

Abstract
Since their divergence from the terrestrial artiodactyls, cetaceans have fully adapted to an aquatic lifestyle, which represents one of the most dramatic transformations in mammalian evolutionary history. Numerous morphological and physiological characters of cetaceans have been acquired in response to this drastic habitat transition, such as thickened blubber, echolocation, and ability to hold their breath for a long period of time. However, knowledge about the molecular basis underlying these adaptations is still limited. The sequence of the genome of Tursiops truncates provides an opportunity for a comparative genomic analyses to examine the molecular adaptation of this species. Here, we constructed 11,838 high-quality orthologous gene alignments culled from the dolphin and four other terrestrial mammalian genomes and screened for positive selection occurring in the dolphin lineage. In total, 368 (3.1%) of the genes were identified as having undergone positive selection by the branch-site model. Functional characterization of these genes showed that they are significantly enriched in the categories of lipid transport and localization, ATPase activity, sense perception of sound, and muscle contraction, areas that are potentially related to cetacean adaptations. In contrast, we did not find a similar pattern in the cow, a closely related species. We resequenced some of the positively selected sites (PSSs), within the positively selected genes, and showed that most of our identified PSSs (50/52) could be replicated. The results from this study should have important implications for our understanding of cetacean evolution and their adaptations to the aquatic environment.
Keywords: Tursiops truncates, aquatic adaptation, positive selection, branch-site model
Introduction
Cetaceans diverged from artiodactyls approximately 50 million years ago (Meredith et al. 2011), and their habitat transition, from land to an aquatic environment, represents one of the most dramatic transformations in mammalian evolutionary history. These adaptation inevitably posed challenges for the ancient cetaceans, which had originally been adapted for terrestrial life, with locomotion (navigation) and detection of prey being major ones. For locomotion, they needed to confront the considerable obstacle provided by water, whose density is much higher than air. To overcome drag, cetaceans have evolved some extreme changes in morphology and physiology, including a streamlined form and a modified skeletal system (Fish et al. 2007; Reidenberg 2007). In addition, most cetaceans possess a thick layer of blubber, which increases their buoyancy (Struntz et al. 2004). For foraging in water, these mammals constantly need to hunt at night or in deep water, therefore, it is vital for them to possess superior capabilities of long-time diving and locating prey. It is striking that some cetacean species have acquired an ability to echolocate that has enabled them to use sound to locate prey or escape obstacles when navigating (Cranford et al. 1996). Moreover, cetaceans have elevated levels of myoglobin in their skeletal muscles (Noren et al. 2001; Wright and Davis 2006), which vastly increases their ability to retain oxygen, allowing for longer time between breaths. They also use glycolysis metabolism to compensate for insufficient levels of oxygen (Butler and Jones 1997), which potentially supports the energy supply for their long dives.
Considering the significant phenotypic modifications in cetaceans, it should be expected that these modifications were shaped by natural selection, and conferred a selective advantage, as they adapted to the new aquatic environment. What are the underlying molecular mechanisms for these innovations? Positive Darwinian selection is one of the major driving forces for adaptive evolution and species diversification, which had been widely investigated in many species (Kosiol et al. 2008; Lefebure and Stanhope 2009; Shen et al. 2010; Oliver et al. 2011; Wissler et al. 2011; McGowen et al. 2012). A few studies have focused on the adaptive evolution of marine mammals (McClellan et al. 2005; Wang et al. 2009); however, as the complete genomes of marine mammals were not available at that time, the data sets analyzed in these previous studies were limited to only a few genes (e.g., cytB and HoxD) (McClellan et al. 2005; Wang et al. 2009). Whole-genome-wide identifications of positively selected genes (PSGs) along the marine mammal lineage should greatly help us understand the genetic bases underlying adaptive evolution in marine mammals. The genome sequence of the bottlenose dolphin (Tursiops truncates) provides an opportunity to conduct this analysis.
A series of evolutionary models for testing positive selection have been developed in the past decade, including the branch model, the site model, and the branch-site model (Yang 1998; Yang et al. 2000; Yang and Nielsen 2002; Yang et al. 2005; Zhang et al. 2005). In the first two models, positive selection is inferred only if the dN/dS average over all sites or all branches is significantly greater than 1. Positive selection, however, often operates episodically on only a small number of sites on a few lineages (Yang and Nielsen 2002), limiting the power of detecting positive selection by the branch and site models. The branch-site model, a more powerful model, was developed to address this issue (Yang and Nielsen 2002; Zhang et al. 2005) and has been widely used in screens for positive selection (cf. Bakewell et al. 2007; Kosiol et al. 2008; Studer et al. 2008; Shen et al. 2010).
Here, we constructed whole-genome ortholog gene sets among five mammalian species, including dolphin (T. truncates), cow (Bos Taurus), dog (Canis familiaris), panda (Ailuropoda melanoleuca), and human (Homo sapiens) and identified PSGs along the dolphin lineage with the improved branch-site model to build a database of genes that might be correlated with aquatic adaptation in the dolphin. As the current release of the dolphin genome has only 2.59× coverage, there are limitations for comparative genomic analyses, especially the detection of positive selection. Sequencing errors, problems with annotation, alternative splicing, amino acid repeats, and frameshift mutations could generate a higher rate of false positive with the branch-site model (Mallick et al. 2009; Schneider et al. 2009; Markova-Raina and Petrov 2011), therefore, generating accurate alignments is an essential step in the inference of positive selection. The Prank software (Loytynoja and Goldman 2005, 2008) was recently reported as being able to generate much more accurate alignments than other traditional aligners (Fletcher and Yang 2010; Markova-Raina and Petrov 2011), thus we used this algorithm to align all the genes used in this study. Moreover, we resequenced some of the candidate PSS regions to confirm their reliability. We show that most (50/52) of our identified PSSs are reliable. Through a functional clustering analysis of the dolphin PSGs, we found that they are enriched for categories such as lipid transport and localization, ATPase activity, perception of sound, and muscle contraction clusters.
Material and Methods
Coding region sequences of individual genes from the genomes of the dolphin and other species were downloaded from Ensembl (version 66, March 2012) using the BioMart tool (Vilella et al. 2009). The species used here for comparison with dolphin include cow (B. Taurus, UMD3.1), dog (C. lupus familiaris, CanFam_2.0), panda (A. melanoleuca, ailMel1), and human (H. sapiens, GRCh37.p6). A phylogenetic tree of these species is shown in figure 1, which is derived from Murphy et al. (2007). To predict homologs among the five genomes, we used the Ensembl inferences (Vilella et al. 2009). For each pair of these genomes, only those that Ensembl annotated as one2one orthologous genes were retrieved and analyzed in the following step. If a gene had multiple transcripts, then the longest one was chosen. After these treatments, we obtained 12,057 gene sets.
Fig. 1.—

Phylogenetic tree used in cthis study. The accepted phylogeny of the species used for the screen for PSGs. The bold line represents the dolphin lineage, the lineage where positive selection was detected.
The Prank program (Loytynoja and Goldman 2005, 2008) was used to align all the gene sets. Because Prank performs much better at the codon level than at the amino acid level (Fletcher and Yang 2010) for protein-coding sequences, all the genes were thus directly aligned at the codon level with the option “-codon.”
After the alignments were generated, we performed a trimming treatment to remove potentially unreliable regions using the Gblocks program (Castresana 2000). The parameters used were the default settings with the sequence type being codon (“-t = c”). In addition, to reduce the effect of uncertain bases on the inference of positive selection, we deleted all positions that had gaps (“-”) and “N” from the alignments. After the trimming process, if the remaining alignment was shorter than 120 bp (40 codons), then the entire alignment was discarded.
In addition to alignment uncertainty, saturation at silent sites (dS) may also bias the inference of positive selection (Smith and Smith 1996). To identify saturation, for each gene, the third codon positions were extracted, and branch lengths on the species tree were estimated using the general time reversible model with PAML (Yang 2007). Branch lengths were used as a proxy for saturation, and genes were removed from the analysis if one or more branches had a length ≥ 1. Our final data set contained 11,838 genes.
For each of the remaining genes, a branch-site evolutionary analysis for positive selection was conducted using codeml from the PAML package (Yang 2007). In this study, the improved branch-site model (Yang and Nielsen 2002; Zhang et al. 2005) was used. This model requires that the branches of the tree be divided in priori into foreground and background lineages. A likelihood ratio test (LRT) compares a model with positive selection on the foreground branch to a null model where no positive selection occurred on the foreground branch and calculates the statistic (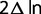 ) to obtain a P value. In this study, genes were inferred to be PSGs only if the P value was less than 0.01. This model can also infer positively selected sites (PSSs) based on an empirical Bayes analysis (Yang et al. 2005). In this study, PSSs were inferred only if their posterior probability was greater than 95%.
) to obtain a P value. In this study, genes were inferred to be PSGs only if the P value was less than 0.01. This model can also infer positively selected sites (PSSs) based on an empirical Bayes analysis (Yang et al. 2005). In this study, PSSs were inferred only if their posterior probability was greater than 95%.
After PSGs were detected, we used the DAVID Functional Annotation tool (Huang da et al. 2009) to investigate their enrichment of gene ontology (GO) terms. During this analysis, the human ortholog of the PSG was as the input against a background of human genes. Within each annotation cluster, DAVID lists the GO terms that are significantly enriched. In this study, we used the approach of McGowen et al. (2012), where only terms with an enrichment score >1.3 were considered meaningful.
To confirm our identified PSSs, we randomly selected 48 PSGs for whom the presence of PSS had been detected and designed polymerase chain reaction (PCR) primers using Primer3 (Rozen and Skaletsky 2000) to directly amplify and sequence these regions using PCR and an Applied Biosystems 3730 DNA Analyzer, respectively. DNA for this study was extracted from the same species of dolphin (T. truncatus). Information on these primers is available in supplementary table S1, Supplementary Material online, and all the segments sequenced in this study were deposited into GenBank with accession numbers from JX856347 to JX856394.
Results
Our analysis began with 12,057 genes that had single copy orthologs in the dolphin, cow, dog, panda, and human genomes. Each of these genes was annotated by Ensembl (version 66) as being one2one orthologous between each pair of species. After our alignment treatments, 219 genes were eliminated because either their final lengths were shorter than 120 bp or they did not pass the synonymous substitution saturation test (see Materials and Methods). Finally, 11,838 genes were tested for positive selection in this study.
First, to determine the overall difference in selective constraints between dolphin and other species, each aligned gene was evaluated for their substitution rates including dN, dS, and dN/dS, under the species tree (fig. 1). The free-ratio model (M1) in PAML (Yang 2007), which allows a separate ω for each branch, was used. We found that 97% of the genes had a dN/dS ratio smaller than 1 on the dolphin lineage, providing support for the overall presence of purifying selection acting at the molecular level over all time. We then compared the mean dN/dS between dolphin and its closely related species. When dS is approximately, or equal to, 0 along a branch, then it always generates a very high dN/dS value, hence, for this comparison, genes with dS <0.0005 were not included. The mean dN/dS along the dolphin lineage was 0.2373, significantly larger than 0.1435 on the cow lineage (P <0.001, Mann–Whitney U test; fig. 2), suggesting that genes evolved faster in the dolphin after their split from artiodactyls. Indeed, by examining the dN/dS for each gene on the dolphin and cow lineages, we found 8,422 genes that have higher dN/dS in the dolphin and only 2,940 genes where it is higher in the cow. In addition, the mean rate of synonymous substitutions along the dolphin lineage was found to be much smaller than those measured for of the other species used in this study (fig. 2), which agrees with a previously reported slowdown in the molecular rate in dolphin (McGowen et al. 2012). The full list of genes, with their substitution traits, is available in supplementary table S2, Supplementary Material online.
Fig. 2.—

Comparisons of rates of evolution at silent and replacement sites in the dolphin and cow lineages. The molecular traits, dN, dS, and dN/dS, on the lineages leading to the dolphin and the cow are presented. For this comparison, genes with dS < 0.0005 were excluded.
Next, we used the codeml program in the PAML package (Yang 2007), with the improved branch-site model (“test2”) (Zhang et al. 2005), to detect signals of positive selection on each alignment. These screens identified 368 genes (3.1% of the total) that show significant evidence of positive selection (P < 0.01) in the dolphin lineage (supplementary table S2, Supplementary Material online). The PSGs were applied to the DAVID Functional Annotation tool (Huang da et al. 2009) to investigate their functional enrichments. Interestingly, we found a number of functional categories that might be correlated with the dolphin-specific traits that were significantly enriched among these dolphin PSGs (table 1), such as GO:0006869: lipid transport, GO:0010876: lipid localization, GO:0007605: sensory perception of sound, GO:0016887: ATPase activity, GO:0006096: glycolysis, GO:0006099: tricarboxylic acid cycle, GO:0050917: sensory perception of umami taste, GO:0003012: muscle system process, and GO:0003774: motor activity. These functional clusters might be related to fat storage, echolocation, energy metabolism, and locomotion in cetaceans.
Table 1.
Some Functional Categories Enriched by Dolphin PSGs
| Category | Gene Number | P | Fold Enrichment |
|---|---|---|---|
| Biological process | |||
| GO:0051693∼actin filament capping | 4 | 0.010194345 | 8.722114765 |
| GO:0007605∼sensory perception of sound | 7 | 0.015587711 | 3.461870293 |
| GO:0043588∼skin development | 4 | 0.021749614 | 6.616776718 |
| GO:0003012∼muscle system process | 9 | 0.024144751 | 2.569908815 |
| GO:0006869∼lipid transport | 8 | 0.031724331 | 2.646710687 |
| GO:0006936∼muscle contraction | 8 | 0.040603444 | 2.508320586 |
| GO:0010876∼lipid localization | 8 | 0.04559503 | 2.444414329 |
| GO:0006096∼glycolysis | 4 | 0.073721144 | 4.082692018 |
| GO:0006099∼tricarboxylic acid cycle | 3 | 0.08174332 | 6.257169288 |
| GO:0046356∼acetyl-CoA catabolic process | 3 | 0.08174332 | 6.257169288 |
| GO:0045927∼positive regulation of growth | 5 | 0.084919683 | 2.998226950 |
| GO:0034380∼high-density lipoprotein particle assembly | 2 | 0.09964683 | 19.18865248 |
| Molecular function | |||
| GO:0003774∼motor activity | 12 | 1.84E−04 | 4.048542176 |
| GO:0003779∼actin binding | 18 | 4.79E−04 | 2.645213139 |
| GO:0016887∼ATPase activity | 15 | 0.010251971 | 2.151545617 |
| GO:0005319∼lipid transporter activity | 5 | 0.041911263 | 1.373626374 |
| GO:0008034∼lipoprotein binding | 4 | 0.035687 | 5.475171323 |
| GO:0070325∼lipoprotein receptor binding | 5 | 0.037857 | 9.581549815 |
| GO:0008236∼serine-type peptidase activity | 8 | 0.079084823 | 2.153157261 |
| GO:0042623∼ATPase activity, coupled | 11 | 0.058341087 | 3.802202308 |
| Cellular component | |||
| GO:0034706∼sodium channel complex | 4 | 0.003404288 | 12.71840796 |
| GO:0016459∼myosin complex | 5 | 0.046770479 | 3.668771527 |
| GO:0015629∼actin cytoskeleton | 11 | 0.056035936 | 1.950313488 |
| GO:0042383∼sarcolemma | 5 | 0.05129551 | 3.559255959 |
To investigate the tissue specificity of these PSGs, we used the Uniprot tissue (UP_tissue) annotation database. All the clustered tissues are reported in figure 3, from which we found that the PSGs are largely expressed in tissues involved with the nervous (199 genes in brain, 20 genes in amygdala, and 5 genes in peripheral nervous system), reproductive (99 genes in testis and 82 genes in placenta), and immune systems (28 genes in spleen). In addition to these tissues that commonly express PSGs in many mammalian lineages (Kosiol et al. 2008), we also found dolphin PSGs that are expressed in the kidney (ZC3H11A, FRAS1, FREM1, FREM2, LIMCH1, DNAH7, PEG3, and ARID5B), nasal polyp (DNAH3, DNAH1, and DNAH7), and salivary gland (MYO7B, RRM2B, CDH24, LIMCH1, DSC2, C1orf168, TEP1, SEMA4A, and SIPA1L2) (fig. 3), suggesting potential functional roles for these tissues in processes requiring aquatic adaptation, such as body fluid equilibrium, breath, and digestion and the absorption of food. A detailed list of the functional categories of the PSGs is available in supplementary table S2, Supplementary Material online.
Fig. 3.—

Tissue-specific expression pattern of dolphin PSGs. Expression of the human orthologs of the dolphin PSGs was used to examine expression patterns. Numbers of PSGs expressed in human tissues are presented. Expression pattern of the human orthologs of the dolphin PSGs was obtained from the DAVID functional annotation. Most of the PSGs are expressed in the brain, a complex tissue, potentially indicating a relationship to the enlarged size of the brain in cetaceans (McGowen et al. 2012). In addition, dolphin PSGs are also enriched in other tissues, including kidney, motor cortex, and salivary gland.
We performed an additional multiple test correction for the PSGs according to the “False Discovery Rate” (FDR) method of Benjamini and Hochberg (1995), even though all the PSGs have passed an LRT with raw P values < 0.01. Briefly, the P values of all genes were first ranked from smallest to the largest, and then each P value was multiplied by the total number of genes (11,838) divided by its rank. After correction, 44 and 101 PSGs were retained at 1% and 5% FDR levels, respectively. Functional classifications of the retained PSGs showed significant GO term enrichment in categories such as ATPase activity and motor activity, at both FDR levels, and the tissue expression patterns of these genes were similar to that obtained above with the complete gene list (available in supplementary table S2, Supplementary Material online).
When the branch-site test for positive selection is significant, then the Bayes empirical Bayes (BEB) procedure (Yang et al. 2005) can be used to calculate the posterior probability of specific sites (codons) being under positive selection. Within the dolphin, PSSs were detected in 125 PSGs. To estimate the effect of sequencing errors, in the low-coverage genome, on our identified PSSs, we randomly chose 48 PSGs that hold one or more PSSs (52 sites in total) and resequenced the candidate gene regions. After comparing our newly generated sequences with the reference dolphin genome, we found only two sites that showed inconsistent base calling, suggesting that our false-positive rate is only 3.8% (2/52). On the basis of this result, we expect that the sequence quality of dolphin genome is at a high level, despite the fact that the coverage is only 2.59×. The average size of the 48 amplified gene regions (containing the 52 resequenced sites) was approximately 390 bp (see supplementary table S1, Supplementary Material online), thus the sequencing error rate (assuming the all inconsistent sites are errors and not just polymorphisms) of the dolphin genome could be estimated to be 0.1% (2/[48 × 390]). Given the potentially high quality of the dolphin genome, and the accurate alignments generated in our analyses, this should have improved our identification of PSGs and PSSs and resulted in a low number of false positives. A full list of genes with PSSs is available in supplementary table S2, Supplementary Material online.
To understand whether the identified PSSs have potential impact on protein structure or function, we searched some of the PSSs against the InterPro database (Apweiler et al. 2001). We first examined the PSGs related to GO:0006869∼lipid transport, where only three genes (APOA2, ANXA1, and ATP8B2) showed evidence of PSSs. As presented in table 2, we found that the PSSs in each of these three genes are located in one or more of the functional domains. We then analyzed another 20 randomly chosen PSGs and found that 15 of them also have PSSs located in their conserved domains (table 2).
Table 2.
Some Functional Domains Located by the Dolphin Positively Selected Sites
| Gene | Domain Hits | Start | End | P | Database | InterPro ID |
|---|---|---|---|---|---|---|
| APOA2 | ApoA-II | 24 | 99 | 4.50E−42 | HMMPfam | IPR006801 |
| ANXA1 | ANNEXIN | 96 | 112 | 5.60E−66 | FPrintScan | IPR001464 |
| ANNEXIN | 2 | 18 | 1.20E−34 | FPrintScan | IPR002388 | |
| ATP8B2 | ATPase_P-type | 811 | 925 | 1.90E−32 | HMMTigr | IPR001757 |
| ACSM3 | AMP binding | 96 | 508 | 8.40E−94 | HMMPfam | IPR000873 |
| C2orf77 | Trichoplein | 148 | 461 | 3.80E−07 | HMMPfam | |
| COL3A1 | Collagen | 959 | 1,015 | 2.70E+08 | HMMPfam | IPR008160 |
| EIF2AK2 | PROTEIN_KINASE_DOM | 267 | 538 | 0 | ProfileScan | IPR000719 |
| EYA4 | EYA-cons_domain | 375 | 644 | 1.00E−111 | HMMTigr | IPR006545 |
| GLB1L2 | GLHYDRLASE35 | 310 | 325 | 4.40E−51 | FPrintScan | IPR001944 |
| Glyco_hydro_35 | 53 | 365 | 2.00E−116 | HMMPfam | IPR001944 | |
| MFSD12 | MFS_2 | 23 | 422 | 6.20E−39 | HMMPfam | |
| NBR1 | PB1 domain | 4 | 85 | 1.20E+14 | HMMSmart | IPR000270 |
| PB1 | 5 | 84 | 2.80E+12 | HMMPfam | IPR000270 | |
| PARP9 | Macro | 335 | 446 | 3.40E−15 | HMMPfam | IPR002589 |
| Appr-1"-p processing enzyme | 318 | 446 | 1.50E−13 | HMMSmart | IPR002589 | |
| PGAM2 | His_Phos_1 | 5 | 191 | 2.40E−41 | HMMPfam | IPR013078 |
| pgm_1: phosphoglycerate mutase 1 family | 5 | 215 | 2.80E−109 | HMMTigr | IPR005952 | |
| Phosphoglycerate mutase family | 5 | 193 | 2.70E−21 | HMMSmart | IPR013078 | |
| RAVER2 | RNA recognition motif | 143 | 216 | 8.40E−12 | HMMSmart | IPR000504 |
| SBF1 | SSF50729 | 895 | 1,039 | 8.50E+15 | superfamily | NULL |
| SCN5A | Ion_trans | 1,241 | 1,469 | 1.10E+53 | HMMPfam | IPR005821 |
| SLC20A2 | Signal peptide | 1 | 21 | NA | SignalPHMM | |
| TEX11 | SPO22 | 161 | 418 | 9.70E+74 | HMMPfam | IPR013940 |
The results obtained in dolphin were compared with those for the cow. Positive selection was detected in the cow lineage, where 242 (2% of the genes) showed significant evidence of positive selection, a number that is smaller than seen in the dolphin. A larger number of genes experiencing positive selection in the dolphin are consistent with our above result that the mean dN/dS in dolphin is much higher than that seen in the cow (fig. 2). For the cow genes, we also performed a functional clustering analysis using the DAVID tool. Although there was some overlap in the biological process and molecular function groups significantly enriched, the most enriched cow PSG categories focused on GO:0006811∼ion transport, GO:0055085∼transmembrane transport, and GO:0042626∼ATPase activity, coupled to transmembrane movement of substances (supplementary table S2, Supplementary Material online), a set that is extremely different from the GO terms enriched in the dolphin.
We also compared our results with those of a recent similar study performed by McGowen et al. (2012), who used the branch model, instead of the branch-site model, to detect PSGs in dolphin. The branch model identifies genes that have dN/dS > 1 as being PSGs. When we compared our results with those from McGowen et al. (2012), we found two major differences: 1) some of the genes identified as having high dN/dS by McGowen et al. did not yield similar values, with the same model, in our analyses. Examples of these genes are SULT2B1, FAM174B, KCNJ2, and CRYGN. A major reason for this difference likely is that different alignments were used. Many factors influence alignment generation, including homology prediction, transcript choice, aligner choice, and species usage. In our study, all the species analyzed are mammals, whereas those used by McGowen et al. ranged more widely and included the chicken. Moreover, McGowen et al. used the program Muscle to align the genes, whereas we used Prank, a program that is reported to be a much more accurate aligner (Fletcher and Yang 2010). The use of different primary treatments of the genomic data might cause differences in the estimation of selective constraints acting on genes. 2) Some of the genes identified by McGowen et al. that had high dN/dS did not pass the LRT during our positive selection detection. Examples of these genes include SULT2B1, CCL24, BAG2, TSPO2, and THAP1. This observation might be a result of the genes having a high dN/dS ratio due to the relaxation of purifying selection, rather than being due to positive selection (Cai and Petrov 2010). Although the gene sets obtained by McGowen et al. (2012) are different from those of our study, the GO term enrichment by PSGs showed fewer differences between the two studies (supplementary table S2, Supplementary Material online). McGowen et al. (2012) also identified genes related to lipid metabolism, lung development, and ATPase activity as being enriched in their analyses, although with a different gene list from that of our analyses.
Adaptive evolution involves more than just changes to amino acid sequence of proteins. Changes in expression, and gain and loss of genes, are also involved in adaptive evolution. Although we could not directly test changes in gene expression levels, we could examine change in expression, we did examine whether change in gene family size had occurred. Change in gene family size could affect both expression level and gain and loss of genes. To address this, we performed an analysis of the size of gene families among our five studied species. According to the Ensembl annotation, and classification of each protein-coding gene, we estimated the size of each gene family in each species and identified gene families that showed expansion and/or contraction specific to the dolphin lineage. Only a few expansion events (e.g., ENSFM00250000002216, ENSFM00540000720241, and ENSFM00550000743164 families, available in the supplementary table S3, Supplementary Material online) were found to have occurred in dolphin lineage, whereas the contractions in family size mainly occurred to genes in the “Olfactory receptor” gene families (e.g., ENSFM00250000000020, ENSFM00320000100072, and ENSFM00430000230074 families).
Discussion
In this study, we have conducted a comparative genomic analysis of the dolphin genome in an attempt to identify the underlying genetic mechanisms for aquatic adaptation in a mammal. We detected 368 PSGs and 1,238 PSSs in the dolphin lineage, which showed significantly enrichment in functions related to specific traits in cetaceans such as fat storage, muscle contraction, sensory perception of sound, and ATPase activity.
Validation of the Low-Coverage Genome
Although the current release of the dolphin’s genome has only 2.59× coverage, we discovered few sequencing errors in the dolphin genes. Comparing our amplified sequences to the draft genome sequence, we calculated an maximal error rate of approximately 0.9 bases per kilobase, a rate similar to that inferred by McGowen et al. (2012). Therefore, sequencing errors should only have a slight influence on the detection of positive selection based on the site sensitive method—branch site model. Combined with our strict alignment generation, including the use of Prank and Gblocks, the final alignments should be of high reliability with few alignment errors.
Adaptive Evolution of Lipid Metabolism Genes and the Evolution of Fat Storage in the Dolphin
Most cetaceans have a thick layer of blubber, which acts as their primary storage location for fat. The thickness of the blubber in whales is approximately 20 cm, being 10 times greater than that of other artiodactyls species (Pond 1978). Blubber provides many benefits to marine mammals, including saving energy by adding buoyancy while they swim. In addition, blubber is also an efficient thermal insulator. People are increasingly interested in genes that are responsible for fat storage (Sohle et al. 2012), because a growing challenge for health care is obesity (Prentice 2006). Here, we have identified some genes that have adaptively evolved and are closely related to lipid metabolism. These adaptively evolving genes belong to the fatty acid/lipid biosynthetic process (e.g., DGAT1, ACSM3, ELOVOL2, and ELOVL5) and lipid transport and localization (e.g., APOA2, START, APOLD1, and PLA2G5).
Fat storage involves the step-wise conversion of exogenous or endogenous fatty acid to diacylglycerol and ultimately triacylglycerols. Fatty acid synthesis is the first step during fat storage with acetyl-CoA being the key substrate. In this study, we observed an acyl-CoA synthetase gene, ACSM3, had three sites (Q158A, S310R, and Y324D) that experienced positive selection in the dolphin. These results suggest that these changes may provide some advantage in generating acetyl-CoA for fat deposit in the dolphin. As presented in table 2, the three PSSs are located in the AMP-binding domain, a domain responsible for the ligase activity. Moreover, two dolphin fatty acid elongase genes (ELOVL2 and ELOVL5) in dolphin were also identified as having PSSs, with ELOVL2 having one site (M264G) and ELOVL5 having three (T21P, N136Y, and H146F).
Diglyceride acyltransferase (DGAT) is an important protein that catalyzes the formation of triglycerides from diacylglycerol and acyl-CoA and whose reaction is the terminal step in triglyceride synthesis. DGAT is essential for the formation of adipose tissue and was also identified as a PSG in the dolphin. In addition, several apolipoprotein genes (APOA2, APOLD1, and LRP1) were also observed to have experienced positive selection, potentially supporting adaptation of lipid transport and localization for fat storage. There is evidence that these genes have roles in elevating lipid content and fat accumulation (Warden et al. 1993; Castellani et al. 2001, 2008; Terrand et al. 2009).
Adaptive Evolution of Locomotion-Related Genes in Cetaceans
Because of the relatively higher density of water compared with air, it is obvious that cetaceans must confront more resistive drag while swimming than other mammals experience during running. To address this issue, cetaceans must use more energy to support muscle contraction. Here, we have observed that 12 and 8 PSGs belong to genes involved in motor activity and muscle contraction, respectively, identifying candidate genes involved in the adaptation of locomotion physiology (table 1).
Muscle contraction is triggered by the flow of specific ions, including the transport of sodium ions into muscle cells. SCN4A, a member of the sodium channel family, plays a key role in the ability of a cell to generate and transmit electrical signals. Previous clinical medicine studies have shown that pathogenic mutations in SCN4A lead to myotonic spasms and hyperkalemia periodic paralysis (McClatchey et al. 1992; Sternberg et al. 2001). Adaptive evolution of the SCN4A gene might help dolphins attain high-speed locomotion. Moreover, some actin cytoskeleton (GO:0015629∼actin cytoskeleton) and myosin complex (GO:0016459∼myosin complex) proteins, key proteins in the muscle system, were also detected to have undergone positive selection.
Adaptation to locomotion in water was essential for foraging by cetaceans, as they need to dive to great depths and for a long time duration predation. This form of locomotion has large energy costs (Croll et al. 2001), thus cetaceans must make full advantage of aspired oxygen to generate energy. In this study, genes for the aerobic (TCA cycle, CS, SDHA, and MDH1B) and anaerobic (glycolysis, HK1, OGDH, PGAM2, and PFKFB1) respiration were identified to have undergone positive selection in dolphin. The genes CS and HK1 are also key rate-limiting enzymes in energy production; therefore, these PSGs may have provided some energetic support for the endurance of long dives by cetaceans.
Genes for Other Cetacean-Specific Traits
In addition to the above PSGs potentially involved in fat storage and locomotion, we also identified genes that were enriched in other functions, such as echolocation and dietary change (Walker et al. 1999). As presented in table 1, seven PSGs were enriched in GO:0007605∼sensory perception of sound category (table 1), with the genes CHD7, BARHL1, and SLC12A2 being closely associated with hearing (Dixon et al. 1999; Li et al. 2002; Vissers et al. 2004). Although the function of these genes in echolocation in the dolphin is unclear, they might provide clues to our understanding of the origin of echolocation in cetaceans.
Unlike their close relatives the artiodactyls, cetaceans have gradually shifted from an herbivorous to a carnivorous diet, with their major food being fish and crustaceans (Walker et al. 1999). The principal nutritional components of these food sources are protein and lipid. In our analysis of the distribution of expression patterns of PSGs, we found several genes that are highly expressed in the salivary gland (fig. 3). Combining this observation with the DAVID functional clustering results, where some of these genes enriched are in category GO:0008236∼serine-type peptidase activity, we expect that these adaptively evolving genes may be in part responsible for the shift in diet during cetacean evolution.
An analysis of the gene family evolution was also conducted in this study, as change in gene number is an additional molecular mechanism underlying adaptive evolution (Zhang et al. 2002). Although only a few expansion events were occurred in the dolphin lineage (supplementary table S3, Supplementary Material online), some have adaptive potential. The dynein domain family (ENSFM00550000743164) may be an example, as it is larger in the dolphin than any of the other species examined. Dynein domain containing genes are involved in muscle function, thus adaptive changes in gene number may have supported adaptation to locomotion in water by cetaceans. Moreover, contraction events were also observed in the dolphin lineage, and these were enriched in “Olfactory receptor” gene families (supplementary table S3, Supplementary Material online). This observation is consistent with previous finding that some genes involved in pheromonal olfaction in cetaceans had been pseudogenized (Yu et al. 2010). However, these results must be treated with caution, as the current low-coverage genome of the dolphin limits the analyses of gene family evolution (Milinkovitch et al. 2010), and thus, an updated high-quality genome would be necessary to conduct further analyses of the genes involved in the adaptation of cetaceans to the aquatic environment.
Conclusion
We conducted a genome-wide scan for PSGs and sites to investigate the genetic bases of aquatic adaptation by the dolphin. Although a limitation of this approach is the low coverage of the dolphin genome, we used strict filters during alignment reconstruction to improve the reliability of the final alignments. These scans, using the improved branch-site model in PAML, revealed 368 PSGs and 1,238 PSSs in the dolphin lineage. The reliability of our results was confirmed by our resequencing data, which showed that 50 of 52 randomly chosen PSSs (belonging to 48 PSGs) could be replicated. Functional clustering analysis showed that the PSGs or genes with PSSs were significantly enriched for functions related to fat storage, muscle contraction, ATP generation, perception of sound, and diet transformation. This study greatly adds to our understanding of the molecular landscape of aquatic adaptation. The low coverage of the dolphin genome, however, limits the detection of other types of genetic mechanism involved in adaption, for example, evolution of gene families and regulatory elements, thus an updated high-coverage genome of the dolphin would greatly help to complement the current analyses and allow a better understanding of the aquatic adaptations of cetaceans. Along with the development of the genome sequencing technologies, the sequencing of the genomes of additional aquatic mammals (including Pinnipeds and Sirenia) should provide more resources for investigating convergent or parallel evolution with respect to aquatic adaptation in mammals.
Supplementary Material
Supplementary tables S1–S3 are available at Genome Biology and Evolution online (http://gbe.oxfordjournals.org/).
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (2007CB411600, 2008GA001) and the Bureau of Science and Technology of Yunnan Province (31061160189).
Literature Cited
- Apweiler R, et al. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakewell MA, Shi P, Zhang J. More genes underwent positive selection in chimpanzee evolution than in human evolution. Proc Natl Acad Sci U S A. 2007;104:7489–7494. doi: 10.1073/pnas.0701705104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57: 289–300. [Google Scholar]
- Butler PJ, Jones DR. Physiology of diving of birds and mammals. Physiol Rev. 1997;77:837–899. doi: 10.1152/physrev.1997.77.3.837. [DOI] [PubMed] [Google Scholar]
- Cai JJ, Petrov DA. Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes. Genome Biol Evol. 2010;2:393–409. doi: 10.1093/gbe/evq019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani LW, Goto AM, Lusis AJ. Studies with apolipoprotein A-II transgenic mice indicate a role for HDLs in adiposity and insulin resistance. Diabetes. 2001;50:643–651. doi: 10.2337/diabetes.50.3.643. [DOI] [PubMed] [Google Scholar]
- Castellani LW, et al. Apolipoprotein AII is a regulator of very low density lipoprotein metabolism and insulin resistance. J Biol Chem. 2008;283:11633–11644. doi: 10.1074/jbc.M708995200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Cranford TW, Amundin M, Norris KS. Functional morphology and homology in the odontocete nasal complex: implications for sound generation. J Morphol. 1996;228:223–285. doi: 10.1002/(SICI)1097-4687(199606)228:3<223::AID-JMOR1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Croll DA, Acevedo-Gutierrez A, Tershy BR, Urban-Ramirez J. The diving behavior of blue and fin whales: is dive duration shorter than expected based on oxygen stores? Comp Biochem Physiol A Mol Integr Physiol. 2001;129:797–809. doi: 10.1016/s1095-6433(01)00348-8. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, et al. Mutation of the Na-K-Cl co-transporter gene Slc12a2 results in deafness in mice. Hum Mol Genet. 1999;8:1579–1584. doi: 10.1093/hmg/8.8.1579. [DOI] [PubMed] [Google Scholar]
- Fish FE, Beneski JT, Ketten DR. Examination of the three-dimensional geometry of cetacean flukes using computed tomography scans: hydrodynamic implications. Anat Rec (Hoboken). 2007;290:614–623. doi: 10.1002/ar.20546. [DOI] [PubMed] [Google Scholar]
- Fletcher W, Yang Z. The effect of insertions, deletions, and alignment errors on the branch-site test of positive selection. Mol Biol Evol. 2010;27:2257–2267. doi: 10.1093/molbev/msq115. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Kosiol C, et al. Patterns of positive selection in six mammalian genomes. PLoS Genet. 2008;4:e1000144. doi: 10.1371/journal.pgen.1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebure T, Stanhope MJ. Pervasive, genome wide positive selection, leading to functional divergence in the bacterial genus Campylobacter. Genome Res. 2009;19: 1224–1232. doi: 10.1101/gr.089250.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, et al. Hearing loss caused by progressive degeneration of cochlear hair cells in mice deficient for the Barhl1 homeobox gene. Development. 2002;129:3523–3532. doi: 10.1242/dev.129.14.3523. [DOI] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005;102:10557–10562. doi: 10.1073/pnas.0409137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loytynoja A, Goldman N. Phylogeny-aware gap placement prevents errors in sequence alignment and evolutionary analysis. Science. 2008;320:1632–1635. doi: 10.1126/science.1158395. [DOI] [PubMed] [Google Scholar]
- Mallick S, Gnerre S, Muller P, Reich D. The difficulty of avoiding false positives in genome scans for natural selection. Genome Res. 2009;19:922–933. doi: 10.1101/gr.086512.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova-Raina P, Petrov D. High sensitivity to aligner and high rate of false positives in the estimates of positive selection in the 12 Drosophila genomes. Genome Res. 2011;21:863–874. doi: 10.1101/gr.115949.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, et al. Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nat Genet. 1992;2:148–152. doi: 10.1038/ng1092-148. [DOI] [PubMed] [Google Scholar]
- McClellan DA, et al. Physicochemical evolution and molecular adaptation of the cetacean and artiodactyl cytochrome b proteins. Mol Biol Evol. 2005;22:437–455. doi: 10.1093/molbev/msi028. [DOI] [PubMed] [Google Scholar]
- McGowen MR, Grossman LI, Wildman DE. Dolphin genome provides evidence for adaptive evolution of nervous system genes and a molecular rate slowdown. Proc Biol Sci. 2012;279:3643–3651. doi: 10.1098/rspb.2012.0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RW, Janecka JE, Gatesy J, et al. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science. 2011;334:521–524. doi: 10.1126/science.1211028. [DOI] [PubMed] [Google Scholar]
- Milinkovitch MC, Helaers R, Depiereux E, Tzika AC, Gabaldon T. 2x genomes—depth does matter. Genome Biol. 2010;11:R16. doi: 10.1186/gb-2010-11-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy WJ, Pringle TH, Crider TA, Springer MS, Miller W. Using genomic data to unravel the root of the placental mammal phylogeny. Genome Res. 2007;17:413–421. doi: 10.1101/gr.5918807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren SR, Williams TM, Pabst DA, McLellan WA, Dearolf JL. The development of diving in marine endotherms: preparing the skeletal muscles of dolphins, penguins, and seals for activity during submergence. J Comp Physiol B. 2001;171:127–134. doi: 10.1007/s003600000161. [DOI] [PubMed] [Google Scholar]
- Oliver TA, et al. Whole-genome positive selection and habitat-driven evolution in a shallow and a deep-sea urchin. Genome Biol Evol. 2011;2:800–814. doi: 10.1093/gbe/evq063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pond CM. Morphological aspects and the ecological and mechanical consequences of fat deposition in wild vertebrates. Ann Rev Ecol Syst. 1978;9:519–570. [Google Scholar]
- Prentice AM. The emerging epidemic of obesity in developing countries. Int J Epidemiol. 2006;35:93–99. doi: 10.1093/ije/dyi272. [DOI] [PubMed] [Google Scholar]
- Reidenberg JS. Anatomical adaptations of aquatic mammals. Anat Rec (Hoboken). 2007;290:507–513. doi: 10.1002/ar.20541. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Schneider A, et al. Estimates of positive Darwinian selection are inflated by errors in sequencing, annotation, and alignment. Genome Biol Evol. 2009;1:114–118. doi: 10.1093/gbe/evp012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YY, et al. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc Natl Acad Sci U S A. 2010;107:8666–8671. doi: 10.1073/pnas.0912613107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JM, Smith NH. Synonymous nucleotide divergence: what is "saturation"? Genetics. 1996;142:1033–1036. doi: 10.1093/genetics/142.3.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohle J, et al. Identification of new genes involved in human adipogenesis and fat storage. PLoS One. 2012;7:e31193. doi: 10.1371/journal.pone.0031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg D, et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. 2001;124:1091–1099. doi: 10.1093/brain/124.6.1091. [DOI] [PubMed] [Google Scholar]
- Struntz DJ, et al. Blubber development in bottlenose dolphins (Tursiops truncatus) J Morphol. 2004;259:7–20. doi: 10.1002/jmor.10154. [DOI] [PubMed] [Google Scholar]
- Studer RA, Penel S, Duret L, Robinson-Rechavi M. Pervasive positive selection on duplicated and nonduplicated vertebrate protein coding genes. Genome Res. 2008;18:1393–1402. doi: 10.1101/gr.076992.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrand J, et al. LRP1 controls intracellular cholesterol storage and fatty acid synthesis through modulation of Wnt signaling. J Biol Chem. 2009;284:381–388. doi: 10.1074/jbc.M806538200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella AJ, et al. EnsemblCompara genetrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;19:327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Walker JL, Potter CW, Macko SA. The diets of modern and historic bottlenose dolphin populations reflected through stable isotopes. Mar Mammal Sci. 1999;15:335–350. [Google Scholar]
- Wang Z, et al. Adaptive evolution of 5'HoxD genes in the origin and diversification of the cetacean flipper. Mol Biol Evol. 2009;26:613–622. doi: 10.1093/molbev/msn282. [DOI] [PubMed] [Google Scholar]
- Warden CH, et al. Evidence for linkage of the apolipoprotein A-II locus to plasma apolipoprotein A-II and free fatty acid levels in mice and humans. Proc Natl Acad Sci U S A. 1993;90:10886–10890. doi: 10.1073/pnas.90.22.10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissler L, et al. Back to the sea twice: identifying candidate plant genes for molecular evolution to marine life. BMC Evol Biol. 2011;11:8. doi: 10.1186/1471-2148-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TJ, Davis RW. The effect of myoglobin concentration on aerobic dive limit in a Weddell seal. J Exp Biol. 2006;209:2576–2585. doi: 10.1242/jeb.02273. [DOI] [PubMed] [Google Scholar]
- Yang Z. Likelihood ratio tests for detecting positive selection and apcplication to primate lysozyme evolution. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002;19:908–917. doi: 10.1093/oxfordjournals.molbev.a004148. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wong WS, Nielsen R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- Yu L, et al. Characterization of TRPC2, an essential genetic component of VNS chemoreception, provides insights into the evolution of pheromonal olfaction in secondary-adapted marine mammals. Mol Biol Evol. 2010;27:1467–1477. doi: 10.1093/molbev/msq027. [DOI] [PubMed] [Google Scholar]
- Zhang J, Nielsen R, Yang Z. Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005;22:2472–2479. doi: 10.1093/molbev/msi237. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang YP, Rosenberg HF. Adaptive evolution of a duplicated pancreatic ribonuclease gene in a leaf-eating monkey. Nat Genet. 2002;30:411–415. doi: 10.1038/ng852. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.