

Abstract
Laboratory-based evolution experiments on microorganisms that do not recombine frequently show two distinct phases: an initial rapid increase in fitness followed by a slower regime. To explore the population structure and the evolutionary tree in the later stages of adaptation, we evolved a very large population (∼3 × 10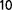 ) of Acinetobacter baylyi bacteria for approximately 2,800 generations from a single clone. The population was maintained in a chemostat at a high dilution rate. Nitrate in limiting amount and as the sole nitrogen source was used as a selection pressure. Analysis via resequencing of genomes extracted from populations at different generations provides evidence that long-term diversity can be established in the chemostat in a very simple medium. To find out which biological parameters were targeted by adaptation, we measured the maximum growth rate, the nitrate uptake, and the resistance to starvation. Overall, we find that maximum growth rate could be a reasonably good proxy for fitness. The late slow adaptation is compatible with selection coefficients spanning a typical range of 10
) of Acinetobacter baylyi bacteria for approximately 2,800 generations from a single clone. The population was maintained in a chemostat at a high dilution rate. Nitrate in limiting amount and as the sole nitrogen source was used as a selection pressure. Analysis via resequencing of genomes extracted from populations at different generations provides evidence that long-term diversity can be established in the chemostat in a very simple medium. To find out which biological parameters were targeted by adaptation, we measured the maximum growth rate, the nitrate uptake, and the resistance to starvation. Overall, we find that maximum growth rate could be a reasonably good proxy for fitness. The late slow adaptation is compatible with selection coefficients spanning a typical range of 10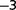 –10
–10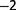 per generation as estimated by resequencing, pointing to a possible subpopulations structuring.
per generation as estimated by resequencing, pointing to a possible subpopulations structuring.
Keywords: Acinetobacter, experimental evolution, resequencing, nitrogen limitation, chemostat, large population
Introduction
Understanding adaptation is important in a wide number of boilogical, ecological, and technological contexts (Toft and Andersson 2010; Jackson et al. 2011). In this framework, controlled laboratory evolution experiments performed on large populations of bacteria can explore adaptation under well-defined selection pressure (Buckling et al. 2009; Conrad et al. 2011; Hindré et al. 2012). Through phenotypic characterization and sequencing, these experiments give a quantitative insight into the genomic adaptation of microbial populations (Barrick and Lenski 2009; Barrick et al. 2009; Tenaillon et al. 2012).
Possibly, one of the best examples of the impact of this approach concerns fundamental ecology. As a result of the “competitive exclusion principle” (Gause 1934; Hardin 1960), it was previously believed that diversity cannot be created and maintained in simple, spatially, and temporally homogeneous environments containing a single resource. The recent in vitro evolution experiment of Maharjan et al. (2006, 2012) has disproven the competitive exclusion principle for large populations in the laboratory and demonstrates that a clonal population of Escherichia coli evolving in a chemostat can diversify into a number of subpopulations.
The mechanisms responsible for this diversity are unclear and include convergent evolution and regulatory degeneracy (Gresham et al. 2008; Wang et al. 2010), cross-feeding (Rosenzweig et al. 1994; Friesen et al. 2004), and frequency-dependent selection (Levin 1988; Doebeli and Ispolatov 2010). Beardmore et al. (2011) recently revealed a fourth mechanism, which explains the emergence and maintenance of diversity in simple environments by the existence of metabolic and physiological trade-offs. A fifth possibility is related to the fact that for such large population-size experiments in the absence of recombination, many beneficial mutations compete to drive adaptation, through “clonal interference” (Gerrish and Lenski 1998). A recent laboratory evolution study (Hughes et al. 2012) found stable competition between coexisting hosts with different plasmid genotypes in plasmid–host adaptation dynamics, suggesting that this diversity plays a role in the evolution of the persistence of drug resistance. Despite recent theoretical and experimental advances (Desai and Fisher 2007; Desai et al. 2007; Park and Krug 2007; Brunet et al. 2008; Good et al. 2012), this regime of adaptation is not completely understood. For example, many open questions concern the dynamics of the advantage (Barrick et al. 2009), the role of epistasis (Chou et al. 2011; Khan et al. 2011), and the structuring of a population into subpopulations of competing clones (Maharjan et al. 2007). A recent study (Maharjan et al. 2012) used large-scale genomic and phenotypic analysis of the chemostat evolution data to identify the extent of diversification. They placed their results in the context of the above theoretical mechanisms of evolutionary divergence and proposed that diversity is unlikely to be explained by any one of the available theoretical models.
A criticism of the earlier study of Maharjan et al. (2006) concerned the relatively short evolutionary time scale of the experiment (26 days), which might lead some to speculate that running similar experiments for longer times would destroy the observed diversity (Conrad et al. 2011). Here, we present an evolution experiment with a different organism, Acinetobacter baylyi, that lasted 124 days (∼2,800 generations), and we report stable and possibly increasing diversity that allows for an analysis of genome adaptation.
We constructed a chemostat culture using a strain of A. baylyi growing in a minimal medium, which in principle allows for a limited number of distinct ecological niches. The bacteria were grown with limited amounts of nitrate as the sole source of nitrogen. A high dilution rate (between 0.6 h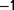 and 0.9 h
and 0.9 h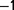 ), which should be the least favorable condition for the onset of diversity (Maharjan et al. 2012) was also used. Our system involves a unique growth medium supply and composed of interconnected alternating tandem chemostats (fig. 1), which prevents bacterial adhesion to the chemostat walls (de Crécy-Lagard et al. 2001). The main advantage of this setup (compared with serial dilution experiments) is that it allows for constant and controlled growth conditions involving very large population sizes. The main drawback is that it is cumbersome, and (with our resources) does not allow for replicate populations in parallel. However, this does not prevent the extraction of relevant information. Acinetobacter baylyi ADP1 has been previously sequenced and annotated (Barbe et al. 2004). We identified the mutations appearing during the experiment by genome resequencing at three late time points during the experiment. Sequencing permitted the determination of the frequencies of the mutations at different points in time and, by genotyping each mutation on isolated clones, resulted in the reconstruction of a partial population tree. These data provide approximate quantitative estimates of the selection coefficient of some mutations. We measured the associated changes in the maximum growth rate and found that the increase is consistent with the estimated selection coefficients.
), which should be the least favorable condition for the onset of diversity (Maharjan et al. 2012) was also used. Our system involves a unique growth medium supply and composed of interconnected alternating tandem chemostats (fig. 1), which prevents bacterial adhesion to the chemostat walls (de Crécy-Lagard et al. 2001). The main advantage of this setup (compared with serial dilution experiments) is that it allows for constant and controlled growth conditions involving very large population sizes. The main drawback is that it is cumbersome, and (with our resources) does not allow for replicate populations in parallel. However, this does not prevent the extraction of relevant information. Acinetobacter baylyi ADP1 has been previously sequenced and annotated (Barbe et al. 2004). We identified the mutations appearing during the experiment by genome resequencing at three late time points during the experiment. Sequencing permitted the determination of the frequencies of the mutations at different points in time and, by genotyping each mutation on isolated clones, resulted in the reconstruction of a partial population tree. These data provide approximate quantitative estimates of the selection coefficient of some mutations. We measured the associated changes in the maximum growth rate and found that the increase is consistent with the estimated selection coefficients.
Fig. 1.—

Left: Diagram of a single reactor during the culture stage. The reactor has been assembled by glass blowing, and incorporates a square cross-section glass tube in the bottom portion, and a screwed plastic cap on top. The entry and exit tubes are welded to the top of the reactor and comprise an air supply connected to a tube that is terminated by a porous glass plug at the bottom of the reactor (generating bubbles that give both aeration and mixing via air lift) (C), a fresh supply of medium (B), a wash fluid entry point (A), a fluid exit, and a biphasic mixture of air and liquid (D). Right: Schematics of the tubing connections during the culture phase in the reactor R1. R1 and R2 are the two reactors of the setup; each is temperature regulated; filtered air (rate adjusted by a flow meter/needle valve) is injected through port (C) of reactor R1. It passes through a porous glass plug and generates bubbles serving for both culture aeration and mixing via air lift within the reactor. The bottle containing the fresh medium is connected to a peristaltic pump; a constant flow of medium arrives at port (B) of reactor R1. A heating resistance heats the medium before entry (B), to prevent backward contamination of the supply of medium; the reactor wash solution (connected to port (A) and not used at this stage of operation of R1) comprises NaOH 1 M and rinse medium (the same solution as the supply of medium to the reactor); a waste bottle is connected to port (D) of reactor R1. It receives a biphasic mixture of air and medium exiting from the reactor by a common path and traps the liquid; the air exiting from the waste bottle is bubbled through bleach in a bubbling bottle, so as to avoid laboratory air contamination. The full arrows indicate the circulation of liquids, and the open arrows indicate the circulation of air. Crosses indicate the pinched-closed sections of tubes. Every week, the content of the operating reactor is transferred to the already cleaned tandem reactor. During the transfer phase (e.g., from R1 to R2), port (D) of R1 is closed and ports (A), (B), and (C) are open; ports (B) and (C) of R2 are closed and ports (A) and (D) are open. Air and medium containing bacteria come from R1 to R2 through port (A). When the transfer is stopped, R2 enters the culture phase, and R1 is rinsed and washed and ready for the next transfer.
Materials and Methods
Bacterial Strain and Medium
We used a strain of A. baylyi obtained from NCIMB (http://www.ncimb.com, last accessed January 3, 2013; NCIMB 11826). The minimal medium (pH 6.8) used in the evolution experiment comprised 50 mM potassium phosphate, 1 mM potassium nitrate, 3 mM potassium citrate, 9 mM citric acid, 1 mM magnesium sulphate, traces of elements (3 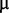 M iron (III) chloride, 1
M iron (III) chloride, 1 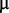 M zinc sulphate, 1
M zinc sulphate, 1 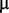 M cobalt chloride, 0.6
M cobalt chloride, 0.6 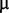 M copper sulphate, 1
M copper sulphate, 1 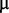 M manganese sulphate, 1
M manganese sulphate, 1 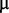 M sodium molybdate, 1
M sodium molybdate, 1 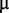 M manganese chloride, and 1
M manganese chloride, and 1 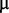 M calcium sulphate), and polypropylene glycol 2,000 at dilution 1:20,000 as antifoaming agent. For the initiation of the evolution experiment, the ancestral strain (ABWT) was plated, a single colony was inoculated in the minimal medium and grown at 35°C until OD reaches 0.1–0.2. A volume of 50 ml of this culture was introduced in the reactor then completed by fresh medium. At various time intervals, samples were snap-frozen as stocks with 10% DMSO. Stocks were later plated and clones isolated. They are named ABGi, where G is the number of generations undergone by the bacteria before sampling, and i is a letter used to identify a clone.
M calcium sulphate), and polypropylene glycol 2,000 at dilution 1:20,000 as antifoaming agent. For the initiation of the evolution experiment, the ancestral strain (ABWT) was plated, a single colony was inoculated in the minimal medium and grown at 35°C until OD reaches 0.1–0.2. A volume of 50 ml of this culture was introduced in the reactor then completed by fresh medium. At various time intervals, samples were snap-frozen as stocks with 10% DMSO. Stocks were later plated and clones isolated. They are named ABGi, where G is the number of generations undergone by the bacteria before sampling, and i is a letter used to identify a clone.
Experimental Evolution in a Chemostat
Two interconnected reactors were used allowing aseptic periodic transfer from one reactor to the other, so that the reactor being not used for growth can be cleaned in situ, preventing long-term biofilm formation and bacteria evading dilution (de Crécy-Lagard et al. 2001). Transfer is made every week. The reactors (150 ml) are made of glass and have been assembled by glass blowing. One entry tube is used for the medium supply with its dripping opening inside the reactor and positioned above the liquid level of the culture. A compressed air input tube is plunged inside the reactor to the bottom. Air bubbles provide for aeration and mixing of the growth medium. An exit tube allows for both air and liquid outflow. Another entry is allowed for the periodic injection of a cleaning solution (1 M NaOH) within the reactor or for the transfer of the bacterial population before reactor cleaning. The reactors are connected to a unique growth medium supply via a flow rate controlling peristaltic pump (Masterflex). Temperature was regulated using a commercial PID regulator (ENDA ETC 442), maintaining the temperature at 35  0.5 °C. During the first 6 days, the dilution rate was kept below 0.5
0.5 °C. During the first 6 days, the dilution rate was kept below 0.5 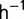 , not to lose the population. Subsequently, it was kept constant between 0.6 h
, not to lose the population. Subsequently, it was kept constant between 0.6 h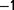 and 0.9 h
and 0.9 h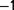 (the reasons for these variations were both practical and related to experiments testing the maximum growth rate that occurs over the evolutionary time).
(the reasons for these variations were both practical and related to experiments testing the maximum growth rate that occurs over the evolutionary time).
Maximum Growth Rate Measurements
The batch cultures were performed in a glass vessel presenting a lower part with a transparent square cross-section, so that an in situ optical density measurement could be performed. This system consisted of 1) a light emitting diode (LED) positioned in close proximity to one flat face of the reactor and emitting light inside the reactor, 2) a first photosensitive detector PD1 placed on the other side of the reactor and facing the LED, and 3) a second photodetector PD2 placed in close proximity of the light emitter. The current of the photodetectors PD1 and PD2 were fed to a logarithmic-ratio amplifier, so as to yield a voltage related to the optical density of the culture. The reference value was obtained when fresh medium was first injected. The system yielded optical density measurements taken approximately every minute. The maximum growth rate in the batch mode for a set of clones was estimated from those measurements.
Genome Resequencing
We detected all events, including IS movements and DNA rearrangements, through genome sequencing. Genome resequencing of two endpoint clones was performed using Illumina platform by GATC-Biotech. The genome of the ancestral clone and the genomes of three sampled populations were resequenced using Illumina platform at GRCF HTS Center at Johns Hopkins University, Baltimore. Sequence alignment against the reference genome (A. baylyi ADP1) was performed using Mozaik (http://bioinformatics.bc.edu/marthlab, last accessed January 3, 2013) or bowtie2 (Langmead et al. 2009). Visualization was performed using Tablet (Milne et al. 2010). Mutation detection was performed using SAMtools (Li et al. 2009) and gigaBayes (http://bioinformatics.bc.edu/marthlab, last accessed January 3, 2013). The de novo genomic assembler velvet (Zerbino and Birney 2008) was also used. These tools are designed to extract allele frequencies and thus are also well adapted for the detection of mutation frequencies in mixed populations.
Sequencing and Reconstruction of the Population Tree
To study the mutational patterns and connect them with the evolutionary dynamics, we resequenced several samples: the ancestral clone (ABWT), two endpoint clones (AB2800a and AB2800b), and three samples of populations at three different time points (AB1900, AB2300, and AB2800) (We adopt the following notations: Clones/populations are labeled by the string AB followed by the number G and the label i, where G is the number of generations undergone by the clone or population and i is a tag to identify an isolated clone.). Sequencing of the ABWT genome revealed that it has differences with respect to the A. baylyi ADP1 genome (Barbe et al. 2004). The list of those differences is given as supplementary data, Supplementary Material online. The mutations that occurred during the experimental evolution from the ancestral ABWT clone are listed in table 1. To infer the order of appearance of the mutations and identify subpopulations carrying different mutations, we performed 378 short polymerase chain reactions (PCRs) on 87 isolated clones targeting the mutation loci, and sequenced the amplified fragments to test for the presence of the mutations, or tested for deletions or insertions by gel electrophoresis (see supplementary data, Supplementary Material online).
Table 1.
Mutations Detected by Analysis of Sequencing Data
| Position on the ADP1 Chromosome | Gene - Region | Product | Mutational Event | Comment | G = 1,900 | G = 2,300 | G = 2,800 |
|---|---|---|---|---|---|---|---|
| SNP | |||||||
| 244223 | glnK (a) | Regulatory protein, for nitrogen assimilation by glutamine synthetase | C  T T |
41 bp upstream of the start codon | ND | 20% | 41% |
| 389458 | citA (a) | Citrate proton symporter | T  C C |
Codon frequencies GAA (0.041)  GAG (0.014) GAG (0.014) |
ND | ND | 29% |
| 1020409 | ACIAD1029 (b) | Putative lipoprotein | T  C C |
100 bp upstream of the start codon | ND | ND | 23% |
| 1903797 | ACIAD1911 | Putative nitrate transporter transmembrane protein | G  A A |
12 bp upstream of the start codon | ND | ND | ND |
| 1903816 | ACIAD1911 (a, b) | Putative nitrate transporter transmembrane protein | C  T T |
31 bp upstream of the start codon | 100% | 100% | 100% |
| 1903844 | ACIAD1911 | Putative nitrate transporter transmembrane protein | G  A A |
59 bp upstream of the start codon | ND | ND | ND |
| 1903876 | ACIAD1911 | Putative nitrate transporter transmembrane protein | A  T T |
91 bp upstream of the start codon | ND | ND | ND |
| 2042174 | ACIAD2054 (a, b) | Conserved hypothetical protein | T  C (S C (S  P) P) |
Nonsynonymous mutation in a gene yet cut by the inversion event | 26% | 65% | 91% |
| 2100627 | ACIAD2113 | Conserved hypothetical protein | A  G G |
239 bp upstream of the start codon | 3% | 41% | 6% |
| 2790910 | ftsH | Cell division protein | G  A (H A (H  Y) Y) |
Nonsynonymous | 33% | 57% | 10% |
| 2891560 | qseB (a, b) | Quorum sensing DNA-binding response regulator | G  A (S A (S  F) F) |
Nonsynonymous | 100% | 100% | 100% |
| Small Indels | |||||||
| 1920605 | ACIAD1931 | Putative magnesium citrate secondary transporter | Ins A | 32 bp upstream of the start codon | ND | ND | 12% |
| 1249430 | csrA (a, b) | Carbon storage regulator |
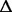 8 bp 8 bp |
Last 15 amino acids (21% of the protein) modified | 100% | 100% | 100% |
| Large Rearrangements | |||||||
| 1230681 | ACIAD1230 (a) | Putative transcriptional regulator | IS insertion | 79 bp upstream of the start codon (B1 branch) | 43% | 32% | 43% |
| 1230766 | ACIAD1230 | Putative transcriptional regulator | IS insertion | Within gene (B5 branch) | 9% | 7% | ND |
| 1230919 | ACIAD1230 | Putative transcriptional regulator | IS insertion | Within gene (B3 branch) | 32% | 53% | 10% |
| 1231207 | ACIAD1230 (b) | Putative transcriptional regulator | IS insertion | Within gene (B2 branch) | 1% | 0.5% | 47% |
| 1860246 | ACIAD1864–ACIAD1865 | Putative transcriptional regulators | IS insertion | Between genes (B4 branch) | 16% | 8% | ND |
| NA | (a, b) | NA | IS insertion | Within a DNA sequence not found in A. baylyi ADP1 | 100% | 100% | 100% |
| 1848721–1858316 | ACIAD1848 ACIAD1861 (a, b) | Putative phage replication initiation factor, putative phage-related protein, and putative phage replication initiation factor | Duplication | Spans ∼10 kb | 100% | 100% | 100% |
| 2041592–2046126 | ACIAD2054, gltL gltK, gltJ, gltI (a, b) | Conserved hypothetical protein and glutamate/aspartate transport proteins | Inversion (4,534 bp) | Cuts gltI and ACIAD2054; quenches glt operon | 100% | 100% | 100% |
| 2046132–2046451 | gltI (b) | Glutamate/aspartate transport protein |
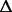 319 bp (named 319 bp (named 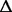 gltI in the text) gltI in the text) |
Deletion in a gene yet cut by the inversion event | ND | ND | 51% |
| 2664637–2726455 | ACIAD2713–ACIAD2778 (a, b) | NA |
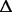 60 kb 60 kb |
58 CDS deleted | 100% | 100% | 100% |
Note.—List of detected mutations occurring in the experiment, separated by type (SNPs, indel, and large rearrangements). The last three columns indicate the frequency of each mutation at the three different sequencing times (G stands for the number of generations); 100% frequencies are marked in boldface. NA, not applicable; ND, not detected ( 1%). The letters (a) and (b) in the second column indicate if the mutation was detected in the endpoint clones AB2800a and AB2800b, respectively. Compared with other laboratory evolution experiments, there is a relevant number of large rearrangements, many of which appear early on and reach very high frequencies.
1%). The letters (a) and (b) in the second column indicate if the mutation was detected in the endpoint clones AB2800a and AB2800b, respectively. Compared with other laboratory evolution experiments, there is a relevant number of large rearrangements, many of which appear early on and reach very high frequencies.
In total, we detected 23 mutations on 15 chromosomal loci, of which 10 and 11 are on the endpoint clones, which is in line with other evolution experiments with similar numbers of generations (Khan et al. 2011; Bachmann et al. 2012). Only four point mutations were found in genes, three of which were nonsynonymous. Remarkably (table 1), large events such as deletions (2), inversions (1), duplications (1), or insertion sequence (IS) transpositions (6) balanced in number small events such as single-nucleotide polymorphisms (SNPs) (11) and indels (2) (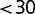 bp) and seem to appear early on during the experiment. To our knowledge, this feature is not common to other experiments of this kind (Barrick et al. 2009; Wang et al. 2010; Tenaillon et al. 2012). Together, these data permitted the reconstruction of a partial evolutionary tree (fig. 2). To this end, an important feature is the detection of multiple co-occurring IS integration events just before 1,000 generations and concomitant with the disappearance of an IS present between ACIAD0290 and ACIAD0291 in ABWT (but not in ADP1). These two types of changes may be related to a single mutational event such as transposition of the latter IS to a new target. However, these changes may also involve two different mutational events. This target locus was variable among different clones as five different loci were identified. This gave birth to five detectable branches (named B1, B2, B3, B4, and B5). This IS insertion was used as a genomic marker to estimate the ratio of the five subpopulations in the sequencing of sampled populations.
bp) and seem to appear early on during the experiment. To our knowledge, this feature is not common to other experiments of this kind (Barrick et al. 2009; Wang et al. 2010; Tenaillon et al. 2012). Together, these data permitted the reconstruction of a partial evolutionary tree (fig. 2). To this end, an important feature is the detection of multiple co-occurring IS integration events just before 1,000 generations and concomitant with the disappearance of an IS present between ACIAD0290 and ACIAD0291 in ABWT (but not in ADP1). These two types of changes may be related to a single mutational event such as transposition of the latter IS to a new target. However, these changes may also involve two different mutational events. This target locus was variable among different clones as five different loci were identified. This gave birth to five detectable branches (named B1, B2, B3, B4, and B5). This IS insertion was used as a genomic marker to estimate the ratio of the five subpopulations in the sequencing of sampled populations.
Fig. 2.—

Inferred evolutionary tree of the population, deduced by combining genome sequencing data analysis and systematic sequencing of PCR fragments performed on 87 isolated clones. The numbers on the left side indicate generations, estimated from the imposed dilution rate. Each vertical line corresponds to a subpopulation (not all subpopulations are displayed). Some relevant mutations are indicated at the time of appearance. N*, N1, N2, and N3 designate C1903816T, C1903797T, C1903844T, and C1903876T, respectively (table 1). Five branches arise before 1,000 generations, and three branches (B1, B2, and B3) are still present at the end of the experiment. For the last three time points, genomes extracted from population samples were resequenced; the sizes of the gray rectangles indicate the estimated relative population percentage of each branch (% values indicated immediately above rectangles) as deduced from genomic data analysis. The labels (a) to (f) designate specific subpopulations as follows: (a) branch B1 with no additional mutation, (b) branch B1 with an additional ACIAD2054 mutation, (c) =(a) with mutated glnk, (d) =(b) with no additional mutation, (e) additional mutation in citA, (f) no additional mutation in citA, (g) deletion in gltI, and (h) deletion in gltI, plus ACIAD1029 mutation.
The dynamics of branch B2 is remarkable. Figure 2 shows the near extinction of the branch over approximately 1,000 generations: at generations 980 and 1,100, one isolated clone out of 10 belongs to B2 (data not shown), whereas at generations 1,900 and 2,300, the sequencing of sampled populations indicates that the B2 branch occupies only approximately 1% of the population. However, the same branch increased to occupy nearly half the population at generation 2,800. Two mutational events appeared after 2,300 generations in the B2 branch and are seemingly associated with this uprise: an SNP upstream of ACIAD1029 and a deletion of 319 bp in the gltI gene (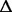 gltI). The frequencies of these two genotypic features at the end of the experiment are 23% and 51%, respectively. Comparing these frequencies to the 47% of the B2 branch at the end of the experiment, we conclude that essentially all B2 bacteria carried the deletion, which is likely linked to a gain in fitness that allows the B2 branch to increase in relative proportion. This deletion event did not appear in the remainder of the population, suggesting that this event might be rare and/or dependent on the mutational background. Notably, the locus of this deletion appears to be highly dynamic during the whole experiment as it involves two other different mutational events that are assumed to give a relevant advantage at two other different times: 1) an early inversion of 4,534 bp and 2) a point mutation (ACIAD2054) shared by several branches, with a frequency increasing from 26% to 91% from generation 1,900 to generation 2,800. However, it must be noted that also some genetic instability may increase the mutation frequency at that specific locus and that nontransitive interactions between subpopulations and negative frequency-dependent selection cannot be ruled out.
gltI). The frequencies of these two genotypic features at the end of the experiment are 23% and 51%, respectively. Comparing these frequencies to the 47% of the B2 branch at the end of the experiment, we conclude that essentially all B2 bacteria carried the deletion, which is likely linked to a gain in fitness that allows the B2 branch to increase in relative proportion. This deletion event did not appear in the remainder of the population, suggesting that this event might be rare and/or dependent on the mutational background. Notably, the locus of this deletion appears to be highly dynamic during the whole experiment as it involves two other different mutational events that are assumed to give a relevant advantage at two other different times: 1) an early inversion of 4,534 bp and 2) a point mutation (ACIAD2054) shared by several branches, with a frequency increasing from 26% to 91% from generation 1,900 to generation 2,800. However, it must be noted that also some genetic instability may increase the mutation frequency at that specific locus and that nontransitive interactions between subpopulations and negative frequency-dependent selection cannot be ruled out.
In brief, the combination of clonal and population sequencing with systematic mutation detection by PCR points to a complex scenario with highly dynamic coevolving subpopulations, even in the later stages of the experiment, where the main metabolic adaptation should have been already accomplished.
The Dynamics of Maximum Growth Rate Is Compatible with the Advantage Estimated from Population Sequencing
During the experiment, the dilution rate was typically kept constant (The dilution rate was below 0.5 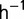 during the first 6 days.) between 0.6 and 0.9 h
during the first 6 days.) between 0.6 and 0.9 h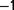 . However, the dilution rate was sometimes transiently increased above the wash-out threshold to measure the maximum growth rate
. However, the dilution rate was sometimes transiently increased above the wash-out threshold to measure the maximum growth rate 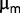 of the population, by real-time monitoring of optical density during the wash out. We assume that this transient increase in dilution rate did not interfere heavily with the population structure because it never washed out more than half of the population and lasted for approximately 1 h only (One may wonder whether this procedure could remove completely some low-frequency mutants. The probability of losing a subpopulation of n bacteria in a wash out of half the population can be estimated as
of the population, by real-time monitoring of optical density during the wash out. We assume that this transient increase in dilution rate did not interfere heavily with the population structure because it never washed out more than half of the population and lasted for approximately 1 h only (One may wonder whether this procedure could remove completely some low-frequency mutants. The probability of losing a subpopulation of n bacteria in a wash out of half the population can be estimated as 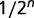 . Let us consider a subpopulation of 20 individuals [which corresponds to a frequency of approximately 10
. Let us consider a subpopulation of 20 individuals [which corresponds to a frequency of approximately 10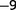 ], the probability of losing this subpopulation is 0.0001%.). We also measured
], the probability of losing this subpopulation is 0.0001%.). We also measured 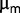 for isolated clones in batch, by fitting the growth curve during the exponential phase. The results are plotted in figure 3. The maximum growth rate in our experiment initially increased rapidly, whereas after approximately 500 generations, it became compatible with a much slower linear increase or plateau. A similar feature appears in other studies of this kind (Barrick et al. 2009; Chou et al. 2011).
for isolated clones in batch, by fitting the growth curve during the exponential phase. The results are plotted in figure 3. The maximum growth rate in our experiment initially increased rapidly, whereas after approximately 500 generations, it became compatible with a much slower linear increase or plateau. A similar feature appears in other studies of this kind (Barrick et al. 2009; Chou et al. 2011).
Fig. 3.—

Maximum growth rate from batch (black circles) and chemostat (open circles) measurements, plotted as a function of time (in generations). Chemostat measurements were performed during the experiment by transiently increasing the dilution rate above the wash-out threshold (open circles). Lines are fits with a hyperbola (dashed: 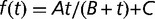 ) and with hyperbolic plus linear function (solid:
) and with hyperbolic plus linear function (solid: 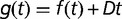 ), as in Barrick et al. (2009). The maximum growth rate initially increases rapidly and drastically slows down after approximately 500 generations. The shaded area indicates the late stages of the experiment. Arrows indicate times of population sequencing. The inset plots the dynamics of frequencies related to some mutational events resulting from resequencing analysis in late stages of the experiment. Each number designates a branch identified by the IS insertion event. 1a, 2a, and 2b designate clearly identified subpopulations validated by PCR on isolated clones. Mutations shared by several branches or not verified by PCR are not represented.
), as in Barrick et al. (2009). The maximum growth rate initially increases rapidly and drastically slows down after approximately 500 generations. The shaded area indicates the late stages of the experiment. Arrows indicate times of population sequencing. The inset plots the dynamics of frequencies related to some mutational events resulting from resequencing analysis in late stages of the experiment. Each number designates a branch identified by the IS insertion event. 1a, 2a, and 2b designate clearly identified subpopulations validated by PCR on isolated clones. Mutations shared by several branches or not verified by PCR are not represented.
Thus, the maximum growth rate increased little in the later stages of the experiment, where the described examples point to a highly dynamic situation. The population sequencing data gave further clues to the population dynamics that occurred between generations 1,900 and 2,800. Some mutation frequencies increase, whereas others decrease, the ratio of subpopulation frequencies not being constant. This indicates that competition between subpopulations still takes place during the latter stage of the experiment. If one assumes that the frequency 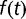 of a mutation is related to the selection coefficient
of a mutation is related to the selection coefficient 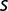 by
by 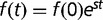 ,
, 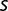 can be roughly estimated by comparing the frequency observed at different generations. Following this reasoning, we obtained seven estimated values of
can be roughly estimated by comparing the frequency observed at different generations. Following this reasoning, we obtained seven estimated values of 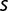 between 0.001 and 0.01 per generation (table 2). If one uses the fit of
between 0.001 and 0.01 per generation (table 2). If one uses the fit of 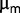 versus number of generations, one estimates values of
versus number of generations, one estimates values of 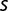 between 0.0075 and 0.0094 per generation, which are of the same order as the values deduced from sequencing. Although both estimates are rough, their agreement suggests that a small increase in
between 0.0075 and 0.0094 per generation, which are of the same order as the values deduced from sequencing. Although both estimates are rough, their agreement suggests that a small increase in 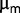 may account for the fairly high population dynamics observed during the last 2,000 generations. It also suggests that
may account for the fairly high population dynamics observed during the last 2,000 generations. It also suggests that 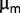 might be a reasonable proxy estimate of fitness. This result is not completely surprising, as the bacteria were subjected to a high dilution rate regime, with a dilution rate of approximately 80% of maximum growth rate during the last 2,500 generations.
might be a reasonable proxy estimate of fitness. This result is not completely surprising, as the bacteria were subjected to a high dilution rate regime, with a dilution rate of approximately 80% of maximum growth rate during the last 2,500 generations.
Table 2.
Selection Coefficient per Generation Estimated from Resequencing Data
| Mutation | Selection Coefficient |
|---|---|
| ACIAD2054 |
 0.004 0.004 |
| ftsH | 0.001 |
| ACIAD2113 | 0.006 |
| glnk |
 0.007 0.007 |
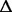 gltI gltI |
0.009 |
| citA |
 0.007 0.007 |
| ACIAD1029 |
 0.006 0.006 |
Notably, some values of 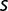 estimated from population sequencing data are still relatively high toward the end of the experiment (e.g., the values of
estimated from population sequencing data are still relatively high toward the end of the experiment (e.g., the values of 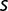 for citA,
for citA, 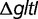 , and ACIAD1029, table 2). This suggests that (on average)
, and ACIAD1029, table 2). This suggests that (on average) 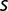 is not decreasing significantly during the late stages of the experiment. Consistent with this picture, the values of
is not decreasing significantly during the late stages of the experiment. Consistent with this picture, the values of 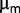 measured late in the experiment appear to be compatible with a slow increase with generation number.
measured late in the experiment appear to be compatible with a slow increase with generation number.
Complementary Contributions to Fitness
Thus, variations in mutation frequencies suggest that the population is still highly dynamic during the last 2,000 generations. Our rough estimates indicate that the increase in 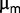 could be sufficient to capture the degree of adaptation during the experiment. Even though the ratio between the dilution rate and
could be sufficient to capture the degree of adaptation during the experiment. Even though the ratio between the dilution rate and 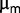 is approximately 0.8, we cannot exclude that other ways of adaptation could be operative, in view of the fact that the chemostat dilution rate is set below
is approximately 0.8, we cannot exclude that other ways of adaptation could be operative, in view of the fact that the chemostat dilution rate is set below 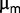 . To illustrate this aspect, let us compare two isolated clones, AB1100h and AB2800b. AB1100h was isolated and identified as belonging to the B2 branch. Its descendants are led to near extinction (between generations 1,900 and 2,300) but later acquire beneficial mutations that make them the largest subpopulation present at the end (fig. 2). It is thus expected that the fitness of AB2800b, an endpoint clone belonging to B2 branch, is larger than the fitness of AB1100h. Because of the uncertainties of the measurements of the maximum growth rate in batch of the two clones (
. To illustrate this aspect, let us compare two isolated clones, AB1100h and AB2800b. AB1100h was isolated and identified as belonging to the B2 branch. Its descendants are led to near extinction (between generations 1,900 and 2,300) but later acquire beneficial mutations that make them the largest subpopulation present at the end (fig. 2). It is thus expected that the fitness of AB2800b, an endpoint clone belonging to B2 branch, is larger than the fitness of AB1100h. Because of the uncertainties of the measurements of the maximum growth rate in batch of the two clones (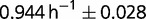 for AB1100h and
for AB1100h and 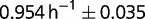 for AB2800b), the possibility remains that other traits of selection might have been involved.
for AB2800b), the possibility remains that other traits of selection might have been involved.
One targeted trait could be nitrate uptake: Bacteria in a chemostat have to manage growth under limited nutrient availability. To measure how bacteria deal with nitrogen limitation, it is necessary to access their affinity for the limiting nutrient (nitrate). To do so, we grew a clone (isolated after 7 days) in a chemostat, up to an equilibrium for different dilution rates, and sampled extracts from the chemostat for each dilution rate. By a series of chemical reactions, we were able to estimate the nitrate concentration in each sample (supplementary data, Supplementary Material online). Plotting the nitrate concentration versus the dilution rate leads in principle to an estimate of the saturation constant. Unfortunately, the concentration was so low ( 2
2 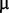 M) that we failed to detect nitrate for all but the highest dilution rates (this value being likely above the wash-out threshold).
M) that we failed to detect nitrate for all but the highest dilution rates (this value being likely above the wash-out threshold).
Thus 1) an increase in nitrate uptake after 7 days could not be measured in our experiment, but 2) we can conclude that bacteria evolved rapidly within 7 days (or were preadapted) to be able to capture  99.8% of the nitrate entering the chemostat, over a wide range of dilution rates. The following additional result is consistent with a good affinity for nitrate: measuring nitrate concentration at different times during growth in batch, we observed that cells grow exponentially until nitrate is no longer detectable, without a smooth transition between exponential and saturation phase (data not shown). In conclusion, if nitrate uptake optimization contributes to fitness, it does so only during the early stages of the experiment.
99.8% of the nitrate entering the chemostat, over a wide range of dilution rates. The following additional result is consistent with a good affinity for nitrate: measuring nitrate concentration at different times during growth in batch, we observed that cells grow exponentially until nitrate is no longer detectable, without a smooth transition between exponential and saturation phase (data not shown). In conclusion, if nitrate uptake optimization contributes to fitness, it does so only during the early stages of the experiment.
Adaptation might also consist of a better management of the low nitrogen level in the chemostat, that is, the evolved cells might be able to grow using lower amounts of nitrogen per cell. For example, an increase in protein turnover has been observed in Pseudomonas putida grown under nitrogen limitation (Hervas et al. 2008). The comparative catabolic activities of the starting strain ABWT and of both endpoint clones AB2800a and AB2800b were examined by the Phenotype MicroArray Services of Biolog, Hayward, CA. The strains were tested on different media (see supplementary data, Supplementary Material online). Some phenotypes were lost when the strains were grown on different Carbon sources, and AB2800a lost more phenotypes than AB2800b. We also compared the catabolic activities of clones AB1100h and AB2800b discussed earlier, but no significant phenotypes were either gained or lost. Overall, the Biolog assay provides evidence of adaptation, but it is difficult to connect these results to a specific parameter that contribute to fitness.
Indirectly, adaptation in the recycling of nitrogen compounds might possibly be revealed by survival in conditions of low nutrients. To test this, we performed starvation experiments with the two clones (AB1100h and AB2800b) discussed earlier. Both clones were grown separately in batch in the selection medium. The end of exponential phase was defined as the beginning of starvation, and samples were then plated to estimate the number of living cells after starvation as a function of time. Starting from approximately 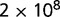 cfu/ml, approximately
cfu/ml, approximately 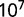 cfu/ml were counted after 100 h of starvation, but no significant differences were observed between the strains. In conclusion, the increase in maximum growth rate
cfu/ml were counted after 100 h of starvation, but no significant differences were observed between the strains. In conclusion, the increase in maximum growth rate 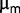 and the appearance of new metabolic phenotypes provide strong evidence of adaptation, but except for
and the appearance of new metabolic phenotypes provide strong evidence of adaptation, but except for 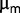 , we failed to identify additional relevant parameters encapsulating fitness.
, we failed to identify additional relevant parameters encapsulating fitness.
Diversity in the Population Appears to Increase Slightly at Later Times
We first focus on the evidence for population diversity during the experiment. After the transposition of the IS that occurs after 1,000 generations, diversity is detected for the whole experiment, because three branches are still present at the end (fig. 2). Each branch evolved with both specific mutations (citA for B1, ACIAD1029 and 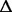 gltI for B2, and ftsH for B3) and mutations that are shared with other branches (glnK and ACIAD2054). The latter likely appeared independently in different branches and are not related to horizontal transfer: In control experiments, the transformation rate in the chemostat was too low to be detected (supplementary data, Supplementary Material online). It is highly probable that diversity took place before 1,000 generations, as observed in short-term evolution experiments with bacteria in minimal medium (Herring et al. 2006; Conrad et al. 2009; Wang et al. 2010; Chou et al. 2011). We lack sequencing data on populations in the early stages of the experiment. However, a clue to diversity is given by the following observation. PCR on clones at the 845th generation were performed to check for the mutation C1903816T. Four other different mutations have been observed in the same locus within a sequence of 80 bp (table 1) in 17 clones. We failed to isolate a clone carrying mutation C1903816T at generation 845, but this allele was later the only one found in the population.
gltI for B2, and ftsH for B3) and mutations that are shared with other branches (glnK and ACIAD2054). The latter likely appeared independently in different branches and are not related to horizontal transfer: In control experiments, the transformation rate in the chemostat was too low to be detected (supplementary data, Supplementary Material online). It is highly probable that diversity took place before 1,000 generations, as observed in short-term evolution experiments with bacteria in minimal medium (Herring et al. 2006; Conrad et al. 2009; Wang et al. 2010; Chou et al. 2011). We lack sequencing data on populations in the early stages of the experiment. However, a clue to diversity is given by the following observation. PCR on clones at the 845th generation were performed to check for the mutation C1903816T. Four other different mutations have been observed in the same locus within a sequence of 80 bp (table 1) in 17 clones. We failed to isolate a clone carrying mutation C1903816T at generation 845, but this allele was later the only one found in the population.
Is diversity constant? The diversity can be roughly estimated by the total number of mutations detected in the population at a given time. It is expected to increase as a function of the time if the number of different lineages increases during the evolution. The number of detected mutations is 15, 16, and 18, respectively, at generations 1,900, 2,300, and 2,800 and thus appears to rise slightly during 1,000 generations. We can estimate the diversity more quantitatively by calculating the Shannon index 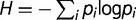 where
where 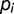 is the frequency of mutation
is the frequency of mutation 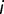 . Note that this is a loose use of this formula, which is a natural definition for frequencies of subpopulations, whereas here we can only access the frequencies of mutations. The indices calculated for generations 1,900, 2,300, and 2,800 are, respectively, 0.88, 1.04, and 1.34, suggesting a slight increase in diversity during the last 1,000 generations.
. Note that this is a loose use of this formula, which is a natural definition for frequencies of subpopulations, whereas here we can only access the frequencies of mutations. The indices calculated for generations 1,900, 2,300, and 2,800 are, respectively, 0.88, 1.04, and 1.34, suggesting a slight increase in diversity during the last 1,000 generations.
It is difficult to judge whether this diversity is large or small. Assuming constant additive 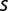 , standard population genetics theories (Desai et al. 2007, Brunet et al. 2008) would predict for our conditions the coexistence on the order of 10 subpopulation fitness classes. However, detailed information on linkage and precise measurements of fitness distributions would be necessary to connect fitness classes and mutation classes. Thus, given this large uncertainty, the observed diversity might well be in the predicted range.
, standard population genetics theories (Desai et al. 2007, Brunet et al. 2008) would predict for our conditions the coexistence on the order of 10 subpopulation fitness classes. However, detailed information on linkage and precise measurements of fitness distributions would be necessary to connect fitness classes and mutation classes. Thus, given this large uncertainty, the observed diversity might well be in the predicted range.
Conclusion
To summarize, we performed a laboratory evolution experiment with bacteria growing in a minimal medium, which focused on its later stages. Sequencing of populations at different evolutionary times shows that the number and type of mutations are compatible with other laboratory evolution experiments, which are characterized by competition between variant clones (Barrick et al. 2009; Bachmann et al. 2012). Interestingly, we found many fixed large rearrangements compared with point mutations.
Also, we found significant and persistent diversity in a population with presumably the same ecotype (Maharjan et al. 2006; Woods et al. 2011). We thus report for the first time that diversity can be established and maintained in a very simple medium for long times, when the proxies for fitness appear to saturate. In this regime, where “adaptation” appears to have slowed down, we observe diversity that is stable or even increasing.
This observed long-term diversity can be due to different mechanisms, which were considered carefully in (Maharjan et al. 2012). Although it is beyond our scope to identify which of these mechanisms were active during our experiment, we can nevertheless formulate some qualitative and hypothetical considerations on the basis of our observations. Metabolic trade-offs (Beardmore et al. 2011) might be witnessed by the results of the biolog assays, where we observed changes in metabolic phenotypes that appeared a priori very distant from the expected target. An example of evolutionary convergence (Maharjan et al. 2012; Tenaillon et al. 2012) possibly can be pointed out by the insertion of an IS upstream or within a putative transcriptional regulator (ACIAD1230), which gave birth to four subpopulations (branches B1, B2, B3, and B5). At the same time, a fifth subpopulation (B4 branch) arose with the insertion of an IS between putative transcriptional regulators, in a totally different region (ACIAD1864–1865). The observation of a significant number of mutations in regulatory regions might also point toward a role played by convergence.
Maharjan et al. (2007) identified an evolved subpopulation of E. coli specialized in the metabolization of by-products of other cells. In our case, reduced forms of nitrogen or amino acids could be cross-fed, so that a role for cross-feeding cannot be excluded. More generally, frequency-dependent selection could be active. Its effect has been observed at low dilution rates and could be ascribed to numerous ecological phenomena, including cross-feeding (Maharjan et al. 2012). In our experiment, the preservation of the B2 branch at low frequency during more than 400 generations could result from a frequency-dependent selection. This needs to be verified in future investigations.
Finally, in terms of basic parameters such as selection coefficient and population size, the results should be compatible with the regime where clonal interference can be observed (Park and Krug 2007; Hughes et al. 2012). However, clonal interference necessarily has to coexist with convergence (availability of alternative evolutionary pathways), and with the observed “diminishing returns” epistasis mechanism (Chou et al. 2011; Khan et al. 2011), according to which the advantage of beneficial mutations depends on their order of appearance.
Overall, we speculate that a possible way for the subpopulations to interact could be indirect through the effect of the limiting amounts of nitrogen in the medium. We explicitly tested for contributions to fitness that, if acquired by one subpopulation would affect the others, such as uptake and resistance to starvation. However, we were not able to identify such an effect. On the other hand, our results suggest that the maximum growth rate 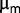 measured in batch might be a sufficiently good proxy for fitness per se, when compared with the selection coefficients estimated from resequencing data. This indicates that the observed subpopulation structure appears consistent with a regime in which competition between beneficial mutations could be relevant. Although both measurements are rough, we can surmise that if there are selected traits that are not recapitulated by
measured in batch might be a sufficiently good proxy for fitness per se, when compared with the selection coefficients estimated from resequencing data. This indicates that the observed subpopulation structure appears consistent with a regime in which competition between beneficial mutations could be relevant. Although both measurements are rough, we can surmise that if there are selected traits that are not recapitulated by 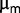 , their associated advantage cannot be very large.
, their associated advantage cannot be very large.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
The authors thank Dominique Schneider, Gilles Fischer, Bianca Sclavi, and John Herrick for critical reading of this manuscript and revision of the text. This work was supported in part by a “Convergences” grant from Université Pierre et Marie Curie, Paris, and a “CNRS-risque” grant.
Literature Cited
- Barbe V, et al. Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res. 2004;32:5766–5779. doi: 10.1093/nar/gkh910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann H, Starrenburg MJ, Molenaar D, Kleerebezem M, van Hylckama Vlieg JE. Microbial domestication signatures of Lactococcus lactis can be reproduced by experimental evolution. Genome Res. 2012;22:115–124. doi: 10.1101/gr.121285.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Lenski RE. Genome-wide mutational diversity in an evolving population of Escherichia coli. Cold Spring Harb Symp Quant Biol. 2009;74:119–129. doi: 10.1101/sqb.2009.74.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature. 2009;461:1243–1247. doi: 10.1038/nature08480. [DOI] [PubMed] [Google Scholar]
- Beardmore RE, Gudelj I, Lipson DA, Hurst LD. Metabolic trade-offs and the maintenance of the fittest and the flattest. Nature. 2011;472:342–346. doi: 10.1038/nature09905. [DOI] [PubMed] [Google Scholar]
- Brunet E, Rouzine IM, Wilke CO. The stochastic edge in adaptive evolution. Genetics. 2008;179:603–620. doi: 10.1534/genetics.107.079319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckling A, Maclean RC, Brockhurst MA, Colegrave N. The Beagle in a bottle. Nature. 2009;457:824–829. doi: 10.1038/nature07892. [DOI] [PubMed] [Google Scholar]
- Chou HH, Chiu HC, Delaney NF, Segra D, Marx CJ. Diminishing returns epistasis among beneficial mutations decelerates adaptation. Science. 2011;332:1190–1192. doi: 10.1126/science.1203799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad TM, Lewis NE, Palsson BO. Microbial laboratory evolution in the era of genome-scale science. Mol Syst Biol. 2011;7:509. doi: 10.1038/msb.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad TM, et al. Whole-genome resequencing of Escherichia coli K-12 MG1655 undergoing short-term laboratory evolution in lactate minimal media reveals flexible selection of adaptive mutations. Genome Biol. 2009;10:R118. doi: 10.1186/gb-2009-10-10-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Crécy-Lagard VA, Bellalou J, Mutzel R, Marlière P. Long term adaptation of a microbial population to a permanent metabolic constraint: overcoming thymineless death by experimental evolution of Escherichia coli. BMC Biotechnol. 2001;1:10–16. doi: 10.1186/1472-6750-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MM, Fisher DS. Beneficial mutation-selection balance and the effect of linkage on positive selection. Genetics. 2007;176:1759–1798. doi: 10.1534/genetics.106.067678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MM, Fisher DS, Murray AW. The speed of evolution and maintenance of variation in asexual populations. Curr Biol. 2007;17:385–394. doi: 10.1016/j.cub.2007.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebeli M, Ispolatov I. Complexity and diversity. Science. 2010;328:494–497. doi: 10.1126/science.1187468. [DOI] [PubMed] [Google Scholar]
- Friesen ML, Saxer G, Travisano M, Doebeli M. Experimental evidence for sympatric ecological diversification due to frequency-dependent competition in Escherichia coli. Evolution. 2004;58:245–260. [PubMed] [Google Scholar]
- Gause GF. The struggle for existence. Baltimore (MD): Williams and Wilkins; 1934. [Google Scholar]
- Gerrish PJ, Lenski RE. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;102(103):127–144. [PubMed] [Google Scholar]
- Good BH, Rouzine IM, Balick DJ, Hallatschek O, Desai MM. Distribution of fixed beneficial mutations and the rate of adaptation in asexual populations. Proc Natl Acad Sci U S A. 2012;109:4950–4955. doi: 10.1073/pnas.1119910109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D, et al. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008;4:e1000303. doi: 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin G. The competitive exclusion principle. Science. 1960;131:1292–1297. doi: 10.1126/science.131.3409.1292. [DOI] [PubMed] [Google Scholar]
- Herring CD, et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet. 2006;38:1406–1412. doi: 10.1038/ng1906. [DOI] [PubMed] [Google Scholar]
- Hervas AB, Canosa I, Santero E. Transcriptome analysis of Pseudomonas putida in response to nitrogen availability. J Bacteriol. 2008;190:416–420. doi: 10.1128/JB.01230-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindré T, Knibbe C, Beslon G, Schneider D. New insights into bacterial adaptation through in vivo and in silico experimental evolution. Nat Rev Microbiol. 2012;10:352–365. doi: 10.1038/nrmicro2750. [DOI] [PubMed] [Google Scholar]
- Hughes JM, et al. The role of clonal interference in the evolutionary dynamics of plasmid-host adaptation. MBio. 2012;3(4):e00077–12. doi: 10.1128/mBio.00077-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RW, Johnson LJ, Clarke SR, Arnold DL. Bacterial pathogen evolution; breaking news. Trends Genet. 2011;27:32–40. doi: 10.1016/j.tig.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Khan AI, Dinh DM, Schneider D, Lenski RE, Cooper TF. Negative epistasis between beneficial mutations in an evolving bacterial population. Science. 2011;332:1193–1196. doi: 10.1126/science.1203801. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BR. Frequency-dependent selection in bacterial populations (and discussion) Philos Trans R Soc Lond B Biol Sci. 1988;319:459–472. doi: 10.1098/rstb.1988.0059. [DOI] [PubMed] [Google Scholar]
- Li H, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan R, Seeto S, Notley-McRobb L, Ferenci T. Clonal adaptive radiation in a constant environment. Science. 2006;313:514–517. doi: 10.1126/science.1129865. [DOI] [PubMed] [Google Scholar]
- Maharjan RP, Seeto S, Ferenci T. Divergence and redundancy of transport and metabolic rate-yield strategies in a single Escherichia coli population. J Bacteriol. 2007;189:2350–2358. doi: 10.1128/JB.01414-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan RP, et al. The multiplicity of divergence mechanisms in a single evolving population. Genome Biol. 2012;13:R41. doi: 10.1186/gb-2012-13-6-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, et al. Tablet—next generation sequence assembly visualization. Bioinformatics. 2010;26:401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SC, Krug J. Clonal interference in large populations. Proc Natl Acad Sci U S A. 2007;104:18135–18140. doi: 10.1073/pnas.0705778104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig RF, Sharp RR, Treves DS, Adams J. Microbial evolution in a simple unstructured environment: genetic differentiation in Escherichia coli. Genetics. 1994;137:903–917. doi: 10.1093/genetics/137.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, et al. The molecular diversity of adaptive convergence. Science. 2012;335:457–461. doi: 10.1126/science.1212986. [DOI] [PubMed] [Google Scholar]
- Toft C, Andersson SG. Evolutionary microbial genomics; insights into bacterial host adaptation. Nature Rev Genet. 2010;11:465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Divergence involving global regulatory gene mutations in an Escherichia coli population evolving under phosphate limitation. Genome Biol Evol. 2010;2:478–487. doi: 10.1093/gbe/evq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods RJ, et al. Second-order selection for evolvability in a large Escherichia coli population. Science. 2011;331(6023):1433–1436. doi: 10.1126/science.1198914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.