

SUMMARY
Systemic lupus erythematosus is a potentially fatal autoimmune disease. Although interleukin-17 (IL-17) has been linked to human lupus and mouse models of this disease, it has not been addressed whether this cytokine plays a critical role in fatal lupus pathology. Here we have demonstrated that increased production of IL-17 cytokines and their signaling via the adaptor protein CIKS (a.k.a. Traf3ip2, Act1) critically contributed to lethal pathology in an FcgammaR2b-deficient mouse model of lupus. Mice lacking IL-17 and especially those lacking CIKS showed greatly improved survival and were largely protected from development of glomerulonephritis. Importantly in this model, potential effects of IL-17 cytokines on antibody production could be distinguished from critical local contributions in kidneys, including recruitment of neutrophils and monocytes. These findings provide the proof of principle that signaling by IL-17 family cytokines mediated via CIKS presents promising therapeutic targets for the treatment of systemic lupus erythematosus, especially in cases with kidney involvement.
INTRODUCTION
Systemic lupus erythematosus (SLE) is one of the most common and devastating systemic autoimmune diseases. In health, various tolerance mechanisms prevent the generation of autoreactive B cells, but once these mechanisms are breached, autoantibodies may be produced, causing immune complex deposition and inflammation. Unless SLE patients receive effective therapies, chronic activation of the immune system and local inflammation may cause permanent organ damage, including glomerulonephritis, leading to renal failure and death.
To elucidate mechanisms underlying the pathogenesis of SLE, we investigated Fcgamma receptor II-b (Fcgr2b)-deficient mice (generated with 129 embryonic stem cells [ESCs], extensively backcrossed to C57BL/6), which develop a fatal lupus-like disease (Bolland and Ravetch, 2000; Daëron et al., 1995). Fcgr2b has been identified as a lupus susceptibility gene in both humans and mice. This inhibitory receptor is thought to help maintain tolerance mechanisms that prevent formation of autoantibody-producing B cells and restrain inflammation in response to immune complex deposition (Baerenwaldt et al., 2011; Smith and Clatworthy, 2010). 129-derived gene variants immediately surrounding the deleted Fcgr2b locus in this mouse background are likely to also play a role in the breach in tolerance, consistent with the multigenic nature of lupus (Boross et al., 2011; Harley et al., 2008; Sato-Hayashizaki et al., 2011).
Interleukin-17 (IL-17, a.k.a. IL-17A) is the signature cytokine of T helper-17 cells, and these cells also produce the closely related IL-17F; both cytokines have been linked to the development of various autoimmune diseases, including multiple sclerosis (MS), rheumatoid arthritis (RA), and SLE. Patients with these diseases express increased amounts of IL-17A and IL-17F (Doreau et al., 2009; Matusevicius et al., 1999; Ziolkowska et al., 2000). Furthermore, IL-17A and IL-17F have been shown to be functionally relevant in the pathogenesis of collagen-induced arthritis (CIA) and experimental autoimmune encephalomyelitis (EAE) in mice; in addition, IL-17C is involved in psoriasis and may also contribute to other diseases, including EAE (Chang et al., 2011; Komiyama et al., 2006; Nakae et al., 2003; Ramirez-Carrozzi et al., 2011). IL-17A, IL-17F, and IL-17C are members of an extended family of IL-17 cytokines (A–F) that signal via heteromeric receptors composed of members of the IL-17 receptor family (RA–RE) (Gaffen, 2009). CIKS (a.k.a. Traf3ip2 or Act1) is an adaptor protein required for signaling by these cytokines (Chang et al., 2011; Gaffen, 2009). Consistent with a role for IL-17 cytokines in disease, the CIKS (Traf3ip2; Act1) adaptor is essential for development of CIA and EAE (Pisitkun et al., 2010; Qian et al., 2007).
Recombinant inbred BXD2 mice develop erosive arthritis and a lupus-like condition. Loss of Il17ra, a receptor chain required for signaling by IL-17 cytokines, abrogates spontaneous germinal center formation in this mouse model, whereas addition of IL-17 exacerbates this process (Hsu et al., 2008). IL-17 also synergizes with “B cell activating factor belonging to the TNF family” to promote the survival and/or proliferation of human B cells and their differentiation into antibody secreting cells in vitro (Doreau et al., 2009). These findings suggest possible roles for IL-17 in autoantibody production; however, it has not been addressed whether such contributions are actually relevant for the fatal outcome in the BXD2 mouse model. Other studies have provided evidence for increased amounts of IL-17 in kidneys of SLE patients and Mrl/lpr and BL6/lpr lupus-prone mice and for a functional role of IL-23 in the latter model (Crispín et al., 2008; Kyttaris et al., 2010). These findings suggest involvement of T helper 17 (Th17) cells and/or IL-17 cytokines in kidney pathology, but it has not been addressed whether IL-17 cytokines are in fact functionally relevant in development of fatal kidney disease in mouse models for lupus.
Here we demonstrate that CIKS (Traf3ip2) adaptor-mediated signaling from IL-17 cytokines, including IL-17A and likely also IL-17C, plays a major role in the development of fatal lupus pathology in Fcgr2b−/− mice. Loss of CIKS, which blocks signaling by all IL-17 cytokines, and to a lesser extent loss of IL-17A, significantly reduced mortality rates, specifically protecting mice from glomerulonephritis, by eliminating an important pathway for chemokine-mediated recruitment of inflammatory cells into kidneys, in particular neutrophils. Contributions of IL-17 cytokines to end-organ pathology were crucial for progression of the disease, and without these contributions autoantibodies alone were largely insufficient to cause severe kidney pathology. Consistent with this, signaling of these cytokines into B cells did not significantly contribute to the course of glomerulonephritis in Fcgr2b−/− mice. Importantly, we detected neutrophil extracellular traps (NETs) in kidneys of Fcgr2b−/− mice, but not in the absence of IL-17 cytokines signaling. We also demonstrated a requirement for CIKS-mediated signaling in a glomerular basement membrane (GBM) antibody-induced model of glomerulonephritis, providing further evidence for a direct role of IL-17 cytokines “downstream” of autoantibodies. These findings identify IL-17 cytokines and the CIKS signaling adaptor as potential therapeutic targets in lupus nephritis.
RESULTS
Loss of the IL-17 Receptor Adaptor CIKS Improves Survival of Fcgr2b−/− Mice
We first examined IL-17 cytokine expression in the Fcgr2b−/− mice at or shortly after the onset of pathology at about 6 months of age. We detected an increased number of IL-17(A) and IL-17F producers among CD3+ T cells, especially in lymph nodes, and to a lesser extent spleens (Figures 1A and 1B; see also Figure S1 available online). The fraction of CD4−CD8−CD3+ (double-negative, DN-T) cells was specifically increased in lymph nodes, a population reportedly expanded in SLE patients and a lupus mouse model (Crispín et al., 2008; Kyttaris et al., 2010) (Figures 1C and 1D). We observed significant increases in IL-17A and/or IL-17F producing CD4+ T cells (Th17 cells) in spleens and IL-17A and/or IL-17F producing DN-T cells in lymph nodes (Figures 1E and 1F; there was also a trend toward an increase in Th17 cells in lymph nodes). The IL-17-producing DN-T cell population in lymph nodes included γδ+ and αβ+ T cells, but not invariant natural killer T (iNKT) cells (Figure S1).
Figure 1. Loss of the IL-17 Signaling Adaptor CIKS Improves Survival of Fcgr2b−/− Mice.

(A) Cells from spleen and lymph node (LN) of WT and Fcgr2b−/− mice (6–8 months old) were gated on CD3+ and stained for intracellular expression of IL-17A and IL-17F. A profile representative of five mice per group is shown.
(B) Percentage of IL-17A and F producing T cells in spleen and LN (n = 5–7).
(C and D) Percentage of CD3+ T subsets in spleen (n = 5–9) and LN (n = 10–16). DN-T cells are CD3+CD4−CD8−.
(E and F) Percentage and numbers of IL-17 producing CD3+ T subsets in spleen (n = 4–7) and LN (n = 7–10). (*p < 0.05, **p < 0.01, ***p < 0.001; mean ± SEM).
(G) Cumulative survival of Fcgr2b−/−.Traf3ip2+/− mice (n = 13) and Fcgr2b−/−.Traf3ip2−/− mice (n = 12); (***p < 0.001). Percent survival rate change = ([% survival of Fcgr2b−/−.Traf3ip2−/− − % survival of Fcgr2b−/−.Traf3ip2+/−] /% survival of Fcgr2b−/−.Traf3ip2−/−) × 100 = ([75 − 7.7]/75) × 100 = 89.7%.
Next we asked whether IL-17 cytokines play a critical role in the fatal outcome in this lupus model. We crossed Fcgr2b−/− mice with Traf3ip2−/− C57BL/6 mice to generate doubly deficient mice (Fcgr2b−/−.Traf3ip2−/−) and littermate controls deficient in Fcgr2b and heterozygous for Traf3ip2 (Traf3ip2 heterozygosity did not alter phenotypes of wild-type [WT] or Fcgr2b−/− mice). Mice were followed for up to 12 months of age, at which time just 7.7% (1 of 13) of Fcgr2b−/− were still alive, whereas 75% (9 of 12) of Fcgr2b−/−.Traf3ip2−/− mice had survived (Figure 1G; an 89.7% improvement of survival rates). This significant difference in survival suggests that Traf3ip2 (CIKS) plays an important role in development of fatal lupus pathology in the Fcgr2b−/− mice.
CIKS Contributes to Germinal Center Formation in Fcgr2b−/− Mice
Unlike WT mice, Fcgr2b−/− mice readily formed spontaneous germinal centers and expanded their plasma cell numbers; however, in mice also lacking Traf3ip2 these phenotypes were substantially, though not completely, reversed (Figures 2A–2E; Figure S2). We also assayed for preswitched (germline) and postswitched immunoglobulin transcripts with semi-quantitative RT-PCR to confirm these findings (Figure S2). We noted increased expression of MHC class II (H2-Ab1) on B cells in Fcgr2b−/− mice, and this expression was significantly reduced in mice also lacking Traf3ip2 (Figures 2F and 2G). Together, these results suggest that IL-17 cytokines signaling via CIKS (Traf3ip2) contribute to spontaneous germinal center B cell formation, plasma cell development, and MHC class II expression on B cells in lupus-prone Fcgr2b−/− mice.
Figure 2. CIKS Contributes to Germinal Center Formation in Fcgr2b−/− Mice.
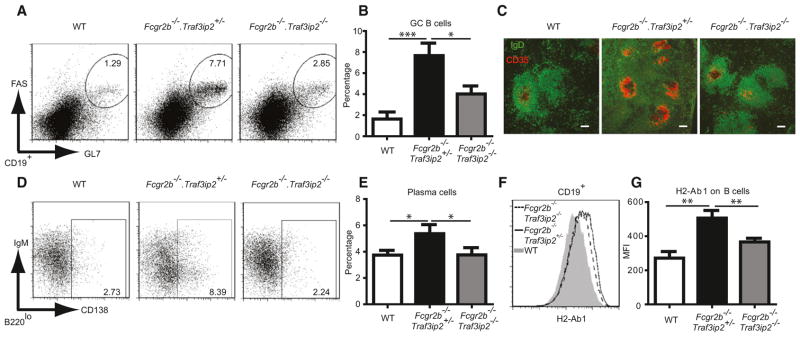
(A and B and D and E) Flow cytometric analysis of spleen cells of WT, Fcgr2b−/−.Traf3ip2+/−, and Fcgr2b−/−.Traf3ip2−/− mice. Representative staining profiles and percentages of (A and B) germinal center B cells (CD19+GL7hiFAShi) and (D and E) plasma cells (B220loCD138+) are shown.
(C) Immunofluorescence staining of spleen sections for IgD (green) and CD35 (red) expression. Scale bar = 100 μm. Data are representative of five mice per group.
(F and G) Representative histogram (F) and mean fluorescence intensity (MFI) (G) for H2-Ab1 on CD19+ cells (*p < 0.05, **p < 0.01, ***p < 0.001; mean ± SEM; n = 7–9).
Loss of CIKS in B Cells Does Not Block Development of Glomerulonephritis
To determine whether IL-17 cytokines directly targeted B cells to promote formation of spontaneous germinal centers in Fcgr2b−/− mice and to cause disease, we generated mice in which Traf3ip2 was conditionally deleted in B cells with Mb1-driven Cre recombinase (Figure S3; Fcgr2b−/−.Traf3ip2fl/fl-Mb1). Although loss of Traf3ip2 in B cells of Fcgr2b−/− mice clearly reduced splenomegaly and total number of B cells and GC B cells, it did not significantly reduce the percentage of GC B cells or plasma cells, although there was a trend toward lower numbers (Figures 3A and 3B; Figure S3). Expression of H2-Ab1 on B cells of Fcgr2b−/−.Traf3ip2fl/fl/Mb1 mice was reduced compared to Fcgr2b−/−.Traf3ip2wt/fl mice. Importantly, however, Fcgr2b−/− mice lacking Traf3ip2 in B cells still developed glomerulonephritis comparable to Traf3ip2 sufficient Fcgr2b−/− mice (Figures 3D and 3E). Thus CIKS (Traf3ip2)-mediated signaling by IL-17 cytokines in B cells in Fcgr2b−/− mice promotes the overall increase in spleen size and total number of B cells but is not required for the increased percentage of GC B cells or for development of glomerulonephritis.
Figure 3. Loss of IL-17 Cytokine Signaling in B Cells Does Not Block Development of Glomerulonephritis.

(A) Flow cytometric analysis of splenocytes of WT, Fcgr2b−/−.Traf3ip2wt/fl, and Fcgr2b−/−.Traf3ip2fl/fl-Mb1 mice, showing percentage and total numbers of CD19+GL7hiFAShi (GC B cells). (*p < 0.05, ***p < 0.001; mean ± SEM; n = 9–11).
(B) Immunofluorescence staining of spleen sections for IgD (green) and CD35 (red) expression. Scale bar = 100 μm. Data representative of five mice per group.
(C) Representative histogram and mean fluorescence intensity (MFI) for H2-Ab1 on CD19+ cells (*p < 0.05; mean ± SEM; n = 9–11).
(D) Upper panels show H&E staining (40×) showing increased glomerular hypercellularity in kidney sections of Fcgr2b−/−.Traf3ip2wt/fl and Fcgr2b−/−. Traf3ip2fl/fl-Mb1 mice (6–8 months old). Lower panels show PAS staining (40X) showing increased numbers of intracapillary leukocytes and double contours of the glomerular basement membrane in kidney sections of Fcgr2b−/−.Traf3ip2wt/fl and Fcgr2b−/−.Traf3ip2fl/fl-Mb1 mice. Data are representative of 8–10 mice per group. Scale bar = 50 μm.
(E) Glomerular scores and interstitial scores of Fcgr2b−/−.Traf3ip2wt/fl and Fcgr2b−/−.Traf3ip2fl/fl-Mb1 mice (mean ± SEM; n = 8–10).
CIKS-Mediated Signaling Is Not Required for Autoantibody Production but Is Critical for End-Organ Pathology in Fcgr2b−/− Mice
Despite reduced numbers of spontaneous germinal centers and splenic plasma cells in Fcgr2b−/−.Traf3ip2−/− as compared to Fcgr2b−/−.Traf3ip2+/− littermates, the titers and staining patterns for anti-nuclear antibodies (ANA) did not differ (Figures 4A and 4B). Similarly, we did not observe any reduction in double-stranded DNA (dsDNA) IgG antibodies (regardless of isotype), nRNP antibodies, total serum IgG or IgG deposition in kidneys in the absence of Traf3ip2 in Fcgr2b−/− mice (Figures 4C and 4D; Figure S4). However, dsDNA antibody amounts do not always correlate with the clinical activities of lupus nephritis (Christensen et al., 2005; Mok, 2010). Indeed, mice deficient in Traf3ip2 exhibited markedly reduced infiltration of inflammatory cells into kidneys (Figure 4E). Glomerular pathology was greatly ameliorated in Traf3ip2 deficient Fcgr2b−/− mice as visualized in H&E and PAS stained sections (Figures 4F and 4G) and quantified as glomerular and interstitial scores (Figures 4H and 4I).
Figure 4. CIKS-Mediated Signaling Is Not Required for Autoantibody Production but Is Critical for End-Organ Pathology in Fcgr2b−/− Mice.

(A) Anti-nuclear antibody (ANA) staining of Hep-2 cell coated slides incubated with diluted serum (1:100) from Fcgr2b−/−.Traf3ip2+/− and Fcgr2b−/−.Traf3ip2−/− mice. Data are representative of ten mice per group. Scale bar = 50 μm.
(B) ANA titers; shown are final dilution factors of serum still ANA positive (mean ± SEM; n = 10).
(C) ELISA assays for dsDNA IgG (mean ± SEM; n = 7–10).
(D) Immunofluorescence staining for IgG deposition in kidney sections from Fcgr2b−/−.Traf3ip2+/− and Fcgr2b−/−.Traf3ip2−/− mice. Data are representative of 7 or 8 mice per group. Scale bar = 50 μm.
(E) H&E staining of kidney sections (20X) showing substantial leukocyte infiltration in the interstitium of Fcgr2b−/−.Traf3ip2+/− but not Fcgr2b−/−.Traf3ip2−/− mice. Scale bar = 100 μm.
(F) H&E staining (40X) showing glomerular hypercellularity with intracapillary proliferation, and periglomerular leukocyte infiltration in kidney sections of Fcgr2b−/−.Traf3ip2+/−, but not Fcgr2b−/−.Traf3ip2−/− mice (6–8 months old).
(G) PAS staining (40X) showing increased numbers of intracapillary leukocytes and double contours of the glomerular basement membrane in Fcgr2b−/−.Traf3ip2+/− but not Fcgr2b−/−.Traf3ip2−/− mice. Data are representative of 7–10 mice per group. Scale bar = 50 μm.
(H and I) Glomerular scores and interstitial scores of Fcgr2b−/−.Traf3ip2+/− and Fcgr2b−/−.Traf3ip2−/− mice (*p < 0.05, **p < 0.01; mean ± SEM; n = 7–10).
(J) Percentage of IFN-γ producing cells from splenocytes evaluated by flow cytometry (*p < 0.05; mean ± SEM; n = 5–7).
Our results suggest that although signaling of IL-17 cytokines via CIKS was able to promote formation of germinal centers in Fcgr2b−/− mice, this was not critical for autoantibody production or IgG deposition. Autoantibody production in this Fcgr2b−/− model is likely to be controlled by other factors. IFN-γ is reported to be critical for autoantibody production and IgG deposition in kidneys of lupus-prone (NZBxNZW)F1 mice as well as for development of lupus nephritis (Haas et al., 1998; Peng et al., 1997; Richards et al., 2001). We observed a significant expansion of IFN-γ producing cells, mainly T cells, in spleens of Fcgr2b−/− mice, irrespective of the presence of Traf3ip2 (Figure 4J; Figure S4). Therefore IFN-γ may promote autoantibody production independent of IL-17 cytokines. These findings suggest that CIKS-mediated signaling by IL-17 cytokines contributes to the fatal outcome in this lupus model in a manner separate from autoantibody formation and IgG deposition.
We also observed increases in CD11b+CD11c− monocytic cells, CD11b+CD11c+ myeloid DCs (mDCs), and CD11c+B220+ plasmacytoid DCs (pDCs) in spleens from Fcgr2b−/− mice; notably, these increases were reversed in the absence of Traf3ip2. Furthermore the shift toward increased numbers of T effector-memory cells (CD4+CD62L−CD44+) in this lupus model was partially reversed in the absence of CIKS (Figure S4). Therefore, CIKS-mediated signaling by IL-17 cytokines in Fcgr2b−/− mice promotes increases in numbers of monocytic cells, mDCs, and pDCs and contributes to activation and/or differentiation of T cells. These latter systemic changes could have had a role in initiating kidney pathology but could also have occurred as a consequence thereof.
Loss of IL-17A Does Not Reduce Formation of Spontaneous Germinal Centers but Does Protect against Fatal Glomerulonephritis in Fcgr2b−/− Mice
The CIKS adaptor is not only required for signaling by IL-17 but also other members of this cytokine family (Chang et al., 2011; Gaffen, 2009). We crossed Fcgr2b−/− mice with Il17a−/− mice to determine whether IL-17 and/or other members of this family might be critical for the pathogenesis in this lupus model. Although only 35.7% (5 of 14) of Fcgr2b−/−.Il17a+/− mice were alive after 12 months, this number increased to 91.7% (11 of 12) of Fcgr2b−/−.Il17a−/− littermates (Figure 5A; a 61.1% improvement of survival rates). This suggests that IL-17(A) plays an important, albeit not exclusive role in the CIKS-dependent fatal outcome of Fcgr2b−/− mice. (Fcgr2b−/−.Il17a+/− mice were somewhat less susceptible to lupus pathology, most likely due to a subtly different BL6 background of Il17a−/− mice; there was no apparent difference between Fcgr2b−/−.Il17a+/+ and Fcgr2b−/−.Il17a+/− mice). The protection afforded by loss of Il17a also failed to correlate with any reduction in ANA or dsDNA antibodies (Figures 5B and 5C). Unexpectedly, Il17a deficient mice failed to show any reduction in the spontaneous germinal centers that developed in Fcgr2b−/− mice (Figure 5D; Figure S5). Consistent with this, immunoglobulin isotype switching, numbers of plasma cells, and H2-Ab1 expression on B cells in the Fcgr2b−/− mice were not changed in the absence of Il17a (Figure S5). Because expression of IL-17B or IL-17C did not change in spleens of Fcgr2b−/− mice compared to WT mice (Figure S5), the increased amounts of IL-17F likely compensated for loss of IL-17A (Figures 1A and 1B; Figure S5), although this remains to be formally proven.
Figure 5. Loss of IL-17A Does Not Reduce Formation of Spontaneous Germinal Centers but Does Protect against Fatal Glomerulonephritis in Fcgr2b−/− Mice.

(A) Cumulative survival of Fcgr2b−/−.Il17a+/− mice (n = 14) and Fcgr2b−/−.Il17a−/− mice (n = 12); (***p < 0.001). Percent survival rate change = ([% survival of Fcgr2b−/−.Il17a−/− − % survival of Fcgr2b−/−.Il17+/−] /% survival of Fcgr2b−/−.Il17a−/−) × 100 = ([91.7 − 35.7]/91.7) × 100 = 61.1%.
(B) ANA staining of Hep-2 cell coated slides incubated with diluted serum (1:100) from Fcgr2b−/−.Il17a+/−, and Fcgr2b−/−.Il17a−/− mice. Data are representative of 10 mice per group. Scale bar = 50 μm.
(C) ELISA assays for dsDNA IgG (mean ± SEM; n = 11).
(D) Representative flow cytometric staining profile for germinal center B cells (CD19+GL7hiFAShi) from mice as shown; data representative of ten mice per group. Immunofluorescence staining of spleen sections for IgD (green) and CD35 (red) expression. Scale bar = 100 μm. Data are representative of five mice per group.
(E) Upper panels show H&E staining (40X) showing glomerular hypercellularity with intracapillary proliferation in kidney sections of Fcgr2b−/−.Il17a+/− but not Fcgr2b−/−.Il17a−/− mice (10–12 months old). Lower panels show PAS staining (40X) showing increased numbers of intracapillary leukocytes and double contours of the glomerular basement membrane in kidney sections of Fcgr2b−/−.Il17a+/− but not Fcgr2b−/−.Il17a−/− mice. Data are representative of 7–9 mice per group. Scale bar = 50 μm.
(F) Glomerular and interstitial scores of Fcgr2b−/−.Il17a+/− and Fcgr2b−/−.Il17a−/− mice (**p < 0.01; mean ± SEM; n = 7–9).
The fact that the survival of Fcgr2b−/− mice was significantly improved in the absence of IL-17, while spontaneous germinal center formation and autoantibody production continued unabated, strongly points to a critical and likely direct role for IL-17-mediated signaling in fatal end-organ pathologies. Consistent with this hypothesis, loss of Il17a significantly reduced inflammatory cell infiltration and glomerulonephritis in kidneys of Fcgr2b−/− mice (Figures 5E and 5F).
Loss of CIKS or IL-17A Prevents Recruitment of Inflammatory Cells into Kidneys of Fcgr2b−/− Mice
We investigated which, if any, of the potential IL-17-producing T cell subsets could be detected in perfused kidneys of Fcgr2b−/− mice. We noted significant increases in numbers of DN-T and CD4+ T cells and, to a lesser degree, CD8+ T cells in kidneys of Fcgr2b−/− mice (Figure 6A). Surprisingly, loss of Traf3ip2 reversed the numbers of DN-T cells (Figure 6A). In contrast, the fraction of DN-T cells in lymph nodes did not change in the absence of Traf3ip2, suggesting that IL-17 cytokines might play a role in recruitment and/or maintenance of these cells in kidneys (Figure 6B). We also performed intracellular staining of cells isolated from kidneys and found that both DN-T cells and CD4+ T cells produced IL-17 (Figure S6). Next we tested for expression of IL-17 cytokines in kidneys of Fcgr2b−/− mice. We detected a significant increase in IL-17 as well as IL-17C, but not IL-17F or IL-17B messenger RNA (mRNA) expression in Fcgr2b−/− mice (Figure 6C). Preferentially increased expression of IL-17 is consistent with the fact that loss of this cytokine afforded significant protection (Figure 5A). Nevertheless, since loss ofTraf3ip2 improved survival rates of Fcgr2b−/− mice more than loss of Il17a (89.7% versus 61.1%), it is likely that IL-17C also contributed to kidney pathology (see Discussion).
Figure 6. Loss of CIKS or IL-17A Prevents Recruitment of Inflammatory Cells into Kidneys of Fcgr2b−/− Mice.

(A) Flow cytometric analysis of cells isolated from kidneys of WT, Fcgr2b−/−.Traf3ip2+/− and Fcgr2b−/−.Traf3ip2−/− mice, showing numbers of CD3+, CD3+ CD4−CD8− (DN-T), CD3+CD4+, and CD3+CD8+ cells (*p < 0.05, **p < 0.01; mean ± SEM; n = 13).
(B) Percentage of DN-T (CD3+CD4−CD8−) in LNs of mice as shown (mean ± SEM; n = 5–8).
(C) Fold change in relative mRNA expression of Il17a, Il17f, Il17b, and Il17c in kidneys of WT and Fcgr2b−/− mice (*p < 0.05, **p < 0.01; mean ± SEM; n = 4–5).
(D and E) Representative staining profiles of cells isolated from kidneys of WT, Fcgr2b−/−.Traf3ip2+/−, and Fcgr2b−/−.Traf3ip2−/− mice showing (D) neutrophils (Ly6g+Ly6c+) and (E) inflammatory monocytes (CD11b+F4/80lo) and macrophages (CD11b+F4/80hi) after gating on Ly6c+Ly6g− cells. Data are representative of 13 mice per group.
(F) Immunofluorescence of kidney sections from Fcgr2b−/−.Traf3ip2+/− and Fcgr2b−/−.Traf3ip2−/− mice (6–8 months old) or Fcgr2b−/−.Il17a+/− and Fcgr2b−/−.Il17a−/− mice (10–12 months old) stained with anti-IgG (green), anti-7/4 (red), and anti-Ly6g (blue) to identify immune complexes, inflammatory monocytes and macrophages (7/4+Ly6g−), and neutrophils (7/4+Ly6g+).
(G) Relative mRNA expression (normalized by actin) of Cxcl1, Cxcl5, Ccl2, Ccl20, and TNF-α in kidney tissues of WT, Fcgr2b−/−.Traf3ip2+/−, and Fcgr2b−/−.Traf3ip2−/− mice (*p < 0.05, **p < 0.01; mean ± SEM; n = 6–8).
(H) Immunofluorescence of kidney sections (genotypes as indicated), stained with anti-MPO (green), anti-LL37 (red), and DAPI (blue). Scale bar = 50 μm. Data are representative of 4–6 mice per group.
We detected increased numbers of neutrophils (Ly6g+Ly6c+) and monocytic cells (Ly6c+Ly6g− CD11b+F4/80lo) in perfused kidneys of Fcgr2b−/− mice; by contrast, these increases were largely erased in the absence of Traf3ip2 or Il17a (Figures 6D and 6E; Figure S6). However, although the absence of Traf3ip2 in this lupus model largely prevented the increase in DN-T cells, this was not the case in mice lacking Il17a (Figure S6); therefore IL-17C could have had a role in recruitment and/or maintenance of these cells in kidneys. To determine the anatomical locations of infiltrating myeloid cells in kidneys of Fcgr2b−/− mice, and to confirm the absence of such cells in mice lackingTraf3ip2 and Il17a, we stained tissue sections of kidneys for neutrophils (7/4+Ly6g+), monocytes and macrophages (7/4+Ly6g−) (Daley et al., 2008; Rosas et al., 2010), and for IgG deposition. We observed substantial foci of infiltrating neutrophils and monocytic cells in Fcgr2b−/− mice; infiltration of these cells extended into interstitial regions. Loss of either Traf3ip2 or Il17a largely prevented infiltration of these cells, while IgG deposition remained (Figure 6F).
Next we examined whether CIKS-mediated signaling might contribute to increased expression of neutrophil- and monocyte-attracting chemokines in kidneys of Fcgr2b−/− mice. We observed CIKS (Traf3ip2)-dependent increases in Cxcl1, Cxcl5, Ccl2, and TNF-α and to a lesser extent Ccl20 mRNA expression (Figure 6G); TNF-α is known to strongly synergize with IL-17 (Hartupee et al., 2007). We extended these analyses to kidney biopsies from lupus nephritis patients and found footprints of IL-17 cytokines and TNF-α in these samples, suggesting that our findings may be relevant for development of lupus nephritis in patients (Figure S6).
Increased ability to form pathogenic neutrophil extracellular traps (NETs) and impairment in their degradation have been associated with lupus nephritis in humans (Hakkim et al., 2010). To test whether neutrophils recruited into kidneys of Fcgr2b−/− mice might develop NETs in tissue, we performed immunofluorescence staining and detected NETs in kidneys of Fcgr2b−/− mice, but not in mice also deficient in Traf3ip2 (Figure 6H).
These results indicate that CIKS-mediated signaling by IL-17 cytokines in kidneys of Fcgr2b−/− mice is involved in recruitment of DN-T cells, monocytic cells as well as neutrophils, with the latter cells able to form pathogenic NETs. These findings may explain the role of these cytokines in the development of fatal end-organ pathology.
CIKS Deficiency Protects Mice from the Development of Glomerular Basement Membrane Antibody-Induced Glomerulonephritis
The data suggest a critical role for CIKS-mediated signaling by IL-17 cytokines in development of glomerulonephritis, apparently acting downstream of autoantibodies. To directly test whether IL-17 cytokines can contribute to the pathogenic effects of autoantibodies, we challenged CIKS sufficient and deficient mice with glomerular basement membrane antibodies in an established model of accelerated nephrotoxic nephritis that is associated with glomerular damage (Vielhauer et al., 2005). Although IL-17 has been proposed to have a pathogenic role in similar models, it has also been suggested to have a protective role (Odobasic et al., 2011; Paust et al., 2012). We found an increase in expression of Il17a, but not Il17c or Il17f in kidneys of WT mice after treatment with CFA and GBM antibodies (Figure S7). The numbers of neutrophils, monocytes, and macrophages were also increased; by contrast, Traf3ip2-deficient mice showed significantly lower counts of neutrophils and monocytes and a trend toward lower counts of macrophages (Figures 7A–7D). These findings correlated well with decreased renal pathology scores (Figures 7E–7G) and inflammatory cell infiltrations (Figures 7H and 7I) inTraf3ip2-deficient mice. Of note, we only detected an increase in CD4+ but not DN-T cells in kidneys in this model (Figure S7). These data demonstrate that CIKS-mediated signaling is essential for disease in a model of anti-GBM mediated glomerulonephritis. This finding is consistent with a major role for IL-17 cytokines in renal injury at a step downstream of autoantibody production.
Figure 7. CIKS Deficiency Protects Mice from the Development of Antibody-Induced Glomerulonephritis.

(A) Representative staining profiles of cells isolated from kidneys of WT and Traf3ip2−/− mice showing neutrophils (Ly6g+Ly6c+). Data are representative of ten mice per group.
(B–D) Flow cytometric analysis of infiltrating cells in kidneys of WT and Traf3ip2−/− mice showing (B) neutrophils (Ly6g+Ly6c+), (C) inflammatory monocytes (Ly6c+Ly6g− CD11b+F4/80lo) and (D) macrophages (Ly6c+Ly6g− CD11b+F4/80hi). (*p < 0.05, **p < 0.01; mean ± SEM; n = 10).
(E–G) Histology scores showing percentage of (E) abnormal glomeruli, (F) crescent, and (G) interstitial scores of WT and Tra3ip2−/− mice (**p < 0.01, ***p < 0.001; mean ± SEM; n = 10).
(H and I) H&E staining (H) showing inflammatory cell infiltration in kidney sections of WT but not Traf3ip2−/− mice. PAS staining (I) showing glomerular hypercellularity, glomerular deposition of PAS stained material, and crescentric glomerunlonephritis of WT but not Traf3ip2−/− mice. Data are representative of ten mice per group. Scale bar = 50 μm.
DISCUSSION
Increased amounts of IL-17 in serum of SLE patients have been correlated with disease activity; in addition, increased production of IL-17 is prominent in two mouse models of lupus (Hsu et al., 2008; Kozyrev et al., 2008; Nalbandian et al., 2009). As shown here, elevated expression of IL-17 was also observed in lupus-prone Fcgr2b−/− mice, in particular in lymph nodes and kidneys. Beyond these observations, however, the work presented establishes that IL-17 cytokines and their signaling via the adaptor Traf3ip2 (CIKS, Act1) play critical roles in the ultimately fatal outcome in a lupus disease model: loss of Il17a or Traf3ip2 profoundly improved survival in Fcgr2b−/− mice, correlating with greatly ameliorated kidney pathology.
Lack of Traf3ip2 in Fcgr2b−/− mice afforded greater protection than lack of Il17a (89.7% versus 61.1% improvement in survival rates, respectively). Although this could be due to slight genetic background differences, it is more likely that in addition to major contributions from IL-17, IL-17C too may have played a role, given that it also signals via CIKS (Chang et al., 2011; Gaffen, 2009) and given that its expression was significantly increased in kidneys of Fcgr2b−/− mice as well. Alternatively, it remains at least theoretically possible that CIKS could have made contributions independent of IL-17 cytokines (Valente et al., 2012).
Although absence of CIKS led to reduced spontaneous germinal center formation in this model, surprisingly, lack of this adaptor (or of IL-17) did not lead to a noticeable reduction in ANA, dsDNA antibodies, or immunoglobulin deposition in kidneys. Aside from germinal centers, autoantibodies could also have arisen from extrafollicular B cells (Sweet et al., 2010). Importantly, loss of Il17a or Traf3ip2 did prevent the influx of inflammatory myeloid-derived cells into kidneys of Fcgr2b−/− mice. Therefore, the present model allowed us to separate potential inputs of IL-17 cytokines to autoantibody production from other, more direct contributions of these cytokines to kidney pathology. In further support of a critical local role of IL-17 cytokines in kidney disease, we demonstrated local production of IL-17 by infiltrating Th17 and DN-T cells, and of IL-17C, most likely by epithelial cells; in addition, we detected increased expression of TNF-α, known to synergize with IL-17 cytokines (Hartupee et al., 2007). The presence of these cytokines may represent a primary means for attracting inflammatory cells into kidneys via induced expression of chemoattractant genes such as Cxcl1, Cxcl5, Ccl2, and Ccl20 (Ramirez-Carrozzi et al., 2011; Ruddy et al., 2004; Shahrara et al., 2010).
Only loss of Traf3ip2, but not Il17a, notably reduced spontaneous germinal center formation. Given that we observed elevated amounts of IL-17F, but not IL-17B or IL-17C in spleens of Fcgr2b−/− mice, it is reasonable to suggest that IL-17F could have compensated for lack of IL-17 to promote spontaneous germinal center formation. Loss of Traf3ip2 in B cells largely reversed increases in spleen size, total GC B cells, and MHC Class II expression on B cells, but not the propensity of B cells to form germinal centers in spleens of Fcgr2b−/− mice, suggesting that IL-17 cytokines may also affect B cells indirectly.
Loss of IL-17RA in BXD2 mice caused definite reductions in autoantibodies and immunoglobulin deposition; in contrast, the absence of CIKS in Fcgr2b−/− mice did not, although spontaneous germinal center formation was reduced in both contexts. It is not clear why production of autoantibodies was more dependent on IL-17 cytokines in the BXD2 model than in the Fcgr2b−/− model. It has been reported that IFN-γ production is critical for autoantibody production in lupus-prone (NZBxNZW) F1 mice (Haas et al., 1998), and expression of this cytokine was elevated in Fcgr2b−/− mice, independent of IL-17 cytokine signaling; thus, IFN-γ may have been responsible for autoantibody production.
The local production of IL-17 in kidneys of Fcgr2b−/− mice from both CD4+ T cells and DN-T cells was likely instrumental in recruitment of neutrophils and monocytes, and thus the ensuing inflammation. The mechanisms leading to local IL-17 production remain to be determined. Interestingly, kidney infiltration of DN-T cells was dependent on CIKS-mediated signaling, but not IL-17, suggesting the possibility that IL-17C may have targeted these cells to exacerbate an already initiated, IL-17-dependent inflammatory process in Fcgr2b−/− mice. IL-17C has also been reported to enhance IL-17 production from infiltrating CD4+ T cells (Chang et al., 2011). Our data suggest that local production of IL-17 cytokines is critical for kidney pathology, consistent also with the observation that CIKS was required for glomerulonephritis and recruitment of neutrophils and monocytes into kidneys in the GBM antibody-induced model of glomerulonephritis. Of note, whereas CD4+ T cells numbers and Il17a expression were elevated in kidneys of Fcgr2b−/− mice and GBM antibody-treated mice, DN-T cells and Il17c were only elevated in Fcgr2b−/− mice.
Prior reports have suggested that the presence of IL-17-producing Th17 and DN-T cells in kidneys of SLE patients and in the MRL/lpr and B6/lpr mouse models (Crispín et al., 2008; Zhang et al., 2009). Consistent with this, IL-23 receptor deficiency in B6/lpr mice mitigates glomerulonephritis in that model (Kyttaris et al., 2010). However, IL-23 is known to stabilize pathogenic Th17 cells and thus boost expression of not only IL-17 but also several other cytokines, including IFN-γ (Oppmann et al., 2000); furthermore, it can restrain regulatory T cell activity (Izcue et al., 2008). Indeed, loss of the IL-23 receptor in B6/lpr mice reduces autoantibody production and immune complex deposition in kidneys (Kyttaris et al., 2010). The present findings directly demonstrate the importance of specifically IL-17 cytokines in the development of fatal kidney pathology, likely including IL-17C, the expression of which is not known to be controlled by IL-23.
Our studies identify IL-17 cytokines and the CIKS-mediated signaling pathway as potential therapeutic targets in SLE, especially in lupus nephritis, and also in anti-GBM disease. Biologic therapies directed at B cells in mice and patients have met with limited success, especially in more severe cases of lupus nephritis (Navarra et al., 2011; Sanz and Lee, 2010); thus, there is great need for new treatment options. The present findings validate IL-17 cytokines and CIKS as promising targets. Because these cytokines have also been implicated in other local inflammatory conditions, including RA, MS, and psoriasis, therapies targeting them or their signaling pathways may have wide-ranging benefits.
EXPERIMENTAL PROCEDURES
Mice
Traf3ip2−/−, Fcgr2b−/−, Il17a−/−, and Mb1-cre mice have been described (Claudio et al., 2009; Hobeika et al., 2006; Nakae et al., 2002; Takai et al., 1996). Mice were intercrossed to generate animals with compound deficiency of Fcgr2b and Traf3ip2, Fcgr2b and Il17a, and control littermates. To generate Traf3ip2 floxed mice, exon 2 was flanked by loxP sites (Ozgenes, Australia) (Figure S3). WT/Flox mice were crossed with Mb1-cre mice to generate mice with B cells specific Traf3ip2 deletion. The experimental mice were used at the age of 6–8 months (Fcgr2b−/−.Traf3ip2 strain) and 10–12 months (Fcgr2b−/−.Il17a strain). Mice were bred and housed in a facility at the National Institute of Allergy and Infectious Diseases (NIAID), and all experiments were performed with the approval of the NIAID Animal Care and Use Committee and in accordance with all relevant institutional guidelines.
Glomerular Basement Membrane Antibody Induced Glomerulonephritis
The protocol for GBM Ab-induced glomerulonephritis has been described (Rosenkranz et al., 1999; Vielhauer et al., 2005). Briefly, mixed rabbit IgG at a final concentration of 0.1 mg/ml in CFA (2.5 mg/ml) was injected subcutaneously on day −3, and then heat inactivated anti-GBM serum was intravenously injected (100 μl) via tail vein on day 0. The 8- to 10-week-old mice were followed up and sacrificed to collect tissues for analysis on day 14.
Cellular Analysis
Splenocytes were isolated as previously described (Pisitkun et al., 2010) and stained with the following antibodies: B220(RA3-6B2), GL7(Ly-77), FAS(Jo2), CD138(281-2), IgM(II/41), IAb(AF6-120.1), CD4(L3T4), CD44(IM7), CD11b(M1/70), Ly6c(AL-21), CD3e(145-2C11), CD11c(HL3), and CD62L(Ly-22) (BD Biosciences); CD19(eBio1D3), ICOS(7E.17G9), F4/80(BM8), CD8a (53-6.7), and IL17A(eBio17B7) (eBioscience); Ly6g (IA8) (Biolegend), CD1dtetramer( PBS57) (NIH tetramer facility), and Aqua (Invitrogen). Cells were stimulated with PMA (5 ng/ml), Ionomycin (500 ng/ml), and Golgi stop (BD) for 4 hr before intracellular staining. Data were collected with a FACSCanto instrument (BD Biosciences).
In Vivo Immunoglobulin Isotype Switching
B cells were isolated from splenocytes using anti-CD19 beads (Miltenyi). RNA isolation from B cells and complimentary DNA (cDNA) synthesis were performed as described (Pisitkun et al., 2010). RT-PCR for immunoglobulin transcription was performed by using primers and condition as described (Muramatsu et al., 2000).
Autoantibody Analysis
Anti-nuclear antibodies (ANA) were assayed with Hep-2 cells (BION). Slides were mounted with Vectashield (Vector Labs) and visualized using a fluorescence microscope. Mouse dsDNA IgG was detected by ELISA using the dsDNA IgG kit (5120) (Alpha Diagnostic International).
Histological Analysis
Kidneys were fixed in 4% paraformaldehyde. Tissue sections were stained with H&E or Periodic acid–Schiff (PAS) and visualized by Olympus BX50. Histological scores were evaluated as described (Chan et al., 1997). We analyzed kidney pathology from these mice compared to their control littermates at between 6–8 months (Fcgr2b−/−.Traf3ip2−/−) and 10–12 months (Fcgr2b−/−.Il17a−/−) of age. In GBM induced glomerunlonephritis, the histology scores were determined as described (Odobasic et al., 2011).
Immunofluorescence
Frozen sections (5 μm thickness) were fixed in acetone, and blocked with 1% BSA in PBS. Diluted antibodies were incubated for 1 hr. Endogenous biotin was blocked using Streptavidin-Biotin blocking kit (Vector Labs). The following antibodies were used: IgD(11-26c.2a) and CD35(8C12) (BD Biosciences), IgG(A11029, Invitrogen), 7/4(Cedarlane), Ly6g(IA8, Biolegend), MPO(2D4, Abcam), and LL37(LS-B2687 CAMP, LS Biosciences). Slides were mounted with Vectashield with or without DAPI (Vector Labs) and visualized using a Leica AF6000LX fluorescence microscope.
Isolation of Single Cells from the Kidney for FACS Analysis
Isolation of single cells from the kidneys was modified from a described procedure (Gunaratne et al., 2010). In brief, perfusion of the kidneys was performed with 20 ml of prewarmed HBSS while the mice were anesthesized with Avertin (0.4–0.75 mg/g of body weight). The perfused kidneys were digested with collagenase B (0.230 U/ml) (Roche Diagnostics) in 10% FCS in IMEM for 20 min (37°C). Single cells of kidneys were isolated the same way as splenocytes (see above) and cell suspensions were subjected to density separation to eliminate the epithelial or tubular cells of the kidneys using Lympholyte-M (Cedarlane).
Quantitative Real-Time PCR Analysis
RNA was purified using TRIzol (Invitrogen), cDNA synthesis, and quantitative real-time PCR was performed as described (Pisitkun et al., 2010). Control RNA of human kidneys was purchased from Ambion, Agilent, and Origene (CR560857, CR560624). The mouse or human primers (Taqman) for actin, IL-17, IL-17F, IL-17C, IL-17B, TNF-α, CXCL1, CXCL5, CCL2, and CCL20 were obtained from Applied Biosystems.
Statistical Analysis
Gehan-Breslow-Wilcoxon test was performed with GraphPad Prism 5 to determine statistical significance of survival curves. Student’s t test (one-tailed) was used for other comparisons.
Supplementary Material
Acknowledgments
We thank Yoichiro Iwakura for providing Il17a−/− B6 mice, Michael Reth for providing Mb1-cre mice, Trairak Pisitkun for providing the technical support for isolation of single cell suspensions from kidneys, and Lily Koo and Steven M. Becker for help with fluorescence microscopy. We are most thankful to Rachel Caspi and Silvia Bolland for critical reading of the manuscript and suggestions and James E. Balow for discussion. We greatly appreciate the constructive inputs provided by members of the Siebenlist laboratory and we are grateful to Anthony S. Fauci for continued support. This research was supported by the Intramural Research Programs of NIAID and NIDCR, NIH.
Footnotes
Supplemental Information includes seven figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2012.08.014.
References
- Baerenwaldt A, Lux A, Danzer H, Spriewald BM, Ullrich E, Heidkamp G, Dudziak D, Nimmerjahn F. Fcγ receptor IIB (FcγRIIB) maintains humoral tolerance in the human immune system in vivo. Proc Natl Acad Sci USA. 2011;108:18772–18777. doi: 10.1073/pnas.1111810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13:277–285. doi: 10.1016/s1074-7613(00)00027-3. [DOI] [PubMed] [Google Scholar]
- Boross P, Arandhara VL, Martin-Ramirez J, Santiago-Raber ML, Carlucci F, Flierman R, van der Kaa J, Breukel C, Claassens JW, Camps M, et al. The inhibiting Fc receptor for IgG, FcγRIIB, is a modifier of autoimmune susceptibility. J Immunol. 2011;187:1304–1313. doi: 10.4049/jimmunol.1101194. [DOI] [PubMed] [Google Scholar]
- Chan O, Madaio MP, Shlomchik MJ. The roles of B cells in MRL/lpr murine lupus. Ann NY Acad Sci. 1997;815:75–87. doi: 10.1111/j.1749-6632.1997.tb52046.x. [DOI] [PubMed] [Google Scholar]
- Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity. 2011;35:611–621. doi: 10.1016/j.immuni.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J Exp Med. 2005;202:321–331. doi: 10.1084/jem.20050338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E, Sønder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daëron M, Latour S, Malbec O, Espinosa E, Pina P, Pasmans S, Fridman WH. The same tyrosine-based inhibition motif, in the intracytoplasmic domain of Fc gamma RIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. 1995;3:635–646. doi: 10.1016/1074-7613(95)90134-5. [DOI] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, Fabien N, Cochat P, Pouteil-Noble C, Trolliet P, et al. Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009;10:778–785. doi: 10.1038/ni.1741. [DOI] [PubMed] [Google Scholar]
- Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunaratne R, Braucht DW, Rinschen MM, Chou CL, Hoffert JD, Pisitkun T, Knepper MA. Quantitative phosphoproteomic analysis reveals cAMP/vasopressin-dependent signaling pathways in native renal thick ascending limb cells. Proc Natl Acad Sci USA. 2010;107:15653–15658. doi: 10.1073/pnas.1007424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas C, Ryffel B, Le Hir M. IFN-gamma receptor deletion prevents autoantibody production and glomerulonephritis in lupus-prone (NZB x NZW)F1 mice. J Immunol. 1998;160:3713–3718. [PubMed] [Google Scholar]
- Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010;107:9813–9818. doi: 10.1073/pnas.0909927107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40:204–210. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartupee J, Liu C, Novotny M, Li X, Hamilton T. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, Reth M. Testing gene function early in the B cell lineage in mb1-cre mice. Proc Natl Acad Sci USA. 2006;103:13789–13794. doi: 10.1073/pnas.0605944103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, Yi J, Guentert T, Tousson A, Stanus AL, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–175. doi: 10.1038/ni1552. [DOI] [PubMed] [Google Scholar]
- Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y, Maloy KJ, Powrie F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Kozyrev SV, Abelson AK, Wojcik J, Zaghlool A, Linga Reddy MV, Sanchez E, Gunnarsson I, Svenungsson E, Sturfelt G, Jönsen A, et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:211–216. doi: 10.1038/ng.79. [DOI] [PubMed] [Google Scholar]
- Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. 2010;184:4605–4609. doi: 10.4049/jimmunol.0903595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusevicius D, Kivisäkk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- Mok CC. Biomarkers for lupus nephritis: a critical appraisal. J Biomed Biotechnol. 2010;2010:638413. doi: 10.1155/2010/638413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- Nalbandian A, Crispín JC, Tsokos GC. Interleukin-17 and systemic lupus erythematosus: current concepts. Clin Exp Immunol. 2009;157:209–215. doi: 10.1111/j.1365-2249.2009.03944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, León MG, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- Odobasic D, Gan PY, Summers SA, Semple TJ, Muljadi RC, Iwakura Y, Kitching AR, Holdsworth SR. Interleukin-17A promotes early but attenuates established disease in crescentic glomerulonephritis in mice. Am J Pathol. 2011;179:1188–1198. doi: 10.1016/j.ajpath.2011.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Paust HJ, Turner JE, Riedel JH, Disteldorf E, Peters A, Schmidt T, Krebs C, Velden J, Mittrücker HW, Steinmetz OM, et al. Chemokines play a critical role in the cross-regulation of Th1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. 2012;82:72–83. doi: 10.1038/ki.2012.101. [DOI] [PubMed] [Google Scholar]
- Peng SL, Moslehi J, Craft J. Roles of interferon-gamma and interleukin-4 in murine lupus. J Clin Invest. 1997;99:1936–1946. doi: 10.1172/JCI119361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun P, Claudio E, Ren N, Wang H, Siebenlist U. The adaptor protein CIKS/ACT1 is necessary for collagen-induced arthritis, and it contributes to the production of collagen-specific antibody. Arthritis Rheum. 2010;62:3334–3344. doi: 10.1002/art.27653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12:1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- Richards HB, Satoh M, Jennette JC, Croker BP, Yoshida H, Reeves WH. Interferon-gamma is required for lupus nephritis in mice treated with the hydrocarbon oil pristane. Kidney Int. 2001;60:2173–2180. doi: 10.1046/j.1523-1755.2001.00045.x. [DOI] [PubMed] [Google Scholar]
- Rosas M, Thomas B, Stacey M, Gordon S, Taylor PR. The myeloid 7/4-antigen defines recently generated inflammatory macrophages and is synonymous with Ly-6B. J Leukoc Biol. 2010;88:169–180. doi: 10.1189/jlb.0809548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz AR, Mendrick DL, Cotran RS, Mayadas TN. P-selectin deficiency exacerbates experimental glomerulonephritis: a protective role for endothelial P-selectin in inflammation. J Clin Invest. 1999;103:649–659. doi: 10.1172/JCI5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76:135–144. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–337. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Hayashizaki A, Ohtsuji M, Lin Q, Hou R, Ohtsuji N, Nishikawa K, Tsurui H, Sudo K, Ono M, Izui S, et al. Presumptive role of 129 strain-derived Sle16 locus in rheumatoid arthritis in a new mouse model with Fcγ receptor type IIb-deficient C57BL/6 genetic background. Arthritis Rheum. 2011;63:2930–2938. doi: 10.1002/art.30485. [DOI] [PubMed] [Google Scholar]
- Shahrara S, Pickens SR, Mandelin AM, 2nd, Karpus WJ, Huang Q, Kolls JK, Pope RM. IL-17-mediated monocyte migration occurs partially through CC chemokine ligand 2/monocyte chemoattractant protein-1 induction. J Immunol. 2010;184:4479–4487. doi: 10.4049/jimmunol.0901942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10:328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, Christensen SR, Harris ML, Shupe J, Sutherland JL, Shlomchik MJ. A new site-directed transgenic rheumatoid factor mouse model demonstrates extrafollicular class switch and plasmablast formation. Autoimmunity. 2010;43:607–618. doi: 10.3109/08916930903567500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T, Ono M, Hikida M, Ohmori H, Ravetch JV. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- Valente AJ, Clark RA, Siddesha JM, Siebenlist U, Chandrasekar B. CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2012;53:113–124. doi: 10.1016/j.yjmcc.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vielhauer V, Stavrakis G, Mayadas TN. Renal cell-expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J Clin Invest. 2005;115:1199–1209. doi: 10.1172/JCI23348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. 2009;183:3160–3169. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.