

Abstract
Rationale
Short-term β-adrenergic stimulation promotes contractility in response to stress but is ultimately detrimental in the failing heart because of accrual of cardiomyocyte death. Endogenous cardiac progenitor cell (CPC) activation may partially offset cardiomyocyte losses, but consequences of long-term β-adrenergic drive on CPC survival and proliferation are unknown.
Objective
We sought to determine the relationship between β-adrenergic activity and regulation of CPC function.
Methods and Results
Mouse and human CPCs express only β2 adrenergic receptor (β2-AR) in conjunction with stem cell marker c-kit. Activation of β2-AR signaling promotes proliferation associated with increased AKT, extracellular signal-regulated kinase 1/2, and endothelial NO synthase phosphorylation, upregulation of cyclin D1, and decreased levels of G protein–coupled receptor kinase 2. Conversely, silencing of β2-AR expression or treatment with β2-antagonist ICI 118, 551 impairs CPC proliferation and survival. β1-AR expression in CPC is induced by differentiation stimuli, sensitizing CPC to isoproterenol-induced cell death that is abrogated by metoprolol. Efficacy of β1-AR blockade by metoprolol to increase CPC survival and proliferation was confirmed in vivo by adoptive transfer of CPC into failing mouse myocardium.
Conclusions
β-adrenergic stimulation promotes expansion and survival of CPCs through β2-AR, but acquisition of β1-AR on commitment to the myocyte lineage results in loss of CPCs and early myocyte precursors.
Keywords: adrenergic receptors, adrenergic regulation, adult stem cells, cardiac progenitor cells, heart failure
β-adrenergic signaling regulates cardiac inotropy, lusitropy, and chronotropy,1,2 mediated primarily by β1 and β2 adrenergic receptor (AR) subtypes present on adult cardiomyocytes. β1-ARs regulate cardiac contractility by signaling through Gαs and adenylyl cyclase,3 but chronic stimulation increases myocyte apoptosis and necrosis.4,5 In contrast, β2-AR signaling antagonizes cardiomyocyte death6,7 and also exerts widespread influence over multiple cell types, including tissue progenitors and precursor cells, by regulating mobilization, differentiation, and proliferation.8–12 Stimulation of β2-AR also promotes DNA synthesis via activation of extracellular signal-regulated kinase and phosphoinositide 3 kinase pathways in cardiac fibroblasts,13,14 but the influence of β-adrenergic signaling on the cardiac progenitor cell (CPC) population remains unknown.
The postnatal heart undergoes substantial proliferation in the face of adrenergic drive after birth15 during which neonatal cardiomyocytes predominantly express β2-AR. However, cardiomyocyte maturation leads to dominant expression of β1-AR concomitant with recession of postnatal adrenergic stimulation in the adult myocardium.16,17 Cardiomyopathic injury reactivates the adrenergic system that, although initially adaptive, eventually becomes cardiotoxic in the chronic setting.18 The seemingly counterintuitive administration of pharmacological β-blockade, a negative inotrope, in patients with heart failure as the current standard of care improves cardiac performance and resensitizes the adrenergic system.19,20 Cardiac homeostasis is intimately tied to adrenergic signaling throughout our lifetime, but the impact of adrenergic stimulation on CPC survival and proliferation has not been assessed. Because CPCs contribute to ongoing myocyte replacement,21 consequences of β-adrenergic drive on CPC biology have important implications for the reparative and regenerative potential of the pathologically injured myocardium.
Potentiation of myocardial function in the damaged heart depends on a delicate balance between promoting contractility without impairing cardiomyocyte survival and regeneration. Deciphering molecular signals that impair cellular replacement are an integral part of developing novel interventional therapeutic strategies to enhance myocardial healing. Recent studies assert that reactivation and involvement of endogenous repair processes are a critical part of stem cell–mediated therapy,22,23 and the deleterious consequences of long-term β-adrenergic hyperactivity are indisputable.1,18 In the present study, we reveal a direct relationship between the β-AR system and CPC biology. Results demonstrate that adrenergic drive is a double-edged sword that promotes CPC survival and proliferation via β2-AR before lineage commitment but transitions into a deleterious stimulus on acquisition of β1-AR upon maturation and sensitization to catecholamine stress. Therefore, the β-AR system is an important factor in CPC biology, which may account, in part, for the beneficial effects of long-term β-adrenergic blockade as a pharmacological intervention to enhance CPC response in the failing heart.
Methods
Cell Culture and Differentiation
CPCs are isolated from syngeneic male FVB mice and cultured for 3 weeks in cardiac stem cell media.24 Differentiation is induced in CPCs by coculture with neonatal cardiomyocytes in a ratio of 1:40.25 Human CPCs from Institutional Review Board–exempt samples of patients receiving left ventricular assist device implantation were isolated and cultured in human CPC media (details provided in the Online Data Supplement).
Lentiviral Transduction
Fluorescent ubiquitination-based cell cycle indicator (FUCCI) CPCs are transduced with Lv-PgK1-Fucci Orange (MKO2) and Lv-PgK1-Fucci Green (Az), whereas CPCs expressing green fluorescent protein (GFP; CPCe) are transduced with Lv-egfp to create stable cell lines (details provided in the Online Data Supplement).
Pharmacological Treatments
CPCs are serum starved overnight and treated with specific β2-agonist fenoterol (FEN; 1 μmol/L; Sigma Aldrich) for 2 hours in serum-free medium before CyQuant labeling, metabolic assays, and protein analysis. Serum-free preconditioning was performed to eliminate the effect of serum on CPC proliferation to rule out any additional factors that may influence CPC proliferation and concentrate only on the effect of the specific β2-agonist (FEN) on CPC proliferation. Optimal FEN concentration with the maximum proliferation was determined by performing a dose–response treatment. Specific β2-antagonist ICI 118, 551 (0.3 μmol/L; TOCRIS biosciences) and β1-antagonist metoprolol (1 μmol/L; Sigma Aldrich) are used for manipulation of adrenergic stimulation. Inhibitors of phosphoinositide 3 kinase LY294002 (20 μmol/L; Sigma Aldrich) and NO synthase L-NG-nitroarginine methyl ester (L-NAME) (100 μmol/L; Sigma Aldrich) are used to assess signal transduction downstream of adrenergic stimulation. FEN-induced proliferation in FUCCI CPCs is determined by serum-starving the cells overnight, followed by FEN treatment for 2 hours the following day in serum-free medium. Cells were allowed to recover for 6 hours in full medium before analysis. Control CPCs were kept in serum-free medium for 2 hours and then for 6 hours in recovery medium. CPCs treated with full medium were used to establish a positive control and were treated for 2 hours in full serum medium, followed by additional 6 hours of full medium treatment (see Online Data Supplement Methods). Isoproterenol (ISO; 1 μmol/L) treatment of CPCs to induce catecholamine stress was performed for 6 hours before flow cytometric analysis.
siRNA Transfection
Small interfering RNA (siRNA; 20 μmol/L) transfection is performed with cells plated in 6-well dishes and transfected with 3 μL of siRNA and 12 μL of Hi Perfect transfection reagent (Qiagen) in 85 μL of DMEM/F12 without serum.26
CyQuant and Metabolic Assay
CyQuant and 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide assay of CPCs are performed by plating cells in quadruplicate (2000 cells/well) in 96-well plates, followed by incubation with CyQuant (Invitrogen) or 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide reagent (Sigma, St Louis, Mo), as previously described.22
Immunoblots
Immunoblotting is performed as described.26 Details regarding sample preparation and antibodies are provided in the Online Data Supplement.
Quantitative Real-Time Quantitative Reverse Transcriptase Polymerase Chain Reaction
Total RNA is isolated from frozen heart or cultured cells using Quick-RNA MiniPrep (Zymo Research) and reverse-transcribed into cDNA using iScript cDNA Synthesis kit (Bio-Rad). Quantitative reverse transcriptase polymerase chain reaction is performed on all samples in triplicate using iQ SYBR Green (Bio-Rad) according to the manufacturer’s instructions. Primer sequences are provided in Online Table I.
Flow Cytometry
Cell death is measured by Annexin V staining (BD Biosciences) according to the manufacturer’s instructions. Cytometry is performed using a BD FACSAria Flow Cytometer (BD Biosciences).
Animal Studies
Institutional Animal Care and Use Committee approval was obtained for all animal studies. Detailed experimental procedures are provided in the Online Data Supplement.
Immunohistochemistry
Immunocytochemistry, terminal deoxynucleotidyl transferase dUTP nick end labeling assays, and immunohistochemistry are performed as previously described26 with additional information given in the Online Data Supplement.
Echo-guided Injection
Echocardiographically guided injections are performed using a Vevo770 imaging system (Visual Sonics, ON, Canada).27
Catecholamine Assay
Plasma epinephrine and norepinephrine levels are determined by ELISA performed on mouse plasma samples using the BI-CAT EIA kit from ALPCO Diagnostics (Windham, NH), with detailed methods provided in the Online Data Supplement.
Statistics
Statistical analysis is performed using Student t test. Comparison of >2 groups is performed by 1-way ANOVA or 2-way ANOVA with Bonferroni post hoc test. P<0.05 is considered statistically significant. Error bars represent SEM. Statistical analysis is performed using Graph Pad prism v 5.0 software.
Results
CPCs Express β2-AR in Conjunction With c-kit
c-kit+ CPCs expressed β2-AR but not β1-AR as evidenced by immunolabeling of adult mouse heart (3-month-old) sections (Figure 1A and 1B), as well as cultured CPCs (Figure 1C and 1D). Expression of β2-AR and without β1-AR in cultured CPCs normalized to expression in heart was confirmed by immunoblot analysis (Figure 1E) and quantitative reverse transcriptase polymerase chain reaction (Figure 1F). Comparable findings are observed with human CPCs isolated from patients receiving left ventricular assist device where β2-AR is observed in the absence of β1-AR expression by immunoblot (Figure 1G). CPCs expressing c-kit+/β2-AR+ were identified in myocardial sections of infarction (Online Figure IA) or hearts of tropomodulin overexpressing transgenic (TOT) mice (Online Figure IB) and sham hearts (Online Figure IC).
Figure 1. Expression of β-adrenergic receptors on mouse cardiac progenitor cells (CPCs).

A and B, Myocardial sections from mouse heart show c-kit+ CPCs (red) are β1-AR (−) and β2-AR (+) (green) and nuclei (white) (n=3). C and D, Cultured CPCs express c-kit (red) and β2-AR (green) but not β1-AR. Nuclei are shown in blue. Scale bar, 40 μm. E, Expression of β-adrenergic receptors by immunoblot (n=4). F, Quantitative reverse transcriptase polymerase chain reaction validation of β-adrenergic receptors on mouse heart and CPCs (n=4) shows expression of β1-AR in the heart but not in CPCs, whereas β2-AR is expressed in the heart and CPCs. G, Immunoblot analysis of β-adrenergic receptors 1 and 2 in human CPC cell lines.
β2-AR Mediates CPC Proliferation
CPCs were modified to express FUCCI reporter genes to monitor cell cycle progression from G1 to S/G2/M by measuring red versus green fluorescence, respectively28 (Online Figure IIA–IIE). FUCCI CPCs show significant (P<0.004) increases in cell cycle entry after 1 μmol/L FEN treatment (11.1%) compared with vehicle-treated controls (4.2%), representing 2.6-fold increase over controls (Figure 2A). DNA synthesis indicative of proliferation is also increased in CPCs after treatment with FEN (1 μmol/L) at day 3 (P<0.001) relative to vehicle-treated CPCs as measured by CyQuant assay (Figure 2B). Conversely, proliferation is significantly reduced at 1 day (P<0.001) or 3 days (P<0.001) if FEN-mediated β2-AR stimulation is blocked by ICI 118, 551 antagonist (Figure 2B). Similarly, metabolic activity is increased in CPCs after FEN treatment at day 3 (P<0.001) and significantly declines with ICI 118, 551 treatment (P<0.001; Figure 2C). Treatment with metoprolol, a specific β1-AR antagonist, did not affect cell proliferation or metabolic activity in the presence or absence of FEN (data not shown). Silencing of β2-AR with siRNA decreases proliferation as measured by CyQuant and 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide assay compared with treatment with scrambled control (Online Figure IIIA–IIIC). Collectively, these results demonstrate an adrenergic-mediated proliferative response in CPCs via β2-AR stimulation.
Figure 2. β2-adrenergic receptor (AR) signaling mediates cardiac progenitor cell (CPC) proliferation.

A, Analysis of cell proliferation in fluorescent ubiquitination-based cell cycle indicator–infected CPCs after treatment with fenoterol (FEN; 1 μmol/L) in serum-free medium for 2 hours followed by 6 hours of recovery in full serum medium. CPCs kept in serum-free medium without FEN (0 μmol/L) for 2 hours and full serum (FM)–treated groups were used as controls (n=5). B, CyQuant assay. CPCs treated with FEN (1 μmol/L) show increased proliferation as measured by CyQuant assay compared with cells treated with β2-AR antagonist ICI 118, 551 (n=3). C, Metabolic activity increased in CPCs stimulated with FEN compared with cells treated with β2-AR antagonist (n=3). D and E, FEN treatment upregulates AKT phosphorylation, ERK phosphorylation, and cyclin D1 and downregulates G protein–coupled receptor kinase 2 (GRK2), whereas treatment with a β2-specific antagonist ICI 118, 551 blocks the effect of FEN stimulation. *P<0.05, **P<0.01, and ***P<0.001 significance for CPCs vs FEN; #P<0.05, ##P<0.01, and ###P<0.001 significance for FEN vs FEN+ICI 118, 551 (n=4). F, Increased endothelial NO synthase (eNOS) phosphorylation in FEN-treated CPCs blocked in the presence of LY294002 and L-NAME (n=3). *P<0.05, **P<0.01, and ***P<0.001 significance for CPCs vs FEN; #P<0.05, ##P<0.01, and ###P<0.001 significance for FEN vs FEN+LY-294002; and ΦP<0.05, ΦΦ P<0.01, and ΦΦΦP<0.001 significance for FEN vs FEN+L-NAME. NS indicates nonsignificant.
The underlying molecular signaling resulting from FEN stimulation of CPCs was assessed in the presence or absence of ICI 118, 551. AKT and extracellular signal-regulated kinase 1/2 phosphorylation increases 3.7-fold and 3.3-fold, respectively, with FEN exposure that was blunted to only 1.4-fold and 1.3-fold increases, respectively, with introduction of ICI 118, 551 (Figure 2D and 2E). Similarly, cyclin D1 levels significantly increase (P<0.01) 2.4-fold with FEN but only 1.4-fold with FEN+ICI 118, 551 compared with vehicle-treated cells. Also, FEN-treated CPCs show a significant decrease (P<0.05) in G protein–coupled receptor kinase 2 (GRK2) levels compared with vehicle treatment (Figure 2D and 2E), corroborating a report of decreased GRK2 levels with cell cycle progression.29 GRK2 levels decrease 49% after FEN but remain elevated if exposed to both FEN and ICI 118, 551. β2-AR signaling in cardiomyocytes promotes protective signaling via an AKT/endothelial NO synthase (eNOS)–dependent pathway30 that can be blocked by pharmacological inhibition of PI3-K (LY-294002) or eNOS (L-NAME). Similar to cardiomyocytes, CPCs show significant increases in eNOS phosphorylation (Figure 2F) after FEN exposure (1.9-fold; P<0.01) that is abrogated by either LY294002 or L-NAME (0.5-fold and 0.7-fold, respectively). Although L-NAME has been shown to affect downstream eNOS signaling, recent published studies support a feedback mechanism of L-NAME,31,32 concurring with our findings of CPCs treated with FEN. Collectively, these results indicate that β2-AR activation promotes cardioprotective signaling in CPCs via the AKT/eNOS pathway in parallel with GRK2 downregulation.
CPCs Acquire β1-AR and Sensitize to Cell Death After Cardiogenic Commitment
Acquisition of a cardiomyocyte phenotype by CPCs inevitably leads to β1-AR expression as maturation progresses toward differentiation, which would correlate with sensitivity to adrenergic-induced cell death. Differentiation stimulus was provided by coculture of neonatal rat cardiomyocytes (NRCM) with CPCs modified to express enhanced GFP (CPCe) to allow fluorescent and immunohistochemical discrimination from NRCM. CPCe exposed to coculture show β1-AR expression concurrent with loss of c-kit as determined by immunocytochemistry (Figure 3A). In addition, CPCe express β2-AR (Figure 3C) after 7 days of coculture and are differentiated from myocytes on the basis of GFP and actinin expression (Figure 3B). β1-AR (Figure 3D) and cardiac troponin T (Figure 3F) mRNAs are detectable, whereas β2-AR mRNA levels decreased in CPCe (Figure 3E) after 7 days of coculture by quantitative reverse transcriptase polymerase chain reaction analysis using validated primers specific to mouse mRNA that do not recognize mRNA of NRCM (Online Figure IIIE–IIIG). Because undifferentiated CPCs do not express cardiac markers and our primers do not bind to rat mRNA, we believe that the cardiac marker expression for β1-AR, β2-AR, and cardiac troponin T may be related to differentiated CPCs. Conversely, selective inhibition of β2-AR by siRNA decreased expression of cardiac troponin T and β1-AR in siβ2-AR–treated CPCs (Online Figure IIID) after coculture with NRCMs, indicating that β2-AR inhibition can reduce CPC commitment to cardiac lineages. The effect of catecholamine stress on CPCe kept in the coculture environment for 1 week with ISO stimulation was assessed by Annexin-V fluorescence-activated cell sorter analysis by colabeling of GFP-expressing CPCs. Acquisition of β1-AR by CPCe after 7 days of coculture (Figure 3A–3F) correlates with significantly increased (17.4%; P<0.001) ISO-induced cell death compared with control vehicle–treated CPCe (1.9%; Figure 3H). Participation of β1-AR is corroborated by a significant decrease in ISO-mediated cell death (8.7%; P<0.001) in the presence of metoprolol, a specific β1-AR antagonist. In the presence of ICI 118, 551, an antagonist of β2-AR, increased cell death (15.1%; Figure 3H) was observed as ISO mediated its effect through enhanced β1-AR stimulation. In comparison, CPCs maintained alone without coculture show no cell death after treatment with ISO (Figure 3G). Furthermore, increased β2-AR mRNA levels were observed in CPCs in concordance with increased survival after commitment in the presence of ISO and metoprolol (Online Figure III I). Similarly, increased β1-AR mRNA level was observed in committed CPCs after ISO and ICI 118, 551 correlating with decreased survival. To further clarify the effect of metoprolol on CPC survival and whether this is because of the inhibition of CPC differentiation, CPCs were cocultured with NRCMs in the presence of metoprolol. No significant change on CPC differentiation was observed in the presence of metoprolol, indicating that metoprolol does not affect CPC differentiation. In parallel, CPCs were adenovirally engineered to express β1-AR (Online Figure IIIH) and subjected to ISO stress to assess β1-AR signaling effect on CPCs without even progressing to a cardiac phenotype. β1-AR–expressing CPCs show a significant increase in cell death (43%; P<0.001) compared with vehicle-treated β1-AR+CPCs (Figure 3I), and this effect is significantly reduced in the presence of metoprolol (22.3%; P<0.001). CPC control infection with adenovirus-GFP shows nonsignificant effects on cell death after ISO treatment concurring with earlier results (Figure 3G). Collectively, these findings indicate that acquisition of β1-AR in the process of lineage commitment correlates with sensitization to adrenergic-induced cell death.
Figure 3. Cardiac progenitor cells (CPCs) acquire β1-adrenergic receptor (AR) after commitment toward cardiac lineages and become sensitized to cell death.

A and B, Immunostaining of green fluorescent protein (GFP)–expressing CPCs in coculture with neonatal rat cardiomyocytes (NRCMs) stained with β1-AR (red), c-kit/sarcomeric actin (blue), and nuclei (white). C, GFP CPCs (green) express β2-AR (red) in coculture with NRCMs (sarcomeric actin; blue) and nuclei (white). Scale bar, 40 μm (n=5). D–F, Real-time polymerase chain reaction analysis for expression of β1-AR, β2-AR, and cardiac troponin T in CPCs after 7 days of coculture. NRCMs were run as control to validate primer specificity to mouse genes (n=4). G, Analysis of cell death in undifferentiated CPCs treated in the presence of isoproterenol (ISO), ICI 118, 551, and metoprolol (n=3). H, Fluorescence-activated cell sorter (FACS) analysis of cell death in differentiated CPCs in a coculture system treated with ISO. *P<0.05, **P<0.01, and ***P<0.001 significance for CPC vs CPC ISO; #P<0.05, ## P<0.01, and ###P<0.001 significance for CPC ISO vs CPC ISO/metoprolol; and ΦP<0.05, ΦΦP<0.01, and ΦΦΦP<0.001 significance for CPC ISO/metoprolol vs CPC ISO/ ICI 118, 551 (n=3). I, CPCs infected with adenovirus (Ad)-β1-AR and treated with ISO show increased cell death as measured by FACS analysis compared with metoprolol-treated CPCs and vehicle-treated controls (n=3). In parallel, AdGFP CPCs showed no effect of ISO-induced stress. *P<0.05, **P<0.01, and ***P<0.001 significance for Ad-β1-AR NT vs Ad-β1-AR CPCs ISO; #P<0.05, ##P<0.01, and ###P<0.001 significance for Ad-β1-AR CPCs ISO vs Ad-β1-AR CPCs ISO/metoprolol.
CPC Survival and Proliferation Are Enhanced by β-Blockade in the Failing Heart
Because adrenergic stimulation of cardiogenic CPCs promotes cell death after β1-AR acquisition (Figure 3), the ability of metoprolol, a selective β1-AR blocker, to protect CPCe was examined in the context of heart failure. The TOT mouse model suffers from catecholamine-induced chronic dilated cardiomyopathy, preventable by administration of nonselective β-blockade.33 Survival of adoptively transferred CPCe in the failing heart was assessed in TOT mice 2 weeks after delivery in the presence or absence of concurrent metoprolol treatment begun 4 weeks before CPCe delivery (Figure 4A). Presence of c-kit+ cells was examined by immunolabeling of myocardial sections to quantify populations of endogenous CPCs and donated CPCe. Metoprolol treatment yields a 3.1-fold increase in CPCe (c-kit+/GFP+) compared with vehicle-treated controls after 2 weeks of transplantation (Figure 4B–4E). In addition, increases in endogenous CPC populations (c-kit+/GFP−) of 3.9-fold were observed in hearts of CPCe/metoprolol-treated hearts, respectively, compared with the CPCe group without metoprolol (Figure 4E). Greater levels of CPCe proliferation are evident in hearts of mice treated with metoprolol confirmed by a 2.7-fold increase in c-kit+/proliferating cell nuclear antigen–positive cells in metoprolol-treated mice compared with vehicle-treated CPCe controls (Online Figure IVA–IVC). Furthermore, CPCs treated with metoprolol for 7 days showed no significant changes in cardiac differentiation markers, ruling out a direct role for metoprolol on CPC differentiation. Collectively, these results support the postulate that selective β1-AR blockade augments CPC survival and proliferation in the failing myocardium.
Figure 4. Blockade of β-adrenergic signaling promotes cardiac progenitor cell (CPC) survival in failing myocardium.

A, Schematic representation of the experimental design. Administration of β1-selective blocker metoprolol was initiated in tropomodulin overexpressing transgenic (TOT) mice except for control group. Mice were transplanted with CPCe 4 weeks after metoprolol administration in control group and metoprolol-treated group. The third group received no CPCe but was given metoprolol till the end of the experiment. Hearts were harvested 2 weeks after cell transplantation. B and C, Increased c-kit+ cells within hearts treated with metoprolol and CPCe compared with CPCe-treated hearts stained with green fluorescent protein (GFP; green), c-kit (red), sarcomeric actin (blue), and nuclei (white). Scale bar, 150 μm (B) and scale bar, 40 μm (C and D). D and E, Quantification of c-kit+/ GFP+ and c-kit+/GFP- cells in TOT mice treated with metoprolol and CPCe. CPCe (n=3), CPCe/ metoprolol (n=4), and metoprolol (n=4). *P<0.05 and **P<0.01 significance for CPCe vs CPCe/ metoprolol. NS indicates nonsignificant.
Increased Survival of β1-Expressing Cells in Failing Hearts With β-Blockade
CPC survival and proliferation are increased in failing hearts with β-blockade (Figure 4), supporting expansion of the resident CPC pool (Figure 4) while increasing the number of committed cells in failing hearts. Acquisition of β1-AR on CPCs is representative of a committed state because c-kit+ CPCs do not express these receptors (Figures 1 and 3). Analysis of CPCe-transplanted TOT hearts with metoprolol treatment revealed significantly more CPCe-expressing β1-AR colocalized with GFP (7.0%) compared with vehicle-treated hearts (1.9%; Figure 5A–5C), indicating increased CPC commitment and survival of myocyte precursors. Similarly, increased β2-AR expression on CPCe was observed in TOT hearts with metoprolol (15.7%) compared with hearts with vehicle-treated controls (3.7%; Figure 5D–5F). In summary, these results indicate that β-blockade leads to enhanced survival of committed CPCs and protection from deleterious effects of β1-AR–mediated signaling, thereby allowing for CPC support of myocardial repair.
Figure 5. Increased survival of cardiac progenitor cells (CPCs) expressing β-adrenergic receptors (AR) in dilated hearts on selective β-blockade.

A–C, Increased survival of β1-AR+ (red) CPCs colocalized with green fluorescent protein (GFP+; green) in hearts with β-blockade compared with vehicle-treated control hearts. An uncommitted GFP+ CPC evidenced by lack of β1-AR expression (inset; yellow arrowhead). D–F, Increased β2-AR+ (red) surviving CPCs colocalized with GFP (green), sarcomeric actin (blue), and nuclei (white). Scale bar, 40 μm. CPCe (n=3), CPCe/metoprolol (n=4), and metoprolol (n=4). *P<0.05 and **P<0.01 significance for CPCe vs CPCe/ metoprolol.
Beneficial Effects of Long-term β1-AR Blockade Include Reduction of Cardiomyocyte Apoptosis and DNA Synthesis in Failing Hearts
Consistent with expected salutary effects of β1-AR blockade, heart: body weight ratio is significantly (P<0.05) decreased by 50% in metoprolol-treated group (Online Figure VA) concurrent with decreased myocyte cross-sectional area compared with vehicle-treated controls (Online Figure VB and VC). Long-term metoprolol treatment of TOT mice for 6 weeks augmented cardiac function measured by echocardiography after 6 weeks, showing 1.7-fold and 1.9-fold increases in ejection fraction and fractional shortening, respectively, compared with vehicle-treated controls (Online Figure VD and VE), whereas no significant change in cardiac function was observed in metoprolol-treated animals (data not shown). Serum levels of epinephrine and norepinephrine were significantly reduced (P<0.001 and P<0.001, respectively) in hearts treated with metoprolol compared with vehicle-treated controls (Online Figure VI). Cardiotoxicity of chronic catecholamine storm in heart failure leads to a vicious cycle of cardiomyocyte death and proliferation34,35 that is interrupted by β1-AR blockade. Metoprolol treatment of TOT mice for 6 weeks leads to significant reduction in cardiomyocyte BrdU+ labeling (39% decrease; P<0.01; Online Figure VIIA–VIIC) and terminal deoxynucleotidyl transferase dUTP nick end labeling+ cells (48% decrease; P<0.05) relative to vehicle-treated controls (Online Figure VIID–VIIF). Collectively, these results show metoprolol-mediated amelioration of cardiomyocyte stress that together validates the efficacy of the 6-week treatment protocol.
Discussion
β-adrenergic signaling is an integral part of cardiac adaptation to physiological demands of cardiac development, as well as acute or pathological stress,15,16 but consequences of adrenergic drive on CPC populations have been previously overlooked. Pathological adrenergic overdrive leads to receptor desensitization that eventually becomes cardiotoxic,18,36 resulting in cardiomyocyte death.36 However, norepinephrine release within bone marrow in cases of stress, inflammation, and tissue repair coincides with increased numbers of progenitors released into the blood circulation.37,38 Similarly, our findings support the concept that catecholamine signaling is beneficial in priming CPCs for response to pathological injury, although simultaneously exerting proapoptotic action on mature myocytes and myocyte precursors upon acquisition of β1-AR expression.
Without pharmacological intervention, the failing heart labors under a catecholamine storm that promotes adrenergic desensitization. β-blockers resensitize the heart toward β-AR signaling associated with improvement in systolic function and regression in myocardial mass.39–41 Although β-blockers are an effective clinical interventional therapy for treatment of failing hearts, relative contributions of underlying mechanisms for salutary effects remain a subject of open debate: myocardial neo-vascularization,42 antioxidant properties,43 and improvement of calcium signaling44 have all been posed as previous explanations. Biological potentiation of CPCs can now be added to this growing list of mechanistic underpinnings of β-blockade, consistent with recent reports of enhanced endothelial progenitor cell activation42 and increased cycling of cardiogenic cells.45
The molecular basis of adrenergic stimulation in CPCs rests with differential expression of β2-AR, but not β1-AR, that can be considered an uncommitted state coincident with c-kit+ expression (Figure 1). Although we observed decreased β2-AR expression in the oldest human patient (84 years), to relate β2-AR expression with age, additional age-matched samples with similar disease causes will be required. Furthermore, uncommitted CPCs actually proliferate in response to adrenergic drive that depends on β2-AR and correlates with salutary AKT/ eNOS phosphorylation (Figure 2). Recent reports demonstrate that both β1-AR and β2-AR couple to the same downstream effectors, such as GRK, regulator of G protein signaling, and so on.46–49 Induction of CPC differentiation by coculture with NRCMs leads to β1-AR acquisition (Figure 3A–3E), consistent with a mature cardiomyocyte phenotype.21 However, once placed in coculture conditions where β1-AR is induced, CPCs now become sensitized to adrenergic stimulation that promotes cell death (Figure 3). Adrenergic stimulation mediates CPC survival and proliferation through differential expression of β-ARs that is altered by β1-specific blockade (Figure 4). Importantly, β1-blockade promotes survival of β1-AR+ CPCs, representative of a committed CPC state (Figure 5). The selection of β1-blockade with metoprolol over nonselective β-antagonism is based on the premise that inhibiting deleterious β1-AR signaling protects CPCs once they become committed and express β1-AR but concomitantly promotes β2-AR signaling leading to increased CPC activation and expansion. Carvedilol is a superior β-blocker than metoprolol with known α1-AR blocking, antioxidant,43 and calcium signaling44 properties in addition to β1-blockade. Because metoprolol is highly specific for β1-AR with no known affinities for other ARs, it represents a better choice to study the differential role of β1-AR on CPC survival versus cell death. It will be interesting to look at the effects of carvedilol treatment on CPC survival in the future, in combination with the role of other ARs and β-ARs. Furthermore, nonspecific β-blockade would impart CPC protection from β1-AR signaling but also limit β2-AR–mediated CPC proliferation. The transition to β1-AR sensitization in the process of lineage specification may provide an important clue for the long-term beneficial effect of β-adrenergic blockade, which in addition to existing cardiomyocyte salvage also allows for improvement of CPC persistence. It is tempting to speculate that the refractory nature of some patients to palliative β-adrenergic blockade may rest with an impaired endogenous CPC population suffering from exhaustion as a result of age, senescence, or chronic stress that incapacitates their cardiogenic potential but requires further validation. Future studies will be needed to assess whether functionally compromised stem cells can be restored by additional interventional approaches in conjunction with β-adrenergic blockade.
The beneficial effects of β-adrenergic blockade in the failing heart are clearly dependent on protection of existing cardiomyocytes from apoptotic stimulation and, based on our findings, may also include potentiation of CPC expansion and survival. Recent evidence supports myocardial regeneration in the heart by activation and commitment of CPCs and a hierarchical organization of their progeny,50 with acquisition of myocyte phenotype as a consequence of sequential generation of various intermediate myocyte precursors and progenitors. Furthermore, sequential expression of β-ARs on CPCs expressing c-kit provides the basis for a disparate cellular response in the face of activated adrenergic signaling (Figure 6). β-adrenergic signaling induces proliferation in CPCs mediated by β2-AR, whereas β-adrenergic signaling becomes proapoptotic in the β1-AR expressing committed CPCs. Furthermore, CPCs do not express β1-AR in an uncommitted state, so β1-AR blockade does not affect β2-AR–mediated CPC expansion. β1-AR–specific blockade protects existing myocytes from chronic β1-AR stimulation, augmenting the cardiac milieu and initiating a positive feedback loop for CPCs increasing survival, and is not related to block or inhibition of CPC differentiation (Figure 6). Age and disease can lead to a significant loss of myocyte reserve within the heart promoting release of paracrine factors21 and activation of signaling cascades50 affecting CPC-mediated reparability. Collectively, adrenergic signaling promotes CPC activation and proliferation in β2-AR–dependent manner, and β1-blockade improves the cardiac environment allowing CPC expansion, proliferation, and acquisition of a committed cardiogenic state. Furthermore, ongoing β1-AR blockade after cardiac commitment prevents catecholamine-induced cell death of lineage committed CPCs (Figure 6) to enhance persistence and engraftment. Reduced β1-AR apoptotic signaling allowing β2-AR to promote prosurvival and proliferation responses improves cardiac function after combined therapy with β2-agonist and β1-blocker,51 potentially as alternate combinatorial therapy for congestive heart failure.52 Synergistic β2-AR stimulation and β1-AR blockade together may be efficacious for CPC expansion in the failing heart, potentially paving the way for a novel therapeutic modality to treat congestive heart failure.
Figure 6. Schematic representation of cardiac stem cell cycling in response to β-adrenergic signaling.
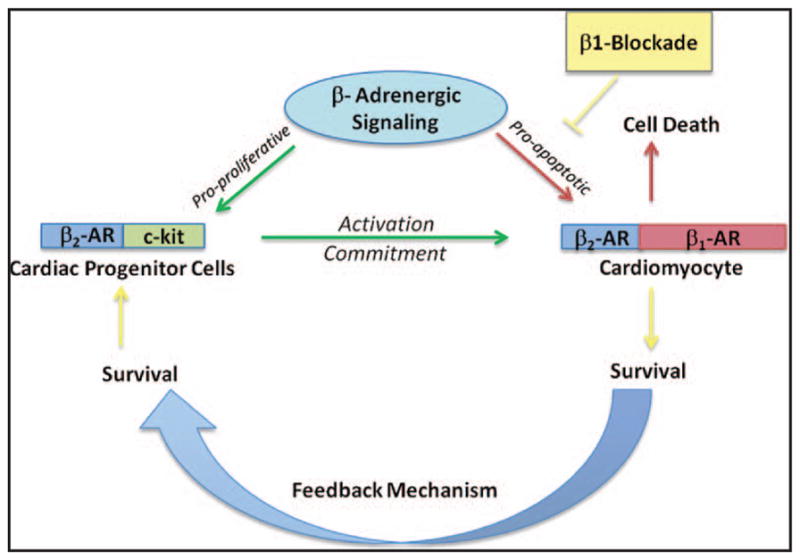
β-adrenergic signaling regulates activation of cardiac progenitor cells (CPCs) promoting enhanced commitment into cardiac phenotypes in heart failure (green arrows). However, signaling through the β-adrenergic system becomes negative at the level of mature cardiomyocyte (red arrows) that expresses both β-receptors, leading to myocyte apoptosis and necrosis. β-blockade protects cardiomyocytes from activated β-adrenergic signaling and enhances cardiomyocyte survival that feedbacks onto preserving CPC population. Persistent cardiomyocyte injury leads to CPC activation and commitment, yet simultaneously kills the newly formed myocytes leading to vicious cycle resulting in exhaustion and depletion of the CPC pool within failing hearts.
Supplementary Material
Novelty and Significance.
What Is Known?
β-adrenergic receptors (β-AR) signaling regulates cardiac contraction and relaxation, but chronic activation of β-AR becomes detrimental leading to the development of heart failure.
Cardiac progenitor cells (CPCs) are capable of differentiating into all 3 cardiac lineages and are able to support myocardial regenerative processes.
What New Information Does This Article Contribute?
CPCs express β2-AR receptor in conjunction with stem cell marker c-kit but no β1-AR in an uncommitted state.
β2-AR promotes CPC proliferation and survival, whereas expression of β1-AR in the process of cardiac lineage commitment sensitizes CPCs to catecholamine-induced cell death.
β1-blockade enhances CPC survival and proliferation in failing hearts.
β-adrenergic receptor signaling regulates cardiac contractility and maintains tissue homeostasis. The heart contains a cardiac progenitor cell pool that forms an integral part of the cardiac regenerative response, yet there is no evidence documenting the effect of β-adrenergic signaling on CPC function. Here, we report that β2-AR promotes CPC survival and proliferation on catecholamine stimulus in an uncommitted state. However, CPCs acquire β1-AR in the process of cardiac lineage commitment that leads to catecholamine-induced cell death. Adoptive transfer of CPCs in combination with β1-blockade demonstrated increased survival and proliferation of transplanted CPCs as a result of protection of the committed CPCs from the deleterious effect of β1-AR signaling. Differential regulation of survival and proliferation mediated by β2-AR versus β1-AR expression in CPCs shows a novel correlation between β-adrenergic signaling and the endogenous myocardial repair processes. Understanding the role of β-adrenergic signaling in regulating CPC function in the heart will provide an important clue in the quest for augmenting endogenous myocardial response to pathological injury and for the development of future stem cell therapy for myocardial infarction.
Acknowledgments
We thank Haruhiro Toko, Mathias Konstandin, Mirko Volkers, Nirmala Hariharan, Brett Collins, Shabana Din, and Travis Cottage of the Sussman laboratory for their helpful discussions and technical support. We wish to thank Brett Hilton of San Diego State University FACS facility for his technical assistance with flow cytometry.
Sources of Funding
M.A. Sussman was supported by National Institutes of Health grants R21HL102714, R01HL067245, R37HL091102, P01HL085577, RC1HL100891, R21HL102613, R21 HL104544, and R01HL105759.
Nonstandard Abbreviations and Acronyms
- AR
adrenergic receptor
- CPC
cardiac progenitor cells
- CPCe
Cardiac progenitor cells expressing green fluorescent protein
- FEN
fenoterol
- FUCCI
fluorescent ubiquitination-based cell cycle indicator
- GFP
green fluorescent protein
- GRK2
G protein–coupled receptor kinase 2
- ISO
isoproterenol
- NRCM
neonatal rat cardiomyocyte
- TOT
tropomodulin overexpressing transgenic
Footnotes
This manuscript was sent to Joseph Wu, Consulting Editor, for review by expert referees, editorial decision, and final disposition.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.112.280735/-/DC1
Disclosures
None.
References
- 1.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 2.Brodde OE, Michel MC. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 1999;51:651–690. [PubMed] [Google Scholar]
- 3.Vaniotis G, Del Duca D, Trieu P, Rohlicek CV, Hébert TE, Allen BG. Nuclear β-adrenergic receptors modulate gene expression in adult rat heart. Cell Signal. 2011;23:89–98. doi: 10.1016/j.cellsig.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwai-Kanai E, Hasegawa K, Araki M, Kakita T, Morimoto T, Sasayama S. alpha- and beta-adrenergic pathways differentially regulate cell type-specific apoptosis in rat cardiac myocytes. Circulation. 1999;100:305–311. doi: 10.1161/01.cir.100.3.305. [DOI] [PubMed] [Google Scholar]
- 5.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao RP, Zhu W, Zheng M, Cao C, Zhang Y, Lakatta EG, Han Q. Subtype-specific alpha1- and beta-adrenoceptor signaling in the heart. Trends Pharmacol Sci. 2006;27:330–337. doi: 10.1016/j.tips.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Foerster K, Groner F, Matthes J, Koch WJ, Birnbaumer L, Herzig S. Cardioprotection specific for the G protein Gi2 in chronic adrenergic signaling through beta 2-adrenoceptors. Proc Natl Acad Sci USA. 2003;100:14475–14480. doi: 10.1073/pnas.1936026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jhaveri DJ, Mackay EW, Hamlin AS, Marathe SV, Nandam LS, Vaidya VA, Bartlett PF. Norepinephrine directly activates adult hippocampal precursors via beta3-adrenergic receptors. J Neurosci. 2010;30:2795–2806. doi: 10.1523/JNEUROSCI.3780-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rider JE, Polster SP, Lee S, Charles NJ, Adhikari N, Mariash A, Tadros G, Stangland J, Smolenski RT, Terracciano CM, Barton PJ, Birks EJ, Yacoub MH, Miller LW, Hall JL. Chronic treatment with clenbuterol modulates endothelial progenitor cells and circulating factors in a murine model of cardiomyopathy. J Cardiovasc Transl Res. 2009;2:182–190. doi: 10.1007/s12265-009-9089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spiegel A, Shivtiel S, Kalinkovich A, Ludin A, Netzer N, Goichberg P, Azaria Y, Resnick I, Hardan I, Ben-Hur H, Nagler A, Rubinstein M, Lapidot T. Catecholaminergic neurotransmitters regulate migration and repopulation of immature human CD34+ cells through Wnt signaling. Nat Immunol. 2007;8:1123–1131. doi: 10.1038/ni1509. [DOI] [PubMed] [Google Scholar]
- 11.Méndez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci. 2010;1192:139–144. doi: 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124:407–421. doi: 10.1016/j.cell.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 13.Kim J, Eckhart AD, Eguchi S, Koch WJ. Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J Biol Chem. 2002;277:32116–32123. doi: 10.1074/jbc.M204895200. [DOI] [PubMed] [Google Scholar]
- 14.Colombo F, Gosselin H, El-Helou V, Calderone A. Beta-adrenergic receptor-mediated DNA synthesis in neonatal rat cardiac fibroblasts proceeds via a phosphatidylinositol 3-kinase dependent pathway refractory to the antiproliferative action of cyclic AMP. J Cell Physiol. 2003;195:322–330. doi: 10.1002/jcp.10251. [DOI] [PubMed] [Google Scholar]
- 15.Collis LP, Srivastava S, Coetzee WA, Artman M. beta2-Adrenergic receptor agonists stimulate L-type calcium current independent of PKA in newborn rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2007;293:H2826–H2835. doi: 10.1152/ajpheart.00101.2007. [DOI] [PubMed] [Google Scholar]
- 16.Morisco C, Zebrowski DC, Vatner DE, Vatner SF, Sadoshima J. Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta(1)-subtype in the rat heart. J Mol Cell Cardiol. 2001;33:561–573. doi: 10.1006/jmcc.2000.1332. [DOI] [PubMed] [Google Scholar]
- 17.Chruscinski A, Brede ME, Meinel L, Lohse MJ, Kobilka BK, Hein L. Differential distribution of beta-adrenergic receptor subtypes in blood vessels of knockout mice lacking beta(1)- or beta(2)-adrenergic receptors. Mol Pharmacol. 2001;60:955–962. doi: 10.1124/mol.60.5.955. [DOI] [PubMed] [Google Scholar]
- 18.Vatner DE, Asai K, Iwase M, Ishikawa Y, Shannon RP, Homcy CJ, Vatner SF. Beta-adrenergic receptor-G protein-adenylyl cyclase signal transduction in the failing heart. Am J Cardiol. 1999;83:80H–85H. doi: 10.1016/s0002-9149(99)00266-0. [DOI] [PubMed] [Google Scholar]
- 19.Zhan DY, Morimoto S, Du CK, Wang YY, Lu QW, Tanaka A, Ide T, Miwa Y, Takahashi-Yanaga F, Sasaguri T. Therapeutic effect of {beta}-adrenoceptor blockers using a mouse model of dilated cardiomyopathy with a troponin mutation. Cardiovasc Res. 2009;84:64–71. doi: 10.1093/cvr/cvp168. [DOI] [PubMed] [Google Scholar]
- 20.Hjalmarson A, Goldstein S, Fagerberg B, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA. 2000;283:1295–1302. doi: 10.1001/jama.283.10.1295. [DOI] [PubMed] [Google Scholar]
- 21.Leri A, Kajstura J, Anversa P. Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology. Circ Res. 2011;109:941–961. doi: 10.1161/CIRCRESAHA.111.243154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Fischer KM, Cottage CT, Wu W, Din S, Gude NA, Avitabile D, Quijada P, Collins BL, Fransioli J, Sussman MA. Enhancement of myocardial regeneration through genetic engineering of cardiac progenitor cells expressing Pim-1 kinase. Circulation. 2009;120:2077–2087. doi: 10.1161/CIRCULATIONAHA.109.884403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang XL, Rokosh G, Sanganalmath SK, et al. Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation. 2010;121:293–305. doi: 10.1161/CIRCULATIONAHA.109.871905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fransioli J, Bailey B, Gude NA, Cottage CT, Muraski JA, Emmanuel G, Wu W, Alvarez R, Rubio M, Ottolenghi S, Schaefer E, Sussman MA. Evolution of the c-kit-positive cell response to pathological challenge in the myocardium. Stem Cells. 2008;26:1315–1324. doi: 10.1634/stemcells.2007-0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey B, Izarra A, Alvarez R, Fischer KM, Cottage CT, Quijada P, Díez-Juan A, Sussman MA. Cardiac stem cell genetic engineering using the alphaMHC promoter. Regen Med. 2009;4:823–833. doi: 10.2217/rme.09.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Z, Völkers M, Din S, et al. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur Heart J. 2011;32:2179–2188. doi: 10.1093/eurheartj/ehq496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Springer ML, Sievers RE, Viswanathan MN, Yee MS, Foster E, Grossman W, Yeghiazarians Y. Closed-chest cell injections into mouse myocardium guided by high-resolution echocardiography. Am J Physiol Heart Circ Physiol. 2005;289:H1307–H1314. doi: 10.1152/ajpheart.00164.2005. [DOI] [PubMed] [Google Scholar]
- 28.Sugiyama M, Sakaue-Sawano A, Iimura T, Fukami K, Kitaguchi T, Kawakami K, Okamoto H, Higashijima S, Miyawaki A. Illuminating cell-cycle progression in the developing zebrafish embryo. Proc Natl Acad Sci USA. 2009;106:20812–20817. doi: 10.1073/pnas.0906464106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Penela P, Rivas V, Salcedo A, Mayor F., Jr G protein-coupled receptor kinase 2 (GRK2) modulation and cell cycle progression. Proc Natl Acad Sci USA. 2010;107:1118–1123. doi: 10.1073/pnas.0905778107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brinks H, Boucher M, Gao E, Chuprun JK, Pesant S, Raake PW, Huang ZM, Wang X, Qiu G, Gumpert A, Harris DM, Eckhart AD, Most P, Koch WJ. Level of G protein-coupled receptor kinase-2 determines myocardial ischemia/reperfusion injury via pro- and anti-apoptotic mechanisms. Circ Res. 2010;107:1140–1149. doi: 10.1161/CIRCRESAHA.110.221010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z, Bakhshi FR, Shajahan AN, Sharma T, Mao M, Trane A, Bernatchez P, van Nieuw Amerongen GP, Bonini MG, Skidgel RA, Malik AB, Minshall RD. Nitric oxide-dependent Src activation and resultant caveolin-1 phosphorylation promote eNOS/caveolin-1 binding and eNOS inhibition. Mol Biol Cell. 2012;23:1388–1398. doi: 10.1091/mbc.E11-09-0811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trouillon R, Kang DK, Park H, Chang SI, O’Hare D. Angiogenin induces nitric oxide synthesis in endothelial cells through PI-3 and Akt kinases. Biochemistry. 2010;49:3282–3288. doi: 10.1021/bi902122w. [DOI] [PubMed] [Google Scholar]
- 33.Plank DM, Yatani A, Ritsu H, Witt S, Glascock B, Lalli MJ, Periasamy M, Fiset C, Benkusky N, Valdivia HH, Sussman MA. Calcium dynamics in the failing heart: restoration by beta-adrenergic receptor blockade. Am J Physiol Heart Circ Physiol. 2003;285:H305–H315. doi: 10.1152/ajpheart.00425.2002. [DOI] [PubMed] [Google Scholar]
- 34.Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci USA. 1998;95:8801–8805. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welch S, Plank D, Witt S, Glascock B, Schaefer E, Chimenti S, Andreoli AM, Limana F, Leri A, Kajstura J, Anversa P, Sussman MA. Cardiac-specific IGF-1 expression attenuates dilated cardiomyopathy in tropomodulin-overexpressing transgenic mice. Circ Res. 2002;90:641–648. doi: 10.1161/01.res.0000013780.77774.75. [DOI] [PubMed] [Google Scholar]
- 36.Bristow MR. beta-adrenergic receptor blockade in chronic heart failure. Circulation. 2000;101:558–569. doi: 10.1161/01.cir.101.5.558. [DOI] [PubMed] [Google Scholar]
- 37.Cottler-Fox MH, Lapidot T, Petit I, Kollet O, DiPersio JF, Link D, Devine S. Stem cell mobilization. Hematology Am Soc Hematol Educ Program. 2003:419–437. doi: 10.1182/asheducation-2003.1.419. [DOI] [PubMed] [Google Scholar]
- 38.Tang Y, Shankar R, Gamelli R, Jones S. Dynamic norepinephrine alterations in bone marrow: evidence of functional innervation. J Neuroimmunol. 1999;96:182–189. doi: 10.1016/s0165-5728(99)00032-6. [DOI] [PubMed] [Google Scholar]
- 39.Hall SA, Cigarroa CG, Marcoux L, Risser RC, Grayburn PA, Eichhorn EJ. Time course of improvement in left ventricular function, mass and geometry in patients with congestive heart failure treated with beta-adrenergic blockade. J Am Coll Cardiol. 1995;25:1154–1161. doi: 10.1016/0735-1097(94)00543-y. [DOI] [PubMed] [Google Scholar]
- 40.Poulsen SH, Jensen SE, Egstrup K. Effects of long-term adrenergic beta-blockade on left ventricular diastolic filling in patients with acute myocardial infarction. Am Heart J. 1999;138:710–720. doi: 10.1016/s0002-8703(99)70187-0. [DOI] [PubMed] [Google Scholar]
- 41.Metra M, Nodari S, Parrinello G, Giubbini R, Manca C, Dei Cas L. Marked improvement in left ventricular ejection fraction during long-term beta-blockade in patients with chronic heart failure: clinical correlates and prognostic significance. Am Heart J. 2003;145:292–299. doi: 10.1067/mhj.2003.105. [DOI] [PubMed] [Google Scholar]
- 42.Sorrentino SA, Doerries C, Manes C, et al. Nebivolol exerts beneficial effects on endothelial function, early endothelial progenitor cells, myocardial neovascularization, and left ventricular dysfunction early after myocardial infarction beyond conventional β1-blockade. J Am Coll Cardiol. 2011;57:601–611. doi: 10.1016/j.jacc.2010.09.037. [DOI] [PubMed] [Google Scholar]
- 43.Koitabashi N, Arai M, Tomaru K, Takizawa T, Watanabe A, Niwano K, Yokoyama T, Wuytack F, Periasamy M, Nagai R, Kurabayashi M. Carvedilol effectively blocks oxidative stress-mediated downregulation of sarcoplasmic reticulum Ca2+-ATPase 2 gene transcription through modification of Sp1 binding. Biochem Biophys Res Commun. 2005;328:116–124. doi: 10.1016/j.bbrc.2004.12.139. [DOI] [PubMed] [Google Scholar]
- 44.Zhou Q, Xiao J, Jiang D, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med. 2011;17:1003–1009. doi: 10.1038/nm.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Musumeci M, Maccari S, Sestili P, Signore M, Molinari P, Ambrosio C, Stati T, Colledge WH, Grace AA, Catalano L, Marano G. Propranolol enhances cell cycle-related gene expression in pressure overloaded hearts. Br J Pharmacol. 2011;164:1917–1928. doi: 10.1111/j.1476-5381.2011.01504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates beta-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- 47.Xiao RP, Zhang SJ, Chakir K, Avdonin P, Zhu W, Bond RA, Balke CW, Lakatta EG, Cheng H. Enhanced G(i) signaling selectively negates beta2-adrenergic receptor (AR)–but not beta1-AR-mediated positive inotropic effect in myocytes from failing rat hearts. Circulation. 2003;108:1633–1639. doi: 10.1161/01.CIR.0000087595.17277.73. [DOI] [PubMed] [Google Scholar]
- 48.Vaughan DJ, Millman EE, Godines V, Friedman J, Tran TM, Dai W, Knoll BJ, Clark RB, Moore RH. Role of the G protein-coupled receptor kinase site serine cluster in beta2-adrenergic receptor internalization, desensitization, and beta-arrestin translocation. J Biol Chem. 2006;281:7684–7692. doi: 10.1074/jbc.M500328200. [DOI] [PubMed] [Google Scholar]
- 49.Chakir K, Zhu W, Tsang S, Woo AY, Yang D, Wang X, Zeng X, Rhee MH, Mende U, Koitabashi N, Takimoto E, Blumer KJ, Lakatta EG, Kass DA, Xiao RP. RGS2 is a primary terminator of β2-adrenergic receptor-mediated G(i) signaling. J Mol Cell Cardiol. 2011;50:1000–1007. doi: 10.1016/j.yjmcc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kajstura J, Gurusamy N, Ogórek B, et al. Myocyte turnover in the aging human heart. Circ Res. 2010;107:1374–1386. doi: 10.1161/CIRCRESAHA.110.231498. [DOI] [PubMed] [Google Scholar]
- 51.Ahmet I, Krawczyk M, Zhu W, Woo AY, Morrell C, Poosala S, Xiao RP, Lakatta EG, Talan MI. Cardioprotective and survival benefits of long-term combined therapy with beta2 adrenoreceptor (AR) agonist and beta1 AR blocker in dilated cardiomyopathy postmyocardial infarction. J Pharmacol Exp Ther. 2008;325:491–499. doi: 10.1124/jpet.107.135335. [DOI] [PubMed] [Google Scholar]
- 52.Talan MI, Ahmet I, Xiao RP, Lakatta EG. β2 AR agonists in treatment of chronic heart failure: long path to translation. J Mol Cell Cardiol. 2011;51:529–533. doi: 10.1016/j.yjmcc.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.