

Summary
Transcription regulatory networks governing the genesis, maturation and maintenance of vertebrate brain serotonin (5–HT) neurons determine the level of serotonergic gene expression and signaling across the lifespan. Recent work suggests alterations in these networks can cause behavioral and physiological pathogenesis in mice. Here, we synthesize findings from vertebrate loss and gain of function studies to build a new model of the transcriptional regulatory networks that specify 5–HT neurons during fetal life, integrate them into CNS circuitry in early postnatal life and maintain them in adulthood. We then describe findings from animal and human genetic studies that support possible altered activity of serotonergic regulatory networks in the etiology of mental illness. We conclude with a discussion of the potential utility of our model, as an experimentally well–defined molecular pathway, to predict and interpret the biological impact of rare human variation that may be discovered in the network.
… “it is possible that the 5–HT in our brains plays an essential part in keeping us sane…”
Sir John Gaddum, 1954
Ever since the heady days of Sir John Gaddum’s animal and self–administration studies of the serotonergic hallucinogen, lysergic acid diethylamide, there has been an incessant quest for an understanding of brain serotonin (5–HT) system function and its importance in behavior and neuropsychiatric disease. An extensive literature that began to appear soon after Gaddum’s and others’ groundbreaking studies of 5–HT 1, 2 has implicated altered 5–HT signaling in the etiology of an apparently disparate group of neurodevelopmental disorders including depression, anxiety, obsessive compulsive disorder, disorders of energy balance, autism, schizophrenia and sudden infant death syndrome 3. The wide range of disorders linked to serotonergic dysfunction is not surprising as the brain 5–HT transmitter system innervates nearly all cytoarchitectonic regions of the brain and spinal cord and has been implicated in the modulation of seemingly every human behavior and physiological process orchestrated by the nervous system. But what is quite striking, in view of this impressive breadth of influence, is that the 5–HT system originates from an exceedingly small proportion of neurons genetically programmed to make and use 5–HT as a neurotransmitter. 5–HT neuron numbers are estimated to be 26,000 in the mouse brain 4 and roughly 300,000 in the human brain 5, 6. The prevailing hypothesis of the role of 5–HT neurons in mental health disorders is that genetic determinants and their interaction with environmental factors program too little or too much 5–HT signaling emanating from these small numbers of neurons at critical developmental stages, which consequently disrupts the developmental trajectories of CNS circuit formation and thereby imposes susceptibility to mental illness 7. As the genetic determinants of early 5–HT system function, it is straightforward to understand why the regulatory programs that implement serotonergic neurogenesis and consequently determine the level of brain 5–HT signaling have gained considerable attention over the past decade for their potential biological relevance to neuropsychiatric disorders 8–11
Here, we review recent progress in understanding the regulatory programs governing 5–HT neuron development and function. We assemble from recent loss and gain of function findings a model of the transcriptional network constituting the 5–HT regulatory programs that direct the generation, maturation and maintenance of brain 5–HT neurons. We then discuss the potential utility of the network, as an experimentally well–defined molecular pathway, in understanding the elusive mechanisms of 5–HT system–related neuropsychiatric disease pathogenesis.
5–HT neuron identity
The small numbers of brain 5–HT neurons express hundreds of genes that encode generic molecular and cellular characteristics shared by all types of neurons such as the ion channels necessary for action potential generation and proteins required for release of neurotransmitter at synapses. However, it is the selection for expression of a serotonergic–type gene battery comprising tryptophan hydroxylase 2 (Tph2), aromatic amino acid decarboxylase (Aadc), plasma membrane serotonin transporter (Slc6a4 or Sert), vesicular monoamine transporter (Slc18a2 or Vmat2), monoamine oxidase A and B (MaoA, B), 5–HT1a autoreceptor (Htr1a), 5–HT1b autoreceptor (Htr1b) and the several genes encoding the synthetic and recycling enzymes for tetrahydrobiopterin cofactor (BH4) synthesis that endows these cells with the specialized capacity to synthesize, transport, autosense, inactivate through reuptake and metabolize 5–HT; hence establishing its physiological function as a neurotransmitter (Fig. 1). Although each of these 5–HT type genes is essential for serotonergic function none of them uniquely define brain 5–HT neuron identity as each of them, even the rate–limiting 5–HT synthetic enzyme, TPH2, is expressed in other neural and non–neural 12 cell types. What determines 5–HT neuron identity, therefore, is the integration of aggregate serotonergic characteristics encoded by the serotonergic–type gene battery with generic neuronal characteristics 8. The set of genetic instructions that coordinates expression of serotonergic–type and neuronal–type characteristics constitutes an intrinsic regulatory program directing brain 5–HT neuron identity. Cell fate regulatory programs comprise complex networks of interacting transcription factors, associated signaling proteins, and the cis regulatory sequences through which combinations of transcription factors control target genes 13. A particular regulatory program specifies and then progressively transforms proliferating neural progenitors into neurons of a particular identity, such as one able to synthesize 5–HT. The regulatory program controlling serotonergic neurogenesis, however, is not sufficient to equip 5–HT neurons with the capacity to exert widespread neuromodulatory influence on CNS circuitry. Additional intrinsic programs, therefore, likely regulate 5–HT neuron maturation during which these neurons are physiologically connected with 5–HT receptor bearing post–synaptic circuitry in widely distributed serotonergic axonal termination zones. Recent findings to be discussed below, also demonstrate that intrinsic serotonergic regulatory programs act in adulthood to maintain serotonergic signaling and plasticity.
Figure 1. Serotonergic–type neuron identity.
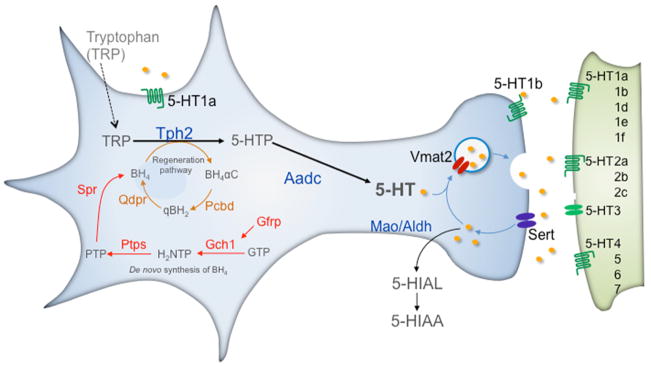
5–HT neurons coexpress a gene battery encoding 5–HT synthetic (Tph2, Aadc, Gch1, Gfrp, Ptps, Qdpr), reuptake (Sert), vesicular transport (Vmat2), autoreceptor signaling (Htr1a, Htr1b) and metabolism (Maoa, Maob) proteins. Tetrahydrobiopterin (BH4), an essential cofactor for Tph2 in the synthesis of 5–HTP, is synthesized (red pathway) de novo from guanosine triphosphate (GTP). It is also recycled through a regeneration pathway (brown). Aldehyde dehydrogenase (Aldh) converts 5–Hydroxyindolealdehyde into 5–Hydroxyindoleacetic acid (5–HIAA). Following release, 5–HT modulates 5–HT neuron firing through somatodendritic Htr1a autoreceptors, 5–HT release from the pre–synaptic terminal through Htr1b autoreceptors and stimulates neurotransmission through post–synaptic 5–HT receptors (5–HT1–7). Tph2, Tryptophan hydroxylase 2; Aadc, Aromatic L–amino acid decarboxylase; Sert, Serotonin transporter; Vmat2, vesicular monoamine transporter 2; Mao A,B, Monoamine oxidases A and B; Gch1, GTP cyclohydrolase 1; Gfrp, GTP cyclohydrolase I feedback regulator; Ptps, 6–pyruvoyl–tetrahydropterin synthase; Spr, sepiapterin reductase; Pcbd, pterin–4 alpha–carbinolamine dehydratase; Qdpr, quinoid dihydropteridine reductase. BH4 synthetic intermediates: H2NTP, 7,8–dihydroneopterin triphosphate; Ptp, 6–pyruvoyl–5,6,7,8–tetrahydropterin; BH4αC, tetrahydrobiopterin 4α–carbinolamine; qBH2, Quinoid–dihydrobiopterin.
Rhombencephalic 5–HT neuron development
The neuroanatomical development and distribution of vertebrate 5–HT neurons is well described in rats, primates and zebrafish with immunohistochemical approaches 14–18 and more recently in mice with genetic based fate mapping 19 (Fig. 2). 5–HT neuron development comprises neurogenesis and maturation. During the relatively brief period, E9.5 to E12, of neurogenesis mouse 5–HT neurons are specified and born in the ventral rhombencephalon (Fig. 2a). These newly born 5–HT neurons will ultimately cluster in several disparate regions of the midbrain, pons, and medulla to form the raphe nuclei (Fig. 2b). The period of 5–HT neuron generation, like that of all neurons, comprises several experimentally recognizable stages (Fig. 2c). During the first stage, the secreted signaling molecules, sonic hedgehog (Shh), and fibroblast growth factors 4 and 8 regionally pattern the mid–hindbrain neuroepithelium along the doroventral and anterior–posterior (AP) axes, respectively, to specify serotonergic progenitors in the ventral hindbrain. The reader is referred to several articles that provide an up to date review of these early patterning events 7–9. The first progenitors committed to a serotonergic fate appear in rhombomere (r) 1 where they generate 5–HT neurons from about E9.5 to E10.5. 5–HT neurons are born about a day later in r2 and r3 because progenitors at these longitudinal levels initially generate visceromotor neurons (vMN) before becoming competent to generate 5–HT neurons 20. A similar motorneuron to 5–HT neuron switch in progenitor fate occurs with similar temporal characteristics in r5–r8 such that caudal 5–HT neurons are born virtually simultaneously with those in the rostral domain. However, there is a 1–2 day delay in the synthesis of 5–HT in these newly born caudal cells 21. The switch to serotonergic neurogenesis is never launched under normal circumstances in r4 so that motor neurogenesis persists in this rhombomere 20. During the next stage progenitors exit the cell cycle to become postmitotic neuronal precursors that do not yet possess serotonergic identity. Serotonergic identity is then acquired through coordinate expression of serotonergic–type gene battery. The newborn 5–HT neurons are phenotypically immature (Fig. 2c) and have not yet been integrated into neural circuitry. Therefore, beginning immediately after the birth of 5–HT neurons and extending at least to the end of the third postnatal week mouse 5–HT neurons undergo a series of complex maturational events 15, 21. These events include cell body migration, dendritic growth, expression of 5–HT autoregulatory pathways, formation of highly collateralized axonal pathways and synaptogenesis.
Figure 2. Neuroanatomical features of 5–HT neuron development.

a) Schematic sagittal view of the developing rodent brain. All rodent 5–HT neurons are born caudal to the mid–hindbrain organizer (MHO) in two longitudinal domains, rostral and caudal, on either side of the floor plate in the ventral rhombencephalon (hindbrain). b) Schematic sagittal view of the adult midbrain, pons and medulla depicting the location of 5–HT neurons clusters (raphe nuclei). Intersectional/substractive fate mapping has shown 19 that all 5–HT neurons in the mature dorsal raphe nucleus (DRN, B4, B6 and B7 groups) are born in r1 (green highlight in a and b). 5–HT neurons in the median raphe nucleus (MRN, B8 and B5 groups) and laterally extending supraleminscal 5–HT neurons, designated the B9 cluster, are derived from serotonergic progenitors in r1, r2 and r3 (green, yellow, blue highlight in a and b). 5–HT neurons born in r5–r8 migrate to form the raphe pallidus (RPa, B1), raphe obscurus (ROb, B2), raphe magnus (RMg, B3) and cell bodies in the ventrolateral medulla (B3). 5–HT neurons in the rostral portion of B3 are born in r5 (red highlight). c) Stages of 5–HT neuron development. Postmitotic 5–HT neuron precursors do not yet possess overt serotonergic identity. Induction of the serotonergic–type gene battery generates immature 5–HT neurons. These newborn neurons are integrated with CNS circuitry during a prolonged maturation stage that produces adult 5–HT neurons.
Intrinsic serotonergic transcription regulatory network
Loss and gain of function studies in mouse and chick have greatly illuminated the idiosyncratic intrinsic roles of eight transcriptional regulators and their complex interactions that progressively restrict the fate of progenitors to a 5–HT phenotype. The proneural bHLH factor, Ascl1 (also known as Mash1), and the homeodomain protein, Nkx2.2, are expressed at the vMN and 5–HT progenitor stages at all rhombomeric levels of the hindbrain and their induction is likely an early response to Shh signaling 22–24. Their expression is extinguished as progenitors exit the cell cycle to become postmitotic precursors. The forkhead box factor, Foxa2, is expressed at a very low level in vMN progenitors but is induced as these progenitors switch to serotonergic neurogenesis 25. It is not expressed in r4 where 5–HT neurons are not generated. Next, expression of the zinc finger proteins Gata2, Gata3, and Insm1 is initiated as serotonergic progenitors transit to the postmitotic precursor stage 24, 26, 27. Finally, expression of the Lim homeodomain, Lmx1b, and then the ETS factor, Pet–1 are induced in postmitotic precursors several hours before 5–HT or TPH2 become detectable 16, 28, 29.
Few of the factors in the network are sufficient by themselves to induce 5–HT cell fate. Misexpression of either Nkx2.2 or Gata2 are sufficient to specify a 5–HT transmitter phenotype but only when they are misexpressed in chick r1 24. In contrast, combined misexpression of Lmx1b and Pet–1 is able to induce ectopic 5–HT–containing cells in the ventral spinal cord. If Nkx2.2 is included with Lmx1b and Pet–1 then ectopic 5–HT–containing cells can be generated in the dorsal cord 30.
Specification of serotonergic progenitors
Nkx2.2
Loss of Nkx2.2 does not affect 5–HT neurons in r1 but 5–HT neuron production is substantially reduced, although not eliminated in all of the posterior rhombomeres 19, 22. The role, if any, of Nkx2.2 in r1 is not clear and it might be compensated by the coexpressed paralog Nkx2.9. However, analysis of Nkx2.2 and 2.9 compound mutants has not been reported. Nkx2.2 regulates 5–HT neuron production in rhombomeres posterior to r1, first, by controlling a temporal switch (Fig. 3) in progenitor fate from one of vMN to 5–HT neurons 20. It does so by regulating the timing of expression of another homeodomain protein, Phox2b, which functions as a proneural factor in vMN generation except in r1 where 5–HT neurons but not vMN are generated. In Phox2b mutants, vMN genesis fails throughout the hindbrain 31 and instead 5–HT neurons are prematurely generated in r2–r3, r5–r8 and ectopic ones are generated in r4 20. In addition to playing an essential role in the vMN/5–HT temporal switch Nkx2.2 is also required to switch on the downstream transcriptional network that directly induces the 5–HT–type gene battery (Fig. 4). In Nkx2.2 mutants, expression of Gata3, Lmx1b, and Pet–1 are greatly reduced posterior to r1 30. Misexpression of Nkx2.2 in the chick hindbrain induces Gata2, Gata3, and 5–HT transmitter phenotype in r1 24. In addition, Nkx6.1 is required for development of 5–HT in chick r1 24.
Figure 3. Serotonergic progenitor specification.
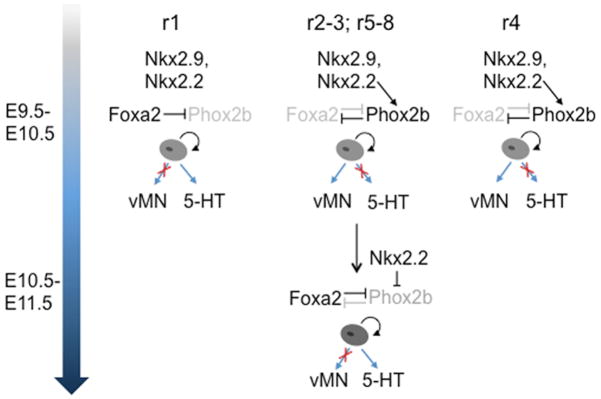
In rhombomere 1 (r1), Foxa2 continually suppresses Phox2b expression to block production of visceral motor neuron (vMN) progenitors and promotes production of 5–HT progenitors. In r2–3, r5–r8, Nkx2.2 initially induces Phox2b, which in turn represses Foxa2. This subcircuit establishes a motoneuron fate of ventral progenitors in r2–r3 and r5–r8. These progenitors produce motor neurons from E9.5 to about E10.5. Starting at about E10.5 a reciprocal temporal cross–repression is initiated whereby Foxa2 now shuts off Phox2b expression. Foxa2 and Nkx2.2 mediated repression of Phox2b switches the fate of ventral progenitors so that a wave of serotonergic neurogenesis follows motor neurogenesis. In r4, Phox2b continually represses Foxa2 thus suppressing the vMN to 5–HT fate switch and prolonging the production of vMNs.
Figure 4. Serotonergic transcription regulatory network.

Stage–specific subcircuits in the network are depicted in colored rectangles with enclosed transcriptional factor gene symbols and arrows or blocked lines leading to other network constituents. Foxa2 is required for Gata3 expression only in r1. In mouse Nkx6.1 and Nkx6.2 do not appear to be required for 5–HT neuron generation but in r4 they suppress 5–HT fate. Nkx6.1 has been shown to be required for Gata2 and Pet–1 expression and production of 5–HT neurons only in chick r1. Chromatin immunoprecipitation experiments indicate direct Gata2 binding (solid blue circle), in vivo, to the Pet–1 upstream regulatory region where conserved Gata2 binding sites are located. Pet–1 induces Htr1a and Htr1b to initiate formation of the two 5–HT autoreceptor pathways in maturing 5–HT neurons (red highlighted box, E14.5). ChIP indicates that Pet–1 binds (solid red circles), in vivo, to the Tph2, Sert, and Pet–1 upstream regions where conserved Pet–1 binding are located. In adult 5–HT neurons (beige highlighted box) Lmx1b and Pet–1 maintain Tph2 and Sert. Lmx1b also maintains Vmat2 expression. Pet–1 expression in adult 5–HT neurons is stabilized through direct positive autoregulation (curved red arrow). Dashed arrows indicates that Gata2 is likely required for Lmx1b expression but this has not been formally demonstrated and Insm–1 may also directly regulate Tph2 but this has not been shown with ChIP.
Foxa2
Foxa proteins are well known for playing multiple roles within single cell lineages 32 and the serotonergic lineage is no exception. Foxa2 acts in a temporally regulated manner to establish serotonergic progenitor domains in the ventral hindbrain and then to activate expression of transcription factors that are directly involved in 5–HT neuron precursor differentiation 25. Foxa2 represses the homeodomain protein, Phox2b, in r1 (Fig. 3) and in so doing permanently suppresses vMN fate while promoting serotonergic fate of the resident progenitors. In more posterior rhombomeres except r4, Phox2b initially represses Foxa2 hence directing a motor neuron fate of resident progenitors up to about E10.5 (Fig. 3). After E10.5 a reciprocal repression occurs in which Foxa2 now represses Phox2b to switch progenitors to a serotonergic fate. In r4, Phox2b permanently represses Foxa2 hence preventing serotonergic neurogenesis (Fig. 3). The mechanism underlying the cross repressive switch is not understood. Ongoing Foxa2 function is required in the same progenitors for activation of Gata2, Lmx1b and Pet–1 (Fig. 4) 25. The loss of Foxa2 at this later stage results in about a 50% loss of 5–HT neurons in the hindbrain.
Ascl1
Despite being expressed in motor neuron progenitors during vMN genesis, loss of Ascl1 does not appear to cause defects in motor neuron number or phenotype 33. Ascl1 function emerges only after Phox2b is shut off in motor neuron progenitors and at this stage it has been shown to perform multiple functions 23. First, Ascl1 performs a proneural role to regulate serotonergic neurogenesis, as expression of all markers (Gata2, Gata3, Pet–1, Lmx1b, 5–HT) of newly generated 5–HT postmitotic precursors is nearly abolished throughout the hindbrain in Ascl1−/− mutants. Second, Ascl1 is required together with Nkx2.2, Foxa2, to induce the inferred (see below) parallel Gata2 and Gata3–dependent 5–HT neuron differentiation networks 23 (Fig. 4). A third function for Ascl1 is to directly activate the expression of the zinc finger protein, Insm1, as mitotically active serotonergic progenitors are exiting the cell cycle 26. Chromatin immunoprecipitation (ChIP) studies show a direct interaction of Ascl1 with a conserved binding site located 4.5kb upstream of the Insm1 transcription start site (Fig. 4). Moreover, Insm1 expression is greatly reduced or eliminated in Ascl1 mutant serotonergic precursors throughout the ventral hindbrain 26.
Insm1
Direct regulation of Insm–1 by Ascl1 suggests a critical role for Insm1 in acquisition of the serotonergic phenotype. In general, loss of function studies confirmed this prediction but also reveal some unexpected nuances that illuminate unique characteristics of Insm1 function 26. First, widely varying deficiencies of 5–HT–positive neurons were detected Insm1 mutants along the AP axis of the hindbrain with levels in r2–r3 being severely affected. Second, although Insm1 loss of function did not change progenitor specification, Lmx1b expression was uniformly and severely reduced. However, deficits in Gata2 and Pet–1 expression were variable along the AP axis. Third, initial Aadc expression was not affected in Insm1 mutants but Tph2 expression was greatly reduced along the entire AP axis. However, by E16.5 Aadc expression was reduced, consistent with the deficit in Pet–1 expression at this stage. These finding suggest a complex network role for Insm1 in which it perhaps directly regulates Tph2 expression while reinforcing Ascl11, Foxa2, Nkx2.2–directed Gata2 expression in a feedforward manner (Fig. 4). Alternatively, because Insm1 plays critical role in Lmx1b expression 26 and loss of Lmx1b results in virtually complete loss of 5–HT and Tph2 expression 30, 34 perhaps Insm1 indirectly regulates Tph2 through direct regulation of Lmx1b expression (Fig. 4).
Gata2
Studies of Gata2’s role in 5–HT development are complicated because all Gata–2−/− embryos die between E9.5 and E11.5 from severe hematopoietic defects 35. However, Craven et al., 2004 were able to show that induction of Pet–1 at E10.5 fails to occur in the rostral hindbrain of Gata2−/− embryos. Furthermore, in contrast to wild type or Gata2+/− explants, no 5–HT can detected in ventral neural tube explants taken from E8 Gata2−/− embryos and analyzed after 5 days in culture 24. At all longitudinal levels, Gata2 is absolutely required for induction of Pet–1 (and presumably Lmx1b) but not Gata3 24. Thus, it can be inferred that Nkx2.2, Ascl1, and Foxa2 induce a Gata3–dependent network and a parallel Gata2–dependent network, which the latter in turn induces Lmx1b and Pet–1 (Fig. 4). Several lines of evidence suggest that Gata2 directly induces Pet–1 expression at the postmitotic precursor stage. First, as described above, Pet–1 expression is completely eliminated in explants of Gata2 mutant neural tubes 24. Second, ChIP studies indicate that Gata2 occupies highly conserved Gata binding sites in the upstream Pet–1 enhancer region. Third, point mutation of these sites greatly diminishes expression of a Pet–1 enhancer regulated transgene reporter in mouse brain 5–HT neurons 36.
Differentiation of postmitotic precursors
Coordinate induction of the serotonergic–type gene battery encoding 5–HT synthesis, reuptake, vesicular transport and metabolism is the final step in serotonergic neurogenesis. Lmx1b, Pet–1 and Gata3 are induced in postmitotic precursors just before initiation of 5–HT synthesis and all are necessary for induction of the battery. None of these factors are dependent on each other for their induction and therefore, they provide parallel transcriptional inputs to induce serotonergic–type gene expression (Fig. 4). In addition, the loss of these factors does not affect progenitors. However, expression of all three of these factors is maintained across the lifespan, which suggests they may be terminal selectors 37 that have distinct functions at different stages of life. Indeed, recent findings summarized below have shown this to be true for Pet–1 and Lmx1b.
Lmx1b
Lmx1b plays numerous critical roles in CNS development including development of the isthmic organizer that induces the mid–hindbrain boundary, mesencephalic dopaminergic neurons and spinal dorsal horn neurons 38. In homozygous Lmx1b mutants 5–HT neuron precursors are generated in normal numbers but virtually all of them fail to activate expression of Tph2, Sert, Vmat2 and 5–HT synthesis 30, 34. Despite being induced normally in Lmx1b mutants, Pet–1 expression begins to decline at E12.5 and by E14.5 Pet–1 expression is no longer detectable 34. Thus, Lmx1b is needed to maintain Pet–1 at later stages of embryogenesis (Fig. 4). Lmx1b and Pet–1 share many targets and therefore Lmx1b’s regulation of Pet–1 suggests that these factors operate in a feedforward manner to regulate the serotonergic–type gene battery.
Neonatal death precluded postnatal analyses of constitutively targeted homozygous Lmx1b mutant mice. However, survival was improved when Lmx1b was conditionally targeted in 5–HT neurons, which permitted study in the postnatal period of the impact of a serotonergic Lmx1b deficiency. Analyses of these targeted mice showed that although mutant 5–HT neuron cell bodies were present in the fetal hindbrain, nearly all of them were missing postnatally 39. These findings verified a cell autonomous function of Lmx1b in 5–HT neurons and demonstrated that it is uniquely required, among the known network factors, for postnatal survival of these cells. The SIM1 transcription factor may also be required for the maintenance or survival of a small subset of 5–HT neurons in the DRN 40.
Although genetic programs appear to specify 5–HT neuron progenitors independent of activity–responsive signaling pathways recent studies in the embryonic Xenopus hindbrain have shown that a portion of 5–HT neuron precursors are dependent on calcium spike activity for serotonergic differentiation 41. Demarque and Spitzer showed using overexpression of sodium channels or potassium channels that increased or decreased calcium spike activity led to almost 30% increases or decreases in the numbers of lmx1b+ and tph+ neurons, respectively. Lmx1b was identified as a node for the activity–dependent regulation of 5–HT neuron number since these alterations were prevented when lmx1b expression was suppressed. The alterations in 5–HT neuron numbers and lmx1b expression also led to changes in swimming behavior of Xenopus larvae 41. Together these findings indicate activity–dependent plasticity of the serotonergic transcriptional network and suggest that an activity–dependent homeostatic mechanism may transcriptionally adjust serotonergic neuromodulation.
Gata3
In Gata3 deficient mice, most precursors fail to initiate 5–HT synthesis in caudal rhombomeres. In contrast, 5–HT neuron differentiation is only modestly affected in the rostral hindbrain suggesting a diminishing caudal to rostral gradient in Gata3 dependency 23, 27. These findings suggest that Gata3 directly regulates Tph2 expression although Gata3 binding sites in the Tph2 regulatory region have not been identified (Fig. 4). Conditional targeting of Gata3 in 5–HT neurons has shown that ongoing Gata3 is also required to maintain normal levels of Tph2, Sert and to a lesser extent Aadc and Vmat2 42.
Pet–1
Pet–1 is the only factor in the transcriptional network whose expression in the brain is restricted to 5–HT neurons 28, 29. FEV (Fifth Ewing Variant), the human orthologue of Pet–1, is expressed specifically in the rhesus macaque 43 and human 44, 45 dorsal and median raphe. Similar to Gata3 and Lmx1b loss of function, constitutive loss of Pet–1 function does not alter the generation of 5–HT neurons precursors 46. However, about 70% of these Pet–1−/− cells fail to initiate 5–HT synthesis. The defect is roughly uniform across all raphe nuclei and leads to an 80% deficiency of 5–HT in the adult ascending and descending 5–HT subsystems 46. These Pet–1 mutant precursors also fail to induce Tph2, Aadc, Sert, Vmat2, and Maob and therefore, Pet–1 is required for induction of all of the genes in the serotonergic–type gene battery (Fig. 4). It is not clear why Pet–1 is largely dispensable for serotonergic differentiation in 30% of 5–HT neuron precursors although recent findings show that the Pet–1 resistant 5–HT neurons constitute a phenotypically distinct ascending serotonergic subsystem that continues to innervate circuitry involved in neuroendocrine and autonomic control of stress responses 47. While other ETS transcripts were detected in dissected adult human raphe tissue 44 whole genome expression profiling failed to identify enriched expression of other ETS genes in flow cytometry sorted E12.5 mouse embryonic 5–HT neurons 48, which if detected would have raised the simple explanation of paralog compensation in Pet–1 resistant 5–HT neurons.
Consistent with its ongoing expression in 5–HT neurons, conditional Pet–1 targeting with an ePet-Cre transgene 49 that is active at the postmitotic stage and causes loss of Pet–1 1–2 days after serotonergic neurogenesis 42, showed that Pet–1 continues to function during the prolonged stage of 5–HT system maturation. First, although Pet–1 resistant 5–HT+ neurons have normal axonal growth characteristics in culture 47, conditionally targeted Pet–1 deficient neurons that lose the ability to synthesize 5–HT have altered axonal innervation patterns in the somatosensory cortex 42. Although these results indicate a role in axonal development it is not yet clear what specific role Pet–1 performs in regulating innervation patterns. The evolutionarily conserved role of different ETS factors in circuit formation suggests several possibilities: AST1 directed axon navigation in C. elegans 50, mouse Pea3 directed axonal branching 51 and mouse ER81 control of target connectivity 52. Second, in situ hybridization analyses and whole cell electrophysiological recordings have shown that during the maturation stage, Pet–1 is required to induce the 5–HT1a and 5–HT1b autoreceptor pathways 42, 53, which do not begin to be expressed in the mouse brain until about E14.5 42, 54. The 5–HT1a autoreceptor pathway was not altered in conditionally targeting Gata3 mice, which places special importance on Pet–1 in this physiologically and clinically relevant regulatory event 55. Conditional targeting of Pet–1 in adult 5–HT neurons with a tamoxifen inducible ePet–CreER transgene showed that Pet–1 is required for maintenance of Tph2 and Sert expression but not expression of other genes whose induction depends on Pet–1 during serotonergic neurogenesis and maturation. Likewise, Ding and colleagues recently reported tamoxifen–induced targeting of Lmx1b in adult 5–HT neurons and showed that Lmx1b is also needed to maintain Tph2 and Sert expression but not Pet–1 and Aadc 56. In contrast to Pet–1, however, Lmx1b was also required to maintain Vmat2 expression suggesting distinct regulatory roles for Pet–1 and Lmx1b in adulthood (Fig 4).
With the exception of Pet–1/DNA interactions, very little is known about the cis–regulatory modules through which factors in the network directly activate transcription of the serotonergic type gene battery. Conserved Pet–1 binding sites have so far been investigated in the upstream regions of Sert, Tph2, Htr1a and Pet–1 28, 42, 53. In vitro DNA binding assays showed that bacterially expressed Pet–1 can bind conserved ETS–like sites present in the upstream regions of Sert and Htr1a genes 28. Further analysis revealed multiple conserved Pet–1 binding sites upstream of the human and rodent Htr1a genes 53. Reporter assays in the HEK293 cell line showed that activity of a 1.5kb Hrt1a promoter construct was substantially reduced when the conserved GGAA core of any of the Pet–1 binding sites were destroyed. However, destruction of a single distal Pet–1 ETS site at –1406 completely abolished promoter activity suggesting a critical role for this site in the regulation of Htr1a 53. Chromatin immunoprecipitation studies with chromatin isolated from embryonic hindbrain have shown that Pet–1 binds, in vivo, to the Tph2, Sert, and Pet–1 upstream regulatory regions where conserved Pet–1 binding sites are located 42. In addition, Pet–1 enhancer activity is dramatically reduced in 5–HT neurons lacking Pet–1 expression in adulthood and therefore ongoing Pet–1 binding is required to maintain its own expression. Together these findings support a model (Fig. 4) in which induction of Pet–1 by the serotonergic transcription regulatory network is subsequently reinforced and stabilized through direct positive autoregulation to maintain Pet–1, Tph2, and Sert expression across the lifespan 42.
In summary, the parallel transcriptional inputs of Gata3, Lmx1b and Pet–1 to the serotonergic–type gene battery constitute a combinatorial code that determines 5–HT neuron identity (Fig. 4). Pet–1 functions as a terminal selector 37 defined as an autoregulated gene that induces and maintains expression of a cell–type specific gene battery across the lifespan through direct interaction with a common DNA regulatory motif. Pet–1, Lmx1b, and Gata3 likely function together as combinatorial terminal selectors, although the latter two factors have not been shown to autoregulate or to directly control the serotonergic–type gene battery. The essential role of Lmx1b but not Gata3 and Pet–1 in all 5–HT neurons suggest Lmx1b forms a distinct combinatorial code with other transcriptional inputs in a subset of 5–HT neurons. In light of the requirement for Lmx1b and Pet–1 in the maintenance of adult 5–HT synthesis and reuptake together with the activity dependent regulation of Lmx1b 41 it will be interesting to determine whether environmental input such as stress or drug exposure alters Pet–1 and/or Lmx1b dependent regulation of serotonergic function in the mammalian brain.
Network alterations and pathogenesis
As alluded to at the beginning of this review, evidence from decades of studies in humans, non–human primates and rodents strongly support an association of altered serotonergic function with behavioral and physiological pathogenesis 55, 57, 58. It is now well understood that genetically engineered alterations in the levels of Sert, Tph2, or Htr1a expression are not compatible with normal emotional and stress–related behaviors, at least in mice 59–61. Many other physiological processes are also disrupted in mice deficient for these genes including growth trajectories, respiration, body weight maintenance, stress susceptibility, cardiovascular function, sleep patterns and maternal behavior 59, 62. These genetic–based animal studies provide a compelling impetus for the quest to find gene variation that impacts serotonergic signaling and risk for neuropsychiatric disease. Numerous variants thought to impact presynaptic serotonergic signaling through intrinsic alterations in brain 5–HT neuron function have been discovered and investigated for association with neuropsychiatric disease or their influence on the function of neural circuitry shaping emotional responses 55, 58, 59, 63. Most of these disease associated 5–HT neuron variants have been tested for functional impact in cultured cell lines and not in brain 5–HT neurons. Yet, an intriguing commonality among the variants with functional impact, in vitro and in some cases in vivo is that they all affect the level of expression or activity of TPH2, SERT, or HTR1A. Widely studied common polymorphisms in the human SERT transcriptional regulatory region affects the level of SERT transcription in cell culture 64, 65 and appears to moderate the impact of stressful life events on risk for adult depression 66. An orthologous polymorphic region in rhesus macaques, likewise, influences stress reactivity in subjects exposed to adverse early rearing experiences 67. Perhaps the most convincing example of disease associated variation in SERT are the multiple rare non–synonymous variants creating gain of function alleles that produce elevated levels of the transporter or enhanced trafficking–independent, 5–HT transport activity. Furthermore, these alleles encode transporters that are no longer sensitive to intracellular signaling pathways that control SERT trafficking and activity 68. Importantly, in a large sample of multiplex autism families, it was shown that these variants in aggregate are significantly associated with autism and rigid compulsive behaviors 69. Despite its apparent very rare frequency 70, another notable example of variation that affects the level of serotonergic activity is a non–synonymous TPH2 single nucleotide coding variant, G1463A 71. The encoded R441H TPH2 variant, exhibits an 80% loss of 5–HT biosynthetic capacity in cell culture and has been associated with late life depression 71. An orthologous mouse knock in allele causes severely reduced brain 5–HT synthesis, increased aggression and anxiety–like behaviors 60. Importantly, the effect of this allele recapitulated in the mouse many of the alterations in the levels of serotonergic biomarker indices that are associated with depression in humans 72, reinforcing the idea that the altered human indices do indeed reflect reduced brain 5–HT levels and neuropsychiatric disease.
In view of the human genetic evidence and abundant animal studies that support altered levels of 5–HT signaling in disease susceptibility a natural but largely unexplored question is whether variation in the serotonergic transcription regulatory network (Fig. 4) impacts the level of 5–HT neuron gene expression and susceptibility to mental illness. So far, variation with such an impact has not been discovered. However, several findings in non–human primates and mice highlight network variation as a potentially important mechanism in neuropsychiatric disease pathogenesis. First, studies of female cynomolgus monkeys have defined a stress sensitive (SS) subgroup, in which reproductive function is impaired, versus highly stress resilient (HSR) individuals who showed no reproductive suppression 43. Interestingly, in SS individuals the level of FEV expression in the dorsal raphe is significantly reduced compared to FEV expression in the HSR dorsal raphe 43. Moreover, the level of expression of TPH2, SERT, and the HTR1A autoreceptor genes were correspondingly decreased in SS DRN compared to the HSR DRN suggesting potentially altered FEV–dependent transcriptional regulation of 5–HT synthesis, reuptake and autosensing in response to stress in female macaques 43, 73. Second, Lmx1b and Pet–1 deficient mice show several emotion and stress–related behavioral abnormalities that parallel those observed in Tph2, Sert, and Htr1a targeted mice including increase aggression, anxiety–like behavior and fear responses 46, 47, 74. Importantly, Pet–1 dependent transcription is required into adulthood to maintain normal anxiety–like behaviors 42 raising the possibility that behavioral pathogenesis might arise from adult onset disruption, genetically or environmentally, of serotonergic transcription. In addition, neonatal growth, respiration, thermoregulation, autonomic control of neonatal cardiac function, nociception and circadian rhythms are disrupted in Lmx1b−/− and Pet–1−/− mice 75–84 suggesting potential broad impact of network alterations. Third, transgenic experiments showed that the extent of transgenic rescue of 5–HT neuron molecular defects and maternal behavior deficits in Pet–1−/− mice is dependent on the level of expression of a FEV BAC transgene 85. These later findings provide evidence in support of the idea that the expression level of serotonergic regulatory network determines the levels of 5–HT neuron gene expression with attendant quantitative effects on 5–HT–modulated behaviors.
Given the direct regulation of Tph2, Sert, and Htr1a genes by Pet–1 and probably also by Lmx1b, naturally occurring human alleles of these two network components are particularly intriguing as a source of potential disease relevant variation. The restricted expression of FEV in the brain suggests that different functional FEV alleles, if they exist, may have 5–HT system specific effects. Positive or negative epistasis 86 with other network alleles may determine whether or not FEV alleles cause deleterious effects on 5–HT system activity. However, naturally occurring functional variation within the serotonergic regulatory network would be expected, in most instances, to exact variable pleiotropic effects because all the others factors in the network are broadly expressed in multiple unrelated lineages within the brain and peripheral tissues. Human genetic studies of LMX1B variation provide an instructive example. Over 100 distinct heterozygous autosomal dominant LMX1B missense, nonsense and deletion mutations have been identified in different cases of Nail Patella Syndrome, a disorder of highly variable expressivity and penetrance that can include nail dysplasia, skeletal malformations, renal defects, sensory deficits, and ocular malformations 38, 87, 88. In support of LMX1B loss of function in NPS, Lmx1b deficient mice display several defining features of the syndrome 89. Many of the human mutations encode homeodomain missense and disrupt sequence specific DNA binding suggesting direct effects on network interactions and downstream transcriptional targets. The likely haploinsufficiency of LMX1B in NPS 90 suggests LMX1B–dependent transcriptional networks are particularly sensitive to the level of LMX1B activity. Given its place, functionally in the serotonergic regulatory network we imagine altered network function in at least some NPS individuals. Although not yet studied in sufficient depth to establish a clear link to neuropsychiatric disease, disrupted LMX1B binding may promote susceptibility to ADHD and major depressive disorder in NPS cases 91. Further study of this important possibility, including quantitative analysis of disease–associated serotonergic biomarker indices in NPS individuals, is of significant interest in the search for the genetic basis of serotonergic dysfunction in neuropsychiatric disorders.
Conclusions and future directions
A detailed understanding of the spatiotemporal expression patterns and functions of eight transcription factors has enabled the construction of a model for the dynamic regulatory network that specifies brain 5–HT neurons (Fig. 4). The network comprises temporally distinct regulatory states each corresponding to a particular stage in the development of 5–HT neuron identity and its maintenance in adulthood. Despite the well–studied effects of specific network perturbations on 5–HT neuron identity as well as a growing understanding of its effects on behavior and physiology several critical gaps in knowledge remain. For example, despite numerous obvious signs of network heterogeneity our understanding of it is insufficient to explain why the phenotype of substantial numbers of 5–HT neurons are resistant to loss of individual transcription factors and how diverse axonal trajectories, cell body migratory routes and physiological properties of different groups of 5–HT neurons are programmed. In addition, nothing is known about the layer of epigenetic transcriptional control within the network and how it responds to environmental signals that may help to shape 5–HT–modulated behaviors. Further study of the network with new genetic–based approaches that provide greater access to 5–HT neurons 49, 92 should help fill in these gaps and provide a better understanding of how it controls 5–HT function.
An additional theme of this review is the potential utility of our network model in helping to advance an understanding of the importance of 5–HT system dysfunction in neuropsychiatric disease pathogenesis. Given the likely essential role of the orthologous human network in the induction of the disease–associated genes, TPH2, SERT, and HTR1A, genetically or epigenetically–driven alterations in expression or function of the network may program altered levels of 5–HT synthesis, reuptake, or autoreceptor function and establish a vulnerability for mental illness. The identification of multiple biologically relevant rare disease–associated variants in SERT, TPH2, and LMX1B is consistent with the now widely recognized fact that significant heterogeneity exists in the allelic architecture of complex human disorders in general and in particular for neuropsychiatric disorders 93–95. As an experimentally well–defined cell–type specific biological pathway the serotonergic regulatory network is a potentially rich source of rare heritable or de novo human variation that, if discovered, can be investigated for functional impact in the context of brain 5–HT system development and neuromodulation. The utility of our model is its power to help interpret the biological significance of network variants within an ensemble of functionally interacting serotonergic transcriptional determinants. Orthologous mutations can be investigated to study their impact on serotonergic differentiation of cultured embryonic stem cells 96, 97 and introduced into the mouse genome to determine their impact on 5–HT modulated behaviors and physiology. Functionally–defined network variants can then be investigated to determine whether they are present in unrelated individuals with particular neuropsychiatric diagnoses 98 as has been accomplished for LMX1B variants in NPS and with SERT variants in autism.
Acknowledgments
We thank R. Blakely (Vanderbilt University) and E. Gilmore (Case Western Reserve University) for valuable discussions and critical comments on the manuscript. Research in the Deneris Lab is supported by grants RO1 MH062723 and the Vanderbilt/NIMH Silvio O Conte Center for Neuroscience Research, P50 MH078028 from the US National Institutes of Health.
References
- 1.Gaddum JH. Ciba Foundation Symposium on Hypertension. Little, Brown and Co; 1954. Drugs antagonistic to 5-hydroxytryptamine; pp. 75–77. [Google Scholar]
- 2.Woolley DW, Shaw E. A Biochemical and Pharmacological Suggestion About Certain Mental Disorders. Proceedings of the National Academy of Sciences of the United States of America. 1954;40:228–231. doi: 10.1073/pnas.40.4.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller CP, Jacobs BL, editors. Handbook of the Behavioral Neurobiology of Serotonin. Academic Press; 2010. [Google Scholar]
- 4.Ishimura K, et al. Quantitative analysis of the distribution of serotonin-immunoreactive cell bodies in the mouse brain. Neurosci Lett. 1988;91:265–270. doi: 10.1016/0304-3940(88)90691-x. [DOI] [PubMed] [Google Scholar]
- 5.Baker KG, et al. Cytoarchitecture of serotonin-synthesizing neurons in the pontine tegmentum of the human brain. Synapse. 1991;7:301–320. doi: 10.1002/syn.890070407. [DOI] [PubMed] [Google Scholar]
- 6.Hornung JP. The human raphe nuclei and the serotonergic system. J Chem Neuroanat. 2003;26:331–343. doi: 10.1016/j.jchemneu.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Gaspar P, Cases O, Maroteaux L. The developmental role of serotonin: news from mouse molecular genetics. Nat Rev Neurosci. 2003;4:1002–1012. doi: 10.1038/nrn1256. [DOI] [PubMed] [Google Scholar]
- 8.Goridis C, Rohrer H. Specification of catecholaminergic and serotonergic neurons. Nat Rev Neurosci. 2002;3:531–541. doi: 10.1038/nrn871. [DOI] [PubMed] [Google Scholar]
- 9.Cordes SP. Molecular genetics of the early development of hindbrain serotonergic neurons. Clinical genetics. 2005;68:487–494. doi: 10.1111/j.1399-0004.2005.00534.x. [DOI] [PubMed] [Google Scholar]
- 10.Scott MM, Deneris ES. Making and breaking serotonin neurons and autism. Int J Dev Neurosci. 2005;23:277–285. doi: 10.1016/j.ijdevneu.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez SP, Gaspar P. Investigating anxiety and depressive-like phenotypes in genetic mouse models of serotonin depletion. Neuropharmacology. 2011 doi: 10.1016/j.neuropharm.2011.08.049. [DOI] [PubMed] [Google Scholar]
- 12.Kim H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nature medicine. 2010;16:804–808. doi: 10.1038/nm.2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben-Tabou de-Leon S, Davidson EH. Gene regulation: gene control network in development. Annu Rev Biophys Biomol Struct. 2007;36:191. doi: 10.1146/annurev.biophys.35.040405.102002. [DOI] [PubMed] [Google Scholar]
- 14.Azmitia EC, Gannon PJ. The primate serotonergic system: a review of human and animal studies and a report on Macaca fascicularis. Adv Neurol. 1986;43:407–468. [PubMed] [Google Scholar]
- 15.Lidov HG, Molliver ME. An immunohistochemical study of serotonin neuron development in the rat: ascending pathways and terminal fields. Brain Res Bull. 1982;8:389–430. doi: 10.1016/0361-9230(82)90077-6. [DOI] [PubMed] [Google Scholar]
- 16.Lillesaar C, Tannhauser B, Stigloher C, Kremmer E, Bally-Cuif L. The serotonergic phenotype is acquired by converging genetic mechanisms within the zebrafish central nervous system. Dev Dyn. 2007;236:1072–1084. doi: 10.1002/dvdy.21095. [DOI] [PubMed] [Google Scholar]
- 17.Wallace JA, Lauder JM. Development of the serotonergic system in the rat embryo: an immunocytochemical study. Brain Res Bull. 1983;10:459–479. doi: 10.1016/0361-9230(83)90144-2. [DOI] [PubMed] [Google Scholar]
- 18.Steinbusch HWM. Distribution of serotonin-immunoreactivity in the central nervous system of the rat-cell bodies and terminals. Neuroscience. 1981;6:557–618. doi: 10.1016/0306-4522(81)90146-9. [DOI] [PubMed] [Google Scholar]
- 19.Jensen P, et al. Redefining the serotonergic system by genetic lineage. Nat Neurosci. 2008;11:417–419. doi: 10.1038/nn2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pattyn A, et al. Coordinated temporal and spatial control of motor neuron and serotonergic neuron generation from a common pool of CNS progenitors. Genes Dev. 2003;17:729–737. doi: 10.1101/gad.255803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lidov HGW, Molliver ME. Immunohistochemical study of the development of serotonergic neurons in the rat CNS. Brain Res Bull. 1982;9:559–604. doi: 10.1016/0361-9230(82)90164-2. [DOI] [PubMed] [Google Scholar]
- 22.Briscoe J, et al. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic Hedgehog signalling. Nature. 1999;398:622–627. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- 23.Pattyn A, et al. Ascl1/Mash1 is required for the development of central serotonergic neurons. Nat Neurosci. 2004;7:589–595. doi: 10.1038/nn1247. [DOI] [PubMed] [Google Scholar]
- 24.Craven SE, et al. Gata2 specifies serotonergic neurons downstream of sonic hedgehog. Development. 2004;131:1165–1173. doi: 10.1242/dev.01024. [DOI] [PubMed] [Google Scholar]
- 25.Jacob J, et al. Transcriptional repression coordinates the temporal switch from motor to serotonergic neurogenesis. Nat Neurosci. 2007;10:1433–1439. doi: 10.1038/nn1985. [DOI] [PubMed] [Google Scholar]
- 26.Jacob J, et al. Insm1 (IA-1) is an essential component of the regulatory network that specifies monoaminergic neuronal phenotypes in the vertebrate hindbrain. Development. 2009;136:2477–2485. doi: 10.1242/dev.034546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Doorninck JH, et al. GATA-3 is involved in the development of serotonergic neurons in the caudal raphe nuclei. J Neurosci. 1999;19:RC12. doi: 10.1523/JNEUROSCI.19-12-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hendricks T, Francis N, Fyodorov D, Deneris E. The ETS Domain Factor Pet-1 is an Early and Precise Marker of Central 5-HT Neurons and Interacts with a Conserved Element in Serotonergic Genes. J Neurosci. 1999;19:10348–10356. doi: 10.1523/JNEUROSCI.19-23-10348.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfaar H, et al. mPet-1, a mouse ETS-domain transcription factor, is expressed in central serotonergic neurons. Dev Genes Evol. 2002;212:43–46. doi: 10.1007/s00427-001-0208-x. [DOI] [PubMed] [Google Scholar]
- 30.Cheng L, et al. Lmx1b, Pet-1, and Nkx2.2 coordinately specify serotonergic neurotransmitter phenotype. J Neurosci. 2003;23:9961–9967. doi: 10.1523/JNEUROSCI.23-31-09961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pattyn A, Hirsch M, Goridis C, Brunet JF. Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development. 2000;127:1349–1358. doi: 10.1242/dev.127.7.1349. [DOI] [PubMed] [Google Scholar]
- 32.Kaestner KH. The FoxA factors in organogenesis and differentiation. Curr Opin Genet Dev. 2010;20:527–532. doi: 10.1016/j.gde.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirsch MR, Tiveron MC, Guillemot F, Brunet JF, Goridis C. Control of noradrenergic differentiation and Phox2a expression by MASH1 in the central and peripheral nervous system. Development. 1998;125:599–608. doi: 10.1242/dev.125.4.599. [DOI] [PubMed] [Google Scholar]
- 34.Ding YQ, et al. Lmx1b is essential for the development of serotonergic neurons. Nat Neurosci. 2003;6:933–938. doi: 10.1038/nn1104. [DOI] [PubMed] [Google Scholar]
- 35.Tsai FY, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- 36.Krueger KC, Deneris ES. Serotonergic transcription of human FEV reveals direct GATA factor interactions and fate of Pet-1-deficient serotonin neuron precursors. J Neurosci. 2008;28:12748–12758. doi: 10.1523/JNEUROSCI.4349-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hobert O, Carrera I, Stefanakis N. The molecular and gene regulatory signature of a neuron. Trends in neurosciences. 2010;33:435–445. doi: 10.1016/j.tins.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai JX, Johnson RL, Ding YQ. Manifold functions of the Nail-Patella Syndrome gene Lmx1b in vertebrate development. Dev Growth Differ. 2009;51:241–250. doi: 10.1111/j.1440-169X.2008.01083.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhao ZQ, et al. Lmx1b is required for maintenance of central serotonergic neurons and mice lacking central serotonergic system exhibit normal locomotor activity. J Neurosci. 2006;26:12781–12788. doi: 10.1523/JNEUROSCI.4143-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osterberg N, et al. Sim1 is a novel regulator in the differentiation of mouse dorsal raphe serotonergic neurons. PloS one. 2011;6:e19239. doi: 10.1371/journal.pone.0019239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demarque M, Spitzer NC. Activity-dependent expression of Lmx1b regulates specification of serotonergic neurons modulating swimming behavior. Neuron. 2010;67:321–334. doi: 10.1016/j.neuron.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C, et al. Pet-1 is required across different stages of life to regulate serotonergic function. Nat Neurosci. 2010;13:1190–1198. doi: 10.1038/nn.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lima FB, et al. Stress sensitive female macaques have decreased fifth Ewing variant (Fev) and serotonin-related gene expression that is not reversed by citalopram. Neuroscience. 2009;164:676–691. doi: 10.1016/j.neuroscience.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maurer P, et al. The Ets transcription factor Fev is specifically expressed in the human central serotonergic neurons. Neurosci Lett. 2004;357:215–218. doi: 10.1016/j.neulet.2003.12.086. [DOI] [PubMed] [Google Scholar]
- 45.Iyo AH, Porter B, Deneris ES, Austin MC. Regional distribution and cellular localization of the ETS-domain transcription factor, FEV, mRNA in the human postmortem brain. Synapse. 2005;57:223–228. doi: 10.1002/syn.20178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hendricks TJ, et al. Pet-1 ETS gene plays a critical role in 5-HT neuron development and is required for normal anxiety-like and aggressive behavior. Neuron. 2003;37:233–247. doi: 10.1016/s0896-6273(02)01167-4. [DOI] [PubMed] [Google Scholar]
- 47.Kiyasova V, et al. A Genetically Defined Morphologically and Functionally Unique Subset of 5-HT Neurons in the Mouse Raphe Nuclei. J Neurosci. 2011;31:2756–2768. doi: 10.1523/JNEUROSCI.4080-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wylie CJ, et al. Distinct transcriptomes define rostral and caudal serotonin neurons. J Neurosci. 2010;30:670–684. doi: 10.1523/JNEUROSCI.4656-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scott MM, et al. A genetic approach to access serotonin neurons for in vivo and in vitro studies. Proc Natl Acad Sci U S A. 2005;102:16472–16477. doi: 10.1073/pnas.0504510102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmid C, Schwarz V, Hutter H. AST-1, a novel ETS-box transcription factor, controls axon guidance and pharynx development in C. elegans. Dev Biol. 2006;293:403–413. doi: 10.1016/j.ydbio.2006.02.042. [DOI] [PubMed] [Google Scholar]
- 51.Livet J, et al. ETS gene PEA3 controls the central position and terminal arborization of specific motor neuron pools. Neuron. 2002;35:877–892. doi: 10.1016/s0896-6273(02)00863-2. [DOI] [PubMed] [Google Scholar]
- 52.Arber S, Ladle DR, Lin JH, Frank E, Jessell TM. ETS gene Er81 controls the formation of functional connections between group Ia sensory afferents and motor neurons. Cell. 2000;101:485–498. doi: 10.1016/s0092-8674(00)80859-4. [DOI] [PubMed] [Google Scholar]
- 53.Jacobsen KX, Czesak M, Deria M, Le Francois B, Albert PR. Region-specific regulation of 5-HT1A receptor expression by Pet-1-dependent mechanisms in vivo. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2010.07161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bonnin A, Peng W, Hewlett W, Levitt P. Expression mapping of 5-HT1 serotonin receptor subtypes during fetal and early postnatal mouse forebrain development. Neuroscience. 2006;141:781–794. doi: 10.1016/j.neuroscience.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 55.Albert PR, Le Francois B, Millar AM. Transcriptional dysregulation of 5-HT1A autoreceptors in mental illness. Mol Brain. 2011;4:21. doi: 10.1186/1756-6606-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song NN, et al. Adult raphe-specific deletion of lmx1b leads to central serotonin deficiency. PLoS One. 2011;6:e15998. doi: 10.1371/journal.pone.0015998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holmes A. Genetic variation in cortico-amygdala serotonin function and risk for stress-related disease. Neurosci Biobehav Rev. 2008;32:1293–1314. doi: 10.1016/j.neubiorev.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waider J, Araragi N, Gutknecht L, Lesch KP. Tryptophan hydroxylase-2 (TPH2) in disorders of cognitive control and emotion regulation: a perspective. Psychoneuroendocrinology. 2011;36:393–405. doi: 10.1016/j.psyneuen.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Murphy DL, et al. How the serotonin story is being rewritten by new gene-based discoveries principally related to SLC6A4, the serotonin transporter gene, which functions to influence all cellular serotonin systems. Neuropharmacology. 2008;55:932–960. doi: 10.1016/j.neuropharm.2008.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beaulieu JM, et al. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105:1333–1338. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Richardson-Jones JW, et al. Serotonin-1A Autoreceptors Are Necessary and Sufficient for the Normal Formation of Circuits Underlying Innate Anxiety. J Neurosci. 2011;31:6008–6018. doi: 10.1523/JNEUROSCI.5836-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alenina N, et al. Growth retardation and altered autonomic control in mice lacking brain serotonin. Proc Natl Acad Sci U S A. 2009;106:10332–10337. doi: 10.1073/pnas.0810793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hariri AR, Holmes A. Genetics of emotional regulation: the role of the serotonin transporter in neural function. Trends Cogn Sci. 2006;10:182–191. doi: 10.1016/j.tics.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 64.Lesch KP, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1531. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- 65.Wendland JR, et al. A novel, putative gain-of-function haplotype at SLC6A4 associates with obsessive-compulsive disorder. Human molecular genetics. 2008;17:717–723. doi: 10.1093/hmg/ddm343. [DOI] [PubMed] [Google Scholar]
- 66.Caspi A, et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- 67.Bennett AJ, et al. Early experience and serotonin transporter gene variation interact to influence primate CNS function. Molecular psychiatry. 2002;7:118–122. doi: 10.1038/sj.mp.4000949. [DOI] [PubMed] [Google Scholar]
- 68.Prasad HC, et al. Human serotonin transporter variants display altered sensitivity to protein kinase G and p38 mitogen-activated protein kinase. Proc Natl Acad Sci U S A. 2005;102:11545–11550. doi: 10.1073/pnas.0501432102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sutcliffe JS, et al. Allelic Heterogeneity at the Serotonin Transporter Locus (SLC6A4) Confers Susceptibility to Autism and Rigid-Compulsive Behaviors. Am J Hum Genet. 2005;77:265–279. doi: 10.1086/432648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blakely RD. Overview: a rare opportunity or just one less reason to be depressed. Neuron. 2005;48:701–702. doi: 10.1016/j.neuron.2005.11.029. author reply 705–706. [DOI] [PubMed] [Google Scholar]
- 71.Zhang X, et al. Loss-of-Function Mutation in Tryptophan Hydroxylase-2 Identified in Unipolar Major Depression. Neuron. 2005;45:11–16. doi: 10.1016/j.neuron.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 72.Jacobsen JP, et al. Deficient serotonin neurotransmission and depression-like serotonin biomarker alterations in tryptophan hydroxylase 2 (Tph2) loss-of-function mice. Molecular psychiatry. 2011 doi: 10.1038/mp.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bethea CL, et al. Effects of citalopram on serotonin and CRF systems in the midbrain of primates with differences in stress sensitivity. Journal of chemical neuroanatomy. 2011;41:200–218. doi: 10.1016/j.jchemneu.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dai JX, et al. Enhanced contextual fear memory in central serotonin-deficient mice. Proc Natl Acad Sci U S A. 2008;105:11981–11986. doi: 10.1073/pnas.0801329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Erickson JT, Shafer G, Rossetti MD, Wilson CG, Deneris ES. Arrest of 5HT neuron differentiation delays respiratory maturation and impairs neonatal homeostatic responses to environmental challenges. Respir Physiol Neurobiol. 2007;159:85–101. doi: 10.1016/j.resp.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Erickson JT, Sposato BC. Autoresuscitation responses to hypoxia-induced apnea are delayed in newborn 5-HT-deficient Pet-1 homozygous mice. J Appl Physiol. 2009;106:1785–1792. doi: 10.1152/japplphysiol.90729.2008. [DOI] [PubMed] [Google Scholar]
- 77.Hodges MR, Best S, Richerson GB. Altered ventilatory and thermoregulatory control in male and female adult Pet-1 null mice. Respir Physiol Neurobiol. 2011;177:133–140. doi: 10.1016/j.resp.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hodges MR, et al. Defects in breathing and thermoregulation in mice with near-complete absence of central serotonin neurons. J Neurosci. 2008;28:2495–2505. doi: 10.1523/JNEUROSCI.4729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hodges MR, Wehner M, Aungst J, Smith JC, Richerson GB. Transgenic mice lacking serotonin neurons have severe apnea and high mortality during development. J Neurosci. 2009;29:10341–10349. doi: 10.1523/JNEUROSCI.1963-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao ZQ, et al. Mice lacking central serotonergic neurons show enhanced inflammatory pain and an impaired analgesic response to antidepressant drugs. J Neurosci. 2007;27:6045–6053. doi: 10.1523/JNEUROSCI.1623-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cummings KJ, et al. Failed heart rate recovery at a critical age in 5-HT-deficient mice exposed to episodic anoxia: implications for SIDS. Journal of applied physiology. 2011;111:825–833. doi: 10.1152/japplphysiol.00336.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cummings KJ, Li A, Deneris ES, Nattie EE. Bradycardia in serotonin-deficient Pet-1−/− mice: influence of respiratory dysfunction and hyperthermia over the first 2 postnatal weeks. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1333–1342. doi: 10.1152/ajpregu.00110.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cummings KJ, Li A, Nattie EE. Brainstem serotonin deficiency in the neonatal period: autonomic dysregulation during mild cold stress. The Journal of physiology. 2011;589:2055–2064. doi: 10.1113/jphysiol.2010.203679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Paulus EV, Mintz EM. Developmental disruption of the serotonin system alters circadian rhythms. Physiology & behavior. 2011;105:257–263. doi: 10.1016/j.physbeh.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 85.Lerch-Haner JK, Frierson D, Crawford LK, Beck SG, Deneris ES. Serotonergic transcriptional programming determines maternal behavior and offspring survival. Nat Neurosci. 2008;11:1001–1003. doi: 10.1038/nn.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Phillips PC. Epistasis--the essential role of gene interactions in the structure and evolution of genetic systems. Nature reviews Genetics. 2008;9:855–867. doi: 10.1038/nrg2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dreyer SD, et al. Mutations in LMX1B cause abnormal skeletal patterning and renal dysplasia in nail patella syndrome. Nature genetics. 1998;19:47–50. doi: 10.1038/ng0598-47. [DOI] [PubMed] [Google Scholar]
- 88.Dunston JA, et al. The human LMX1B gene: transcription unit, promoter, and pathogenic mutations. Genomics. 2004;84:565–576. doi: 10.1016/j.ygeno.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 89.Chen H, et al. Limb and kidney defects in Lmx1b mutant mice suggest an involvement of LMX1B in human nail patella syndrome. Nat Genet. 1998;19:51–55. doi: 10.1038/ng0598-51. [DOI] [PubMed] [Google Scholar]
- 90.Vollrath D, et al. Loss-of-function mutations in the LIM-homeodomain gene, LMX1B, in nail-patella syndrome. Human molecular genetics. 1998;7:1091–1098. doi: 10.1093/hmg/7.7.1091. [DOI] [PubMed] [Google Scholar]
- 91.Lopez-Arvizu C, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in Nail-patella syndrome: potential association with LMX1B loss-of-function. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59–66. doi: 10.1002/ajmg.b.31138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deneris ES. Molecular genetics of mouse serotonin neurons across the lifespan. Neuroscience. 2011 doi: 10.1016/j.neuroscience.2011.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cirulli ET, Goldstein DB. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nature reviews Genetics. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 94.McClellan J, King MC. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 95.State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nature neuroscience. 2011;14:1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dolmazon V, et al. Forced Expression of LIM Homeodomain Transcription Factor 1b Enhances Differentiation of Mouse Embryonic Stem Cells into Serotonergic Neurons. Stem Cells Dev. 2010 doi: 10.1089/scd.2010.0224. [DOI] [PubMed] [Google Scholar]
- 97.Tokuyama Y, Ingram SL, Woodward JS, Bethea CL. Functional characterization of rhesus embryonic stem cell-derived serotonin neurons. Exp Biol Med (Maywood) 2010;235:649–657. doi: 10.1258/ebm.2010.009307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Akil H, et al. Medicine. The future of psychiatric research: genomes and neural circuits. Science. 2010;327:1580–1581. doi: 10.1126/science.1188654. [DOI] [PMC free article] [PubMed] [Google Scholar]