

Abstract
We report a highly diastereo- and enantioselective allylation of azlactones catalyzed by the combination of a metallayclic iridium complex and an optically inactive phosphate anion. The process demonstrates an approach to conduct diastereoselective reactions with prochiral nucleophiles in the presence of metallacyclic allyliridium complexes. The reaction provides access to an array of enantioenriched allylated azlactones containing adjacent tertiary and quaternary carbon centers. Preliminary mechanistic studies suggest that the phosphate and methyl carbonate anions together induce the unusually high diastereoselectivity.
Asymmetric allylic substitution catalyzed by metallacyclic iridium phosphoramidite complexes occurs with high regioselectivity and enantioselectivity with a variety of heteroatom and carbon nucleophiles.1 However, none of these reactions with prochiral nucleophiles occur with high diastereoselectivity.1b, 2 Careful analysis of these reactions revealed that both diastereomers were generally formed with high enantiopurity.3 Thus, the low diastereoselectivity originated from the lack of control of the configuration at the prochiral nucleophiles. Therefore, a novel strategy is needed to control the configuration at the nucleophilic carbon atom for Ir-catalyzed allylic substitutions to be diastereoselective.
We considered that the configuration at the nucleophilic carbon atom might be controlled by the counterion.4 Mechanistic studies of Ir-catalyzed asymmetric allylation reactions have shown that the counterion promoted nucleophilic attack by deprotonation of an acidic pronucleophile5 or desilylation of a silyl enolate.6 However, a counterion effect on the diastereoselectivity of Ir-catalyzed asymmetric allylation reactions has not been reported, presumably due to the lack of appropriate methods to introduce different counterions.7
Here, we report a highly diastereo- and enantioselective allylation of azlactones catalyzed by the combination of a metallacyclic iridium complex and optically inactive phosphate cocatalysts. These reactions reveal a new approach to control diastereoselectivity with iridium catalysts for allylic substitutions.
Because silver salts, such as AgBF4, have been used in the synthesis of metallacyclic allyliridium complexes,8 we envisioned that counterions for control of diastereoselectivity could be introduced into the catalytic system by generating the metallacyclic iridium catalysts in situ from various silver salts (AgX). In this case, the counterion X− would deprotonate the pronucleophile and modulate the degree of stereocontrol at the new chiral center of prochiral nucleophiles (Scheme 1). Because azlactones are valuable pronucleophiles to access biologically important quaternary amino acids9 and have been used previously for allylic substitution,10 we chose to explore the counterion effect on diastereoselectivity in Ir-catalyzed allylic substitutions with this class of reagents.
Scheme 1.
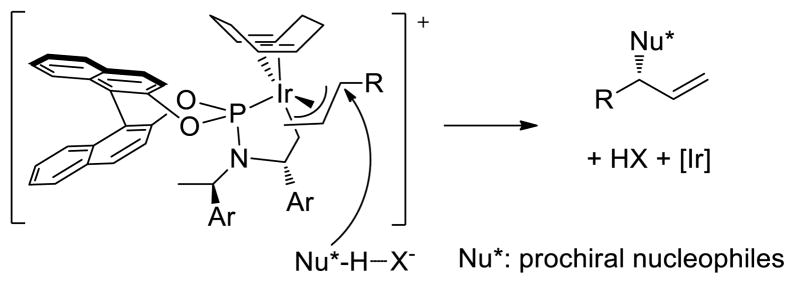
Envisioned Ir-Catalyzed Allylation Assisted by the Counterion X−
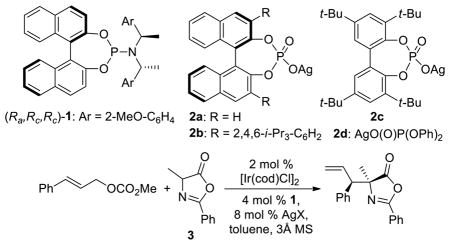
The reaction of cinnamyl methyl carbonate with azlactone 3 derived from alanine was first investigated to reveal potential effects of the ligand and counterion on the Ir-catalyzed allylation reaction (Table 1). Reactions catalyzed by the combination of [Ir(COD)Cl]2, phosphoramidite 1 and AgBF4 formed the branched allylation product in 31% yield with 7:1 dr and 98% ee (entry 1).11 When AgBF4 was replaced with the silver phosphate 2a prepared from (R)-1,1′-bi-2-naphthol (BINOL), the yield of the allylation reaction increased dramatically (83% yield, dr 8:1, 98% ee, entry 2). When 2b, a substituted analog of 2a, was used as the silver salt, the reaction furnished the product with high yield, enantioselectivity and diastereoselectivity (87% yield, dr >20:1, 98% ee, entry 3). Variations of the conditions that gave these high yields and selectivities showed that 8% silver phosphate and 3 Å molecular sieves were integral to obtaining complete conversion of the carbonate (entries 4 and 5). An assessment of the effect of the configuration of the ligand and counterion on the product stereoisomer was conducted. In the presence of ent-1 and the same phosphate 2b, the reaction yielded the enantiomer of the allylation product just described. Moreover, the reaction with ent-1 occurred with only slightly lower diastereoselectivity (88% yield, dr 20:1, −98% ee, entry 6). Therefore, the configuration of the ligand dictated the absolute configuration of the product.
Table 1.
Evaluation of the Effects of Ligand and Counterion on the Ir-Catalyzed Allylation of azlactone 3a
| |||||
|---|---|---|---|---|---|
| Entry | ligand | AgX | Yield(%)b | drc | ee(%)d |
| 1e | 1 | AgBF4 | 31 | 7:1 | 98 |
| 2 | 1 | 2a | 83 | 8:1 | 98 |
| 3 | 1 | 2b | 87 | >20:1 | 98 |
| 4f | 1 | 2b | 57 | >20:1 | 98 |
| 5g | 1 | 2b | 70 | >20:1 | 98 |
| 6 | ent-1 | 2b | 88 | 20:1 | −98 |
| 7 | 1 | 2c | 89(87) | >20:1 | 98 |
| 8 | 1 | 2d | 81 | 18:1 | 98 |
1.00 equiv of cinnamyl carbonate, 2.20 equiv of 3. See the Supporting Information (SI) for experimental details. Absolute configuration of the allylation product was determined by chemical correlation.
Determined by 1H NMR analysis with mesitylene as the internal standard. Numbers in parentheses correspond to isolated yield.
Determined by 1H NMR analysis of crude reaction mixtures.
Determined by chiral HPLC analysis of the major diastereomer.
90% conversion of cinnamyl carbonate.
4 mol % 2b was used. 64% conversion of cinnamyl carbonate.
Without molecular sieves. 80% conversion of cinnamyl carbonate.
Because the configuration of the phosphate in 2b had a negligible effect on the diastereoselectivity of the reaction, an easily accessible silver phosphate 2c derived from an optically inactive biphenol was examined as the source of the counterion.12 The reaction provided the product with yield, diastereoselectivity and enantioselectivity (89% yield, >20:1 dr, 98% ee, entry 7) that were comparable to those of the reactions with the optically active phosphate anions. Furthermore, the reactions conducted with the phosphate containing two phenols in place of the biphenol, AgO(O)P(OPh)2 (2d), furnished the substitution product with only slightly lower yield and diastereoselectivity (entry 8). Because of the easy access to the biphenol12a in the silver phosphate 2c and the high solubility of the silver phosphate 2c in various organic solvents, we conducted our further studies with this silver salt.
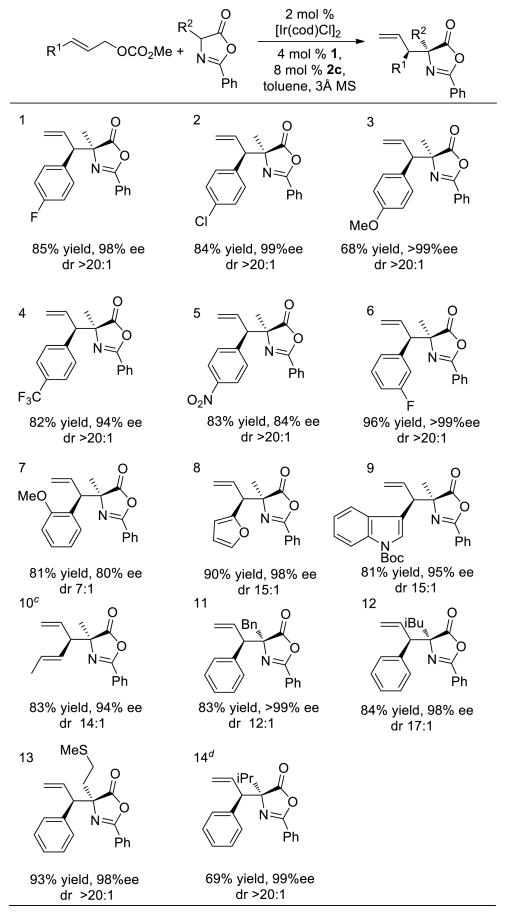
The scope of the reaction under the conditions just described is shown in Table 2. A broad range of para-substituted cinnamyl carbonates possessing diverse electronic properties were examined. The reactions with cinnamyl carbonates containing a halogen in the para position furnished the products in high yields with excellent diastereo- and enantioselectivities (entries 1 and 2). The substrate containing a MeO group in the para position afforded the product in 68% yield with >20:1 dr and 98% ee (entry 3). Although the substrates possessing strong electron-withdrawing groups were less reactive, the reaction still yielded the product with good enantioselectivity (82–83% yield, >20:1 dr, 84–94% ee, entries 4 and 5). The substrate containing a meta-fluoro substituent reacted in high yield and selectivity (96% yield, dr >20:1, >99% ee, entry 6). The lowest selectivities were observed with the ortho-MeO substituted cinnamyl carbonate (81% yield, 7:1 dr and 80% ee; entry 7). Ortho-substituted cinnamyl carbonates typically undergo Ir-catalyzed allylic substitution reactions with lower enantioselectivity than do the meta- and para-substituted isomers.13
Table 2.
|
See the SI for experimental details.
Absolute configurations were assigned by analogy. The diastereomeric ratios were determined by 1H NMR analysis of the crude reaction mixtures. The ee’s were determined by chiral HPLC analysis.
8% linear product was identified.
[Ir(dbcot)Cl]2 was used.
The reactions of electrophiles containing heteroaryl, alkenyl and alkyl substituents were also examined. Carbonates containing furyl and indoly groups reacted like the cinnamyl carbonates (81–90% yield, 15:1 dr, 95–98% ee, entries 8 and 9). Dienyl carbonates also furnished the branched substitution product with high yield (83%) and stereoselectivity (14:1 dr and 94% ee; entry 10). Aliphatic allylic carbonates reacted with low diastereoselectivity.
The reactions of various azlactones derived from natural amino acids were also examined. Azlactones containing benzyl, iso-butyl and thioether groups underwent the reaction smoothly and with high stereoselectivity (entries 11–13). Even the reaction of the azlactone containing a bulky iso-propyl group at the prochiral center proceeded in acceptable yield and high stereoselectivity when [Ir(dbcot)Cl]214 was used as the iridium source (69% yield, >20:1 dr, 99% ee, entry 14). In contrast to the prior allylations of azlactones to give branched products,10b, 10d the current process occurs with allylic carbonates containing alkenyl groups and electron-deficient aryl groups in yields that are comparable to those of the reactions of allylic carbonates containing electron-rich aryl groups.
To gain insight into the mechanism of this reaction, we first conducted experiments to test if the metallacyclic structure present in prior systems is the active form of the catalyst generated with silver phosphates. Reactions conducted with catalytic amounts of the preformed metallacyclic iridium phosphoramidite complex were conducted (Scheme 2). In the absence of phosphoric acid 2e, the product was obtained in 92% yield with 3:1 dr and 98% ee. However, in the presence of 4 mol% of phosphoric acid 2e the product was obtained with a high >20:1 dr and 98% ee. These results imply that: (1) the reactions are catalyzed by a metallacyclic iridium complex and (2) the phosphate anion, not the silver cation is responsible for the high diastereoselectivity.
Scheme 2.

Allylation of 3 Catalyzed by the Preformed Metallacyclic Iridium Catalyst with and without the Phosphoric Acid 2e
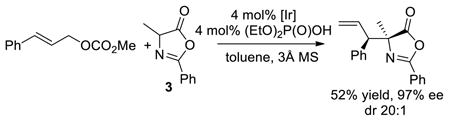
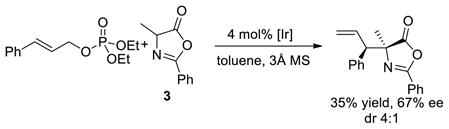
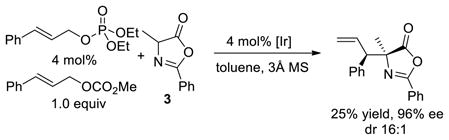
Several experiments were conducted to reveal the origin of the effect of the phosphate anion. First, the reaction of cinnamyl carbonate with azlactone 3 in the presence of 4 mol% diethyl phosphoric acid, along with the analogous reaction of the corresponding allylic phosphate, were conducted. The reaction with added phosphate formed the substitution product in 52% yield with 20:1 dr and 97% ee (eq 1). However, the reaction of cinnamyl phosphate formed the substitution product in only 35% yield, with just 4:1 dr, and 67% ee (eq 2). In contrast, the same reaction of methyl cinnamyl carbonate in the presence of 4 mol% added cinnamyl phosphate gave the product with 16:1 dr and 96% ee (eq 3), albeit in only 25% yield. The difference in diastereomeric ratio between the reaction of the cinnamyl phosphate alone and the reaction with the two cinnamyl electrophiles together suggests that the diastereoselectivity results from the presence of the carbonate and phosphate anions together. This conclusion is consistent with a dependence of the diastereoselectivity of the reaction of cinnamyl electrophiles with azlactone 3 on the identity of the carbonate leaving group (see Table 1 of the SI).
The iridium complexes present in the catalytic system were revealed by NMR spectroscopy. The 31P NMR spectrum of the mixture of [Ir(cod)Cl]2, ligand 1, silver phosphate 2c and cinnamyl carbonate consisted of a major peak at 122.5 ppm. This spectral feature was consistent with the 31P NMR chemical shift of allyliridium complexes containing cyclometallated phosphoramidites characterized previously.8a, b A broad peak between −2.0 and 2.0 ppm in the 31P NMR spectrum was attributed to the phosphate. Therefore, we suggest that the reaction proceeded through a metallacyclic allyliridium complex generated in situ and nucleophilic attack by the azlactones assisted by the phosphate and carbonate.15 The absolute configuration of the allylation product was consistent with nucleophilic attack from the face opposite to iridium moiety.16 Further studies on the details of the mechanism are underway in this laboratory.
 |
(1) |
 |
(2) |
 |
(3) |
To demonstrate the utility of this allylation chemistry, we applied the substitution process to prepare an enantioenriched pyrolidine, which is a member of a family of compounds possessing monoamine reuptake-blocking activity (Scheme 3).17 The reaction of carbonate 6a with azlactone 3 provided the substitution product 6b in 93% yield with 15:1 dr under the standard conditions. The enantiomeric excess of this product was determined to be 94% after 6b was converted to the corresponding methyl ester 6c. Hydroboration and oxidation yielded the terminal alcohol, which was converted to the corresponding tosylate 6d in 57% yield over three steps. Cyclization in the presence of NaH delivered 6e in 95% yield.
Scheme 3.

Synthesis of Pyrrolidine 6ea, b
a(a) 2 mol % [Ir(cod)Cl]2, 4 mol % 1, 8 mol % 2c, toluene, 3Å MS; (b) MeOH, K2CO3; (c) 9-BBN, THF; NaBO3·4H2O; (d) TsCl, triethylamine, DCM; (e) NaH, DMF. bAbsolute configurations were assigned by analogy.
In summary, we have revealed a strategy to achieve high diastereoselectivity for allylic substitution reactions of a prochiral nucleophile catalyzed by the phosphoramidite-derived metallacyclic iridium complex, which had catalyzed enantioselective, but not diastereoselective, reactions previously. This stereoselectivity is achieved by the addition of an optically inactive phosphate anion. Preliminary mechanistic data suggest that the reaction proceeds by the generation of a metallacyclic iridium allylcomplex, and both the carbonate and phosphate contribute to the high diastereoselectivity observed. Considering the multitude of carbon nucleophiles that react with allyliridium complexes and the availability of structurally diverse phosphates, this approach should be widely applicable to the control of diastereoselectivity with iridium catalysts. Studies to expand the scope of the prochiral nucleophiles that undergo similar diastereo- and enantioselective Ir-catalyzed allylation reactions are ongoing.
Supplementary Material
Acknowledgments
We thank the NIH (GM-58108) for support of this work, Johnson-Matthey for gifts of [Ir(cod)Cl]2, and Dr. Klaus Ditrich and BASF for gifts of chiral amines. W. C. thanks Dr. Ming Chen for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.For reviews on Ir-catalyzed asymmetric allylation, see: Hartwig JF, Stanley LM. Acc Chem Res. 2010;43:1461. doi: 10.1021/ar100047x.Tosatti P, Nelson A, Marsden SP. Org Biomol Chem. 2012;10:3147. doi: 10.1039/c2ob07086c.Liu WB, Xia JB, You SL. Top Organomet Chem. 2012;38:155.Hartwig JF, Pouy MJ. Top Organomet Chem. 2011;34:169.Helmchen G. In: Iridium Complexes in Organic Synthesis. Oro LA, Claver C, editors. Wiley-VCH; Weinheim, Germany: 2009. p. 211.For a review on phosphoramidite ligand in asymmetric catalysis, see: Teichert JF, Feringa BL. Angew Chem, Int Ed. 2010;49:2486. doi: 10.1002/anie.200904948.
- 2.For moderately diastereoselective allylations catalyzed by an iridium phosphite complex, see: Kanayama T, Yoshida K, Miyabe H, Takemoto Y. Angew Chem, Int Ed. 2003;42:2054. doi: 10.1002/anie.200250654.Kanayama T, Yoshida K, Miyabe H, Kimachi T, Takemoto Y. J Org Chem. 2003;68:6197. doi: 10.1021/jo034638f.For an intramolecular example with good diastereoselectivity, see: Wu QF, Liu WB, Zhuo CX, Rong ZQ, Ye KY, You SL. Angew Chem, Int Ed. 2011;50:4455. doi: 10.1002/anie.201100206.
- 3.Unpublished results. For two reported examples, see: Dahnz A, Helmchen G. Synlett. 2006:697.Polet D, Alexakis A, Tissot-Croset K, Corminboeuf C, Ditrich K. Chem-Eur J. 2006;12:3596. doi: 10.1002/chem.200501180.
- 4.For a review on chiral counterion strategy, see: Phipps RJ, Hamilton GL, Toste FD. Nature Chem. 2012;4:603. doi: 10.1038/nchem.1405.
- 5.Gnamm C, Förster S, Miller N, Brödner K, Helmchen G. Synlett. 2007:790. [Google Scholar]
- 6.Chen W, Hartwig JF. J Am Chem Soc. 2012;134:15249. doi: 10.1021/ja306850b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.In transition metal catalyzed allylation reactions, counterions were generally introduced by employing different leaving groups, which restricted the scope of counterions.
- 8.For stoichiometric study on silver salt promoted formation of metallacyclic iridium allylcomplex, see: Madrahimov ST, Markovic D, Hartwig JF. J Am Chem Soc. 2009;131:7228. doi: 10.1021/ja902609g.Raskatov JA, Spiess S, Gnamm C, Broedner K, Rominger F, Helmchen G. Chem--Eur J. 2010;16:6601. doi: 10.1002/chem.200903465.For a cycloisomerisation catalyzed by an iridium chiral phosphate complex, see: Barbazanges M, Augé M, Moussa J, Amouri H, Aubert C, Desmarets C, Fensterbank L, Gandon V, Malacria M, Ollivier C. Chem-Eur J. 2011;17:13789. doi: 10.1002/chem.201102723.
- 9.Tanaka M. Chem Pharm Bull. 2007;55:349. doi: 10.1248/cpb.55.349. [DOI] [PubMed] [Google Scholar]
- 10.For a review on azlactones in enantioselective reactions, see: Alba ANR, Rios R. Chem -Asian J. 2011;6:720. doi: 10.1002/asia.201000636.For a Mo-catalyzed asymmetric allylation of azlactones, see: Trost BM, Dogra K. J Am Chem Soc. 2002;124:7256. doi: 10.1021/ja020290e.For a Pd-catalyzed asymmetric allylation of azlactones to form linear products, see: Trost BM, Ariza X. J Am Chem Soc. 1999;121:10727.For an [Ir(cod)Cl]2 catalyzed allylation/aza-Cope sequence with azlactones, see: Kawatsura M, Tsuji H, Uchida K, Itoh T. Tetrahedron. 2011;67:7686.
- 11.Unless otherwise noted, 2.20 equiv 3 was used because methanol formed in the reaction reacted with 3 to form the methyl ester.
- 12.See the SI for the synthesis of 2c. For the preparation of the biphenol, see: Wuennemann S, Froehlich R, Hoppe D. Eur J Org Chem. 2008:684.For an example of the phosphoric acid catalyzed reaction, see: Moreau J, Hubert C, Batany J, Toupet L, Roisnel T, Hurvois JP, Renaud JL. J Org Chem. 2009;74:8963. doi: 10.1021/jo901238y.
- 13.Liu WB, Zheng C, Zhuo CX, Dai LX, You SL. J Am Chem Soc. 2012;134:4812. doi: 10.1021/ja210923k. [DOI] [PubMed] [Google Scholar]
- 14.See the SI for a new route to DBCOT.
- 15.The anion contains both components with an IR band at 3372 cm−1 that is consistent with the stretch of a hydrogen bonded OH group.
- 16.The absolute configuration of the allylation product was determined by comparison of the optical rotation and retention time to a literature value in reference 10d.
- 17.Yoshikawa M, Kamei T. WO2010123006A1. Preparation of Pyrrolidine Compounds as Monoamine Reuptake Inhibitors. 2010
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.