent that can be used to quantify protein and peptide expression in shotgun proteomics experiments. The reagent uses similar chemistry for labeling as other commercially available reagents. The technology was applied to the study of changes in CSF protein expression from AD subjects undergoing IVIg treatment. The subjects had positive clinical outcomes and the observed changes in protein expression were generally consistent with these outcomes and many proteins with a known relationship to AD pathology were characterized. In such quantitative, time-course proteomics studies, a visualization tool to assess individual peptide quantitation relative to protein-level quantitation can provide further insights into the data.
Supplementary Material
Acknowledgments
We thank Baxter Healthcare for providing IVIg for the study. We gratefully acknowledge the National Institutes of Health, the New York State Office of Science, Technology, and Academic Research, and the Institute for the Study of Aging for support of this work.
Abbreviations
- AD
Alzheimer’s disease
- CSF
cerebrospinal fluid
- IVIg
intravenous immunoglobulin
- MMSE
mini mental status examination
References
- 1.Tonge R, Shaw J, Middleton B, Rowlinson R, et al. Validation and development of fluorescence two-dimensional differential gel electrophoresis proteomics technology. Proteomics. 2001;1:377–396. doi: 10.1002/1615-9861(200103)1:3<377::AID-PROT377>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Choe LH, Lee KH. Quantitative and qualitative measure of intralaboratory two-dimensional protein gel reproducibility and the effects of sample preparation, sample load, and image analysis. Electrophoresis. 2003;24:3500–3507. doi: 10.1002/elps.200305614. [DOI] [PubMed] [Google Scholar]
- 3.Lee KH. Proteomics: a technology-driven and technology-limited discovery science. Trends Biotechnol. 2001;19:217–222. doi: 10.1016/s0167-7799(01)01639-0. [DOI] [PubMed] [Google Scholar]
- 4.McDonald WH, Yates JR. Shotgun proteomics: integrating technologies to answer biological questions. Curr Opin Mol Ther. 2003;5:302–309. [PubMed] [Google Scholar]
- 5.Ross PL, Huang YN, Marchese JN, Williamson B, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Dodel RC, Du Y, Depboylu C, Hampel H, et al. Intravenous immunoglobulins containing antibodies against beta-amyloid for the treatment of Alzheimer’s disease. J Neurol Neurosurg Psych. 2004;75:1472–1474. doi: 10.1136/jnnp.2003.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schenk D, Barbour R, Dunn W, Gordon G, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 8.Weksler ME, Relkin N, Turkenich R, LaRusse S, et al. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Exp Gerontol. 2002;37:943–948. doi: 10.1016/s0531-5565(02)00029-3. [DOI] [PubMed] [Google Scholar]
- 9.Aggarwal K, Choe LH, Lee KH. Quantitative analysis of protein expression using amine-specific isobaric tags in Escherichia coli cells expressing rhsA elements. Proteomics. 2005;5:2297–2308. doi: 10.1002/pmic.200401231. [DOI] [PubMed] [Google Scholar]
- 10.Huttlin EL, Hegemen AD, Harms AC, Sussman MR. Prediction of error associated with false-positive rate determination for peptide identification in large-scale proteomics experiments using a combined reverse and forward peptide sequence database strategy. J Proteome Res. 2007;6:392–398. doi: 10.1021/pr0603194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castano EM, Roher AE, Esh CL, Kokjohn TA, et al. Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol Res. 2006;28:155–163. doi: 10.1179/016164106X98035. [DOI] [PubMed] [Google Scholar]
- 12.Blacker D, Wilcox MA, Laird NM, Rodes L, et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19:357–360. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- 13.Puchades M, Hansson SF, Nilsson CL, Andreasen N, et al. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer’s disease. Brain Res Mol Brain Res. 2003;118:140–146. doi: 10.1016/j.molbrainres.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Eder U, Leitner B, Kirchmair R, Pohl P, et al. Levels and proteolytic processing of chromogranin A and B and secretogranin II in cerebrospinal fluid in neurological diseases. J Neural Transm. 1998;105:39–51. doi: 10.1007/s007020050036. [DOI] [PubMed] [Google Scholar]
- 15.Finehout EJ, Franck Z, Choe LH, Relkin N, et al. Cerebrospinal fluid proteomic biomarkers for Alzheimer’s disease. Ann Neurol. 2007;61:120–29. doi: 10.1002/ana.21038. [DOI] [PubMed] [Google Scholar]
- 16.Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data: the protein inference problem. Mol Cell Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Fishman RA. Cerebrospinal Fluid in Diseases of the Nervous System. 2. W. B. Saunders Company; Philadelphia: 1992. [Google Scholar]
- 18.Nilselid AM, Davidsson P, Nagga K, Andreasen N, et al. Clusterin in cerebrospinal fluid: analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem Int. 2006;48:718–728. doi: 10.1016/j.neuint.2005.12.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.