

Abstract
Craniofrontonasal syndrome (CNFS) is an X-linked disorder caused by mutations in the EFNB1 gene in which, paradoxically, heterozygous females are more severely affected than hemizygous males. In this paper, the clinical and molecular studies of a female subject with CFNS are described. A novel de novo c.473T>C (p.M158T) mutation in exon 3 of EFNB1 was demonstrated in this patient. The M158 residue of the Ephrin-B1 protein is highly conserved between species. Our results expand the mutational spectrum exposed by CNFS.
1. Introduction
Craniofrontonasal syndrome (CFNS; OMIM # 304110) [1] is an X-linked syndrome involving developmental malformation with variable clinical expression characterized by severe hypertelorism, depressed nasal bridge and bifid nasal tip, frontal bossing, coronal suture synostosis, corpus callosum agenesis, and occasionally cleft lip or palate [2–4]. This disorder is caused by mutations in the EFNB1 gene, located at Xq13.1, and encoding a ligand of the Ephrin family of receptor protein tyrosine kinases [3]. The most common types of EFNB1 mutations (up to 55%) in CFNS patients are frameshift, nonsense, and splice site mutations that lead to premature termination codons (PTCs). Missense mutations constitute approximately 42% of all EFNB1 mutations, and most of them occur in exons 2 and 3, leading to the substitutions of amino acid residues involved in receptor-ligand interaction and cell signaling, which are critical for cell sorting, migration and adhesion, midline fusion, axon guidance, neural plasticity, and synaptogenesis [5–8]. Here, we describe a sporadic case of CFNS due to a novel EFNB1 mutation occurring in a female Mexican patient.
2. Case Presentation
A 3-month-old girl was referred to the Genetics Department after a frontoorbital advancement surgery due to right unicoronal synostosis and facial dysmorphism. She is the only child of healthy, nonconsanguineous parents. There was no prenatal exposure to teratogenic agents. A structural ultrasound at 24 weeks revealed a nonspecific cranial malformation. The patient was delivered by caesarean section at 38 weeks of pregnancy and had a birth weight of 2,700 g, birth length of 48.5 cm, and an Apgar score of 8/9. Birth examination disclosed plagiocephaly (left frontal bossing and right coronal synostosis), hypertelorism, downslanting palpebral fissures, facial asymmetry, a broad and flattened nasal bridge, a bifid nasal tip, and broad thumbs and halluces with longitudinally split nails (Figure 1). No abnormalities were found in the transfontanellar ultrasound and echocardiogram performed during the first week of life. Cytogenetic study was normal (46,XX[25]). When the patient reached the age of 3 months, cranial computerized tomography (CT) revealed right coronal synostosis and mild compression of the surrounding cerebral parenchyma, which prompted surgery. At present, she has reached adequate development milestones and growth parameters.
Figure 1.
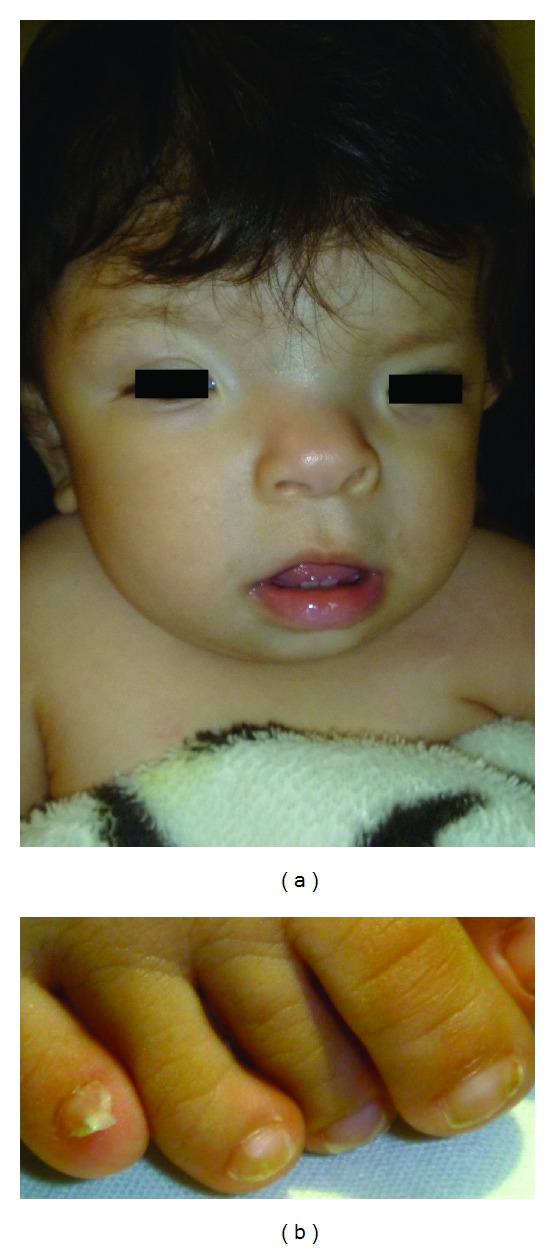
(a) Patient's facial appearance. Hypertelorism, broad and flattened nasal bridge, and bifid nasal tip are evident. (b) Broad toes with longitudinally split nails can be observed.
After obtaining local ethics institutional approval and the informed consent of her parents, genomic DNA was extracted from the patient's peripheral blood leukocytes using a semiautomated Quickgene system (Fujifilm, Tokyo, Japan). The complete EFNB1 coding sequence, including the exon-intron boundaries, was amplified by PCR using primers for the 5 exons (Table 1), and direct automated sequencing was performed using the Big Dye Terminator Cycle Sequencing kit (Applied Biosystems, Foster City, California, USA) in an ABI Prism 3130 Genetic Analyzer (Applied Biosystems).
Table 1.
PCR and sequencing primers for EFNB1.
| Primer name | 5′-3′ orientation | Length of PCR product (bp) |
|---|---|---|
| EFNB1-ex1-F | AAGGCGAGGCGAGCTTTGGT | 318 |
| EFNB1-ex1-R | AAGCCGGAGACAAAATGAGG | |
| EFNB1-ex2-F | TTGTCCGCTTCCCTGGTTCT | 445 |
| EFNB1-ex2-R | ATTGCACCACTTAGAAGCTCC | |
| EFNB1-ex3-F | GCTGAAGCAGAATGGGAGTT | 246 |
| EFNB1-ex3-R | GCCAGGAACATCTGTTCCAA | |
| EFNB1-ex4-F | GGTTACAGTATCCAGGCCAT | 312 |
| EFNB1-ex4-R | GCCCAGCTTGCATTTCTTCA | |
| EFNB1-ex5-F | TGAAGAAATGCAAGCTGGGC | 606 |
| EFNB1-ex5-R | ATACAAAGGTGGGCACAGCT |
Nucleotide analysis disclosed a novel heterozygous transition c.473T>C in exon 3 of EFNB1. This mutation predicted a substitution of methionine (ATG) to threonine (ACG) in the extracellular domain of the protein (p.M158T) (Figure 2(a)). Both parents had a normal sequence (Figures 2(b) and 2(c)).
Figure 2.
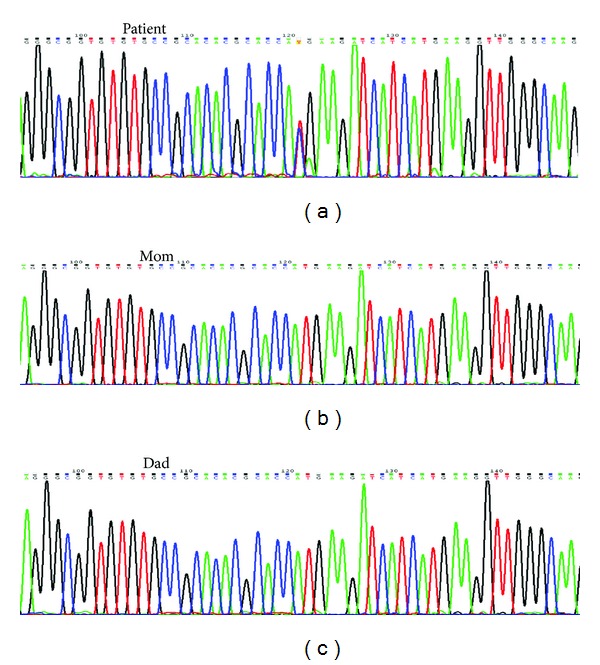
Partial nucleotide sequence of the EFNB1 gene in DNA from patient (a), proband's mother (b), and proband's father (c). (a) A heterozygous T to C transition at nucleotide position c.473 in exon 3 predicting a methionine (ATG) to threonine (ACG) replacement at residue 158 (p.M158T) is shown. (b and c) Normal nucleotide (T) in both parents.
3. Discussion
Our report discussed a patient with clinical characteristics consistent with CNFS and in whom a novel de novo EFNB1 mutation was demonstrated. CFNS shows a phenotypic pattern not usually seen in X-linked disorders, as heterozygous females are more severely affected than hemizygous males. Mutations in EFNB1 are the cause of CFNS in the majority of patients, with a mutation detection rate of 92% [9, 10]. CNFS's clinical manifestations are sex dependent, with multiple skeletal malformations in affected females and mild or no malformations in male carriers. Recently, the severe phenotype in females has been explained through the cellular interference hypothesis; cellular interference is caused by the combination of Ephrin ligand/receptor promiscuity and the consequences of random X inactivation in distinct cellular compartments [3, 11]. Although we have identified a novel de novo mutation, no other new clinical features were found in the physical examination.
The EFNB1 gene encodes Ephrin-B1 protein, a member of the ephrin family of transmembrane ligands for Eph receptors with tyrosine kinase activity. These proteins play a crucial role in cell migration and pattern formation during embryonic development [12]. Missense mutations, such as that demonstrated in our patient, constitute about 42% of EFNB1 mutations. There are non-hotspot mutations in EFNB1 gene; however, most of reported substitutions occur in the extracellular domain, which is encoded by exons 2 and 3, leading to a change in amino acid residues, which are important for receptor-ligand interaction and signaling, and cause loss of function [7].
The replacement from a hydrophobic sulfur amino acid such as methionine for a hydrophilic amino acid, threonine, modifies the polarity of the protein. In silico analysis of this novel missense EFNB1 mutation using PolyPhen-2 software indicated that this change is pathogenic (Figure 3(a)) and predicted to be highly conserved in different species (Figure 3(b)) [13]. The fact that this nucleotide substitution-transition was not found in the NHLBI Exome Sequencing Project (ESP; Exome Variant Server) supports the novel condition of our mutation [14].
Figure 3.

In silico analysis using PolyPhen-2 software shows that change in p.M158T is pathogenic (a) and predicted to be highly conserved in different species (b).
In conclusion, we presented a patient with craniofrontonasal syndrome due to a novel de novo heterozygous transition mutation c.473T>C in exon 3 of EFNB1. Our results expand the EFNB1 mutational spectrum in CNFS patients. The M158 residue (methionine) of the Ephrin-B1 protein is highly conserved between species.
Acknowledgment
The authors thank the Research Unit of the Instituto de Oftalmología “Conde de Valenciana.”
References
- 1.OMIM (TM) Bethesda MD. McKusick-Nathans Institute for Genetic Medicine, John Hopkins University, National Center for Biotechnology Information, National Library of Medicine. Online Mendelian Inheritance in Man, 2008.
- 2.Wieland I, Jakubiczka S, Muschke P, et al. Mutations of the ephrin-B1 gene cause craniofrontonasal syndrome. American Journal of Human Genetics. 2004;74(6):1209–1215. doi: 10.1086/421532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wieacker P, Wieland I. Clinical and genetic aspects of craniofrontonasal syndrome: towards resolving a genetic paradox. Molecular Genetics and Metabolism. 2005;86(1-2):110–116. doi: 10.1016/j.ymgme.2005.07.017. [DOI] [PubMed] [Google Scholar]
- 4.Saavedra D, Richieri-Costa A, Guion-Almeida ML, et al. Craniofrontonasal syndrome: study of 41 patients. American Journal of Medical Genetics A. 1996;61(2):147–151. doi: 10.1002/(SICI)1096-8628(19960111)61:2<147::AID-AJMG8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 5.Twigg SRF, Matsumoto K, Kidd AMJ, et al. The origin of EFNB1 mutations in craniofrontonasal syndrome: frequent somatic mosaicism and explanation of the paucity of carrier males. American Journal of Human Genetics. 2006;78(6):999–1010. doi: 10.1086/504440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Twigg SRF, Kan R, Babbs C, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(23):8652–8657. doi: 10.1073/pnas.0402819101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makarov R, Steiner B, Gucev Z, Tasic V, Wieacker P, Wieland I. The impact of CFNS-causing EFNB1 mutations on ephrin-B1 function. BMC Medical Genetics. 2010;11(1, article 98) doi: 10.1186/1471-2350-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wieland I, Makarov R, Reardon W, et al. Dissecting the molecular mechanisms in craniofrontonasal syndrome: differential mRNA expression of mutant EFNB1 and the cellular mosaic. European Journal of Human Genetics. 2008;16(2):184–191. doi: 10.1038/sj.ejhg.5201968. [DOI] [PubMed] [Google Scholar]
- 9.Wallis D, Lacbawan F, Jain M, et al. Additional EFNB1 mutations in craniofrontonasal syndrome. American Journal of Medical Genetics A. 2008;146(15):2008–2012. doi: 10.1002/ajmg.a.32388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wieland I, Reardon W, Jakubiczka S, et al. Twenty-six novel EFNB1 mutations in familial and sporadic craniofrontonasal syndrome (CFNS) Human Mutation. 2005;26(2):113–118. doi: 10.1002/humu.20193. [DOI] [PubMed] [Google Scholar]
- 11.Zafeiriou DI, Pavlidou EL, Vargìami E. Diverse clinical and genetic aspects of craniofrontonasal syndrome. Pediatric Neurology. 2011;44(2):83–87. doi: 10.1016/j.pediatrneurol.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 12.Klein R. Eph/ephrin signaling in morphogenesis, neural development and plasticity. Current Opinion in Cell Biology. 2004;16(5):580–589. doi: 10.1016/j.ceb.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 13. http://genetics.bwh.harvard.edu/pph2/
- 14. http://evs.gs.washington.edu/EVS/