

Abstract
Deoxynucleoside 5′-triphosphate analogues in which the β,γ-bridging oxygen has been replaced with a CXY group are useful chemical probes to investigate DNA polymerase catalytic and base selection mechanisms. A limitation of such probes has been that conventional synthetic methods generate a mixture of diastereomers when the bridging carbon substitution is non-equivalent (X ≠ Y). We report here a general solution to this long-standing problem with four examples of individual β,γ-CXY dNTP diastereomers: (S)- and (R)-β,γ-CHCl dGTP (12a-1, 12a-2) and (S)- and (R)-β,γ-CHF dGTP (12b-1, 12b-2). Central to their preparation was conversion of the achiral parent bisphosphonic acids to P,C-dimorpholinamide derivatives (7) of their (R)-mandelic acid monoesters (6), which provided access to the individual diastereomers 7a-1, 7a-2, 7b-1, and 7b-2 by preparative HPLC. Selective acidic hydrolysis of the P-N bond then afforded the “ portal ” diastereomers 10, which were readily coupled to morpholine-activated dGMP. Removal of the chiral auxiliary by H2 (Pd/C) afforded the four individual diastereomeric nucleotides (12), which were characterized by 31P, 1H and 19F NMR, and by MS. After treatment with Chelex®-100 to remove traces of paramagnetic ions, at pH ~10 the diastereomer pairs 12a and 12b exhibit discrete Pα and Pβ 31P resonances. The more upfield Pα and more downfield Pβ resonances (and also the more upfield 19F NMR resonance in 12b) are assigned to the (R) configuration at the Pβ-CHX-Pγ carbons, based on the absolute configurations of the individual diastereomers as determined by X-ray crystallographic structures of their ternary complexes with DNA-pol β.
Nucleotide bisphosphonate analogues in which a pyrophosphate bridging oxygen is replaced by a methylene carbon were first described by Myers.1 Subsequently, it was suggested that the bisphosphonate moiety more closely mimics pyrophosphate when the bridging carbon is fluorinated.2 Pα-CXY-Pβ substitution will block nucleotidyl transfer catalyzed by polymerases, whereas Pβ-CXY-Pγ substitution (Fig. 1) produces a dNTP substrate analogue with leaving group properties that can be tuned by the substituents. Conventional synthesis of α,β- or β,γ-CXY nucleotide analogues involves coupling of bisphosphonates with nucleosides or nucleoside monophos-phates, respectively.2c,2d The α,β- and β,γ-analogues where X ≠ Y are therefore obtained as mixtures of two diastereomers due to the generation of a new chiral center at the bisphosphonate bridging carbon (Fig. 1). Several β,γ-CXY nucleotide analogues have been investigated as enzymatic probes for DNA, viral RNA or RNA-directed DNA polymerases,3 but the potential for a stereospecific interaction of these diastereomeric analogues with the enzyme active site has received little attention3c-g,3i until recently.4,5
Figure 1.
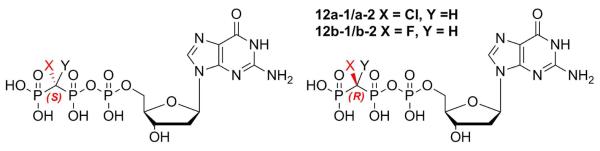
Diastereomeric β,γ-CXY analogues of dGTP.
DNA polymerase β (pol β) is a key polymerase in short-patch base excision repair (BER).6 This is the predominant BER pathway in humans and is crucial for maintaining genome integrity. Pol β has been an extensively studied model for understanding polymerase fidelity.6c,7 In a recent study of the thus far obscure transition state of pol β, we reported a series of α,β- and β,γ-CXY dNTP analogues as structural, functional and fidelity probes.8 The use of a systematically varied β,γ-CXY group to probe for a leaving group effect in the catalytic process revealed a base match-dependent chemical transition step,8b,8c consistent with the findings of Lin et al.9 and Tsai et al.10 using different approaches.
X-ray crystallographic studies of ternary complexes formed from diastereomeric β,γ-CXY dGTP analogue mixtures incubated with binary DNA-pol β crystals have revealed the presence of only one diastereomer in the active site for monofluorinated analogues (X = H, Cl, Me; Y = F), associated with an interaction between the fluorine atom and Arg183.4,11 Other β,γ-monohalogenated (X = H; Y = Cl, Br), monomethylated (X = H, Y = Me) and heterodihalogenated (X = Cl, Y = Br) analogues populated the active site evenly in the crystal complex.4 These observations provided a strong impetus to obtain the individual diastereomers.
Nucleotide analogue stereoisomers resulting from replacing a non-bridging Pα or Pβ oxygen with boron, sulfur or selenium have been isolated by HPLC, or, in the case of ATPαS and ATPβS, by selective enzymatic depletion.12 In contrast, diastereomers where a bridging oxygen is replaced by a CXY group have proven elusive. The diastereomers of α,β-CMe(N3) dATP were recently prepared via the corresponding dADP isomers, which could be separated by preparative RP-C18 HPLC.13 However, efforts to separate the α,β-CH(N3) stereoisomers were unsuccessful.13 (R/S)-β,γ-CH(N3) and (R/S)-β,γ-CMe(N3) dGTP also proved refractory to this method,13 suggesting that both the substituent size and the distance of CXY group from the chiral ribose affects separation. It seemed apparent that preparation of the elusive individual β,γ-CXY stereoisomers, particularly the highly desirable monohalo derivatives,4-5,14 required a new approach, ideally one achieving stereochemical separation at the bisphosphonate level.
Here, we communicate the synthesis, analytically discrete 31P NMR signatures, and absolute configuration assignments of all four diastereomers of β,γ-CHX dGTP (X = F or Cl), based on fixing the chirality at the bridging carbon of the prochiral α-halo bisphosphonate prior to conjugation using a novel chiral auxiliary strategy. After separation, the intermediate is conjugated conventionally with the targeted dNMP. As the incorporated nucleoside suffices to maintain the bisphosphonate stereochemistry, the chiral auxiliary can then be removed reductively under mild, unracemizing conditions.
Synthesis of the chiral bisphosphonate synthons 7a-1/7a-2 and 7b-1/7b-2 is outlined in Scheme 1. The readily available α-halo bisphosphonic acid 1a15 or 1b2a,4a,16 was heated at reflux with trimethylorthoformate to afford the corresponding tetramethyl ester, 2a17 or 2b.18 Monodemethylation using 1 equiv. of NaI in acetone afforded the racemic trimethyl esters 3,19 which were converted to their acid forms on Dowex (H+) and then esterified with (R)-(-)-methyl mandelate using Mitsunobu coupling, giving esters 4 with inversion at the chiral center of the auxiliary.20 These mixtures of diastereomers were subjected to selective silyldemethylation by BTMS21 in anhy-drous CH3CN followed by methanolysis21 to afford the (S)-mandelyl bisphosphonates (5a or 5b) as diastereomer pairs.
Scheme 1.

Synthesis of the chiral bisphosphonate synthons 7.
Attempts to separate the 5 stereoisomers by RP-C18 HPLC were not successful. However, the diastereomers after facile (pH 8) hydrolysis of the carboxylate methyl ester (which proceeded without loss of stereochemistry at the benzyl carbon), the resulting mandelic acid bisphosphonate esters 6a-1 and 6a-2 could be separated on preparative RP-C18 HPLC under isocratic condition. Unfortunately, the α-fluoro bisphosphonate diastereomers 6b-1 and 6b-2 could not be resolved suitably by this method, even on analytical scale. Reasoning that masking both the carboxylic and the phosphonic acid groups might improve chromatographic separability, we converted the 6 diastereomer mixtures to the corresponding P,C-dimorpholinamides (7), which gratifyingly made possible facile preparative isolation of all four individual diasteromers 7a-1, 7a-2, 7b-1 and 7b-2 by preparative HPLC. The 31P NMR spectra of 7a before and after separation are shown in Fig. 2. The relatively more downfield chemical shift in each individual diastereomer corresponds to the benzyl ester phosphonate P nucleus, based on 1H-31P gHMBC (gradient heteronuclear multiple bond correlation) between the downfield phosphorus peak and the benzyl proton.
Figure 2.

31P NMR (202 MHz, D2O) of a) diastereomer mixture of 7a-1/7a-2, pH 9.8; b) Individual diastereomer 7a-1, pH 9.8; c) Individual diastereomer 7a-2, pH 10.0.
Formation of the target nucleotides was carried out by conjugation with a 5′-activated dGMP (Scheme 2). The isolated 7a or 7b stereoisomers were first exchanged on a Dowex H+ column and the pH of the eluate was adjusted to 1 (1 M HCl) to complete hydrolysis of the P-N bond, giving the “portal” monoesters (10). Each of these diastereomers was coupled with dGMP-morpholidate (9)4a (prepared by DCC coupling of dGMP, 8, with morpholine)22 by stirring in anhydrous DMSO for 3 d to afford the nucleoside triphosphate analogues 11. These were purified by strong anion exchange (SAX) HPLC and obtained as triethylammonium salts.4a Removal of the (R)-mandelic acid morpholinamide auxiliary by hydrogenolysis over 10 wt. % Pd/C in 0.1 M TEAB:MeOH (1:1, pH 8) gave the deprotected individual dGTP β,γ-CHCl (12a-1, 12a-2) and β,γ-CHF (12b-1, 12b-2) diastereomers, which were then purified by RP-C18 HPLC and obtained as triethylammonium salts.
Scheme 2.

Synthesis of target diastereomeric nucleotides.
The 19F NMR spectrum of the 12b-1/12b-2 mixture as obtained by conventional synthesis was previously reported to display non-overlapping peaks for the two diastereomers.4a The chemical shifts and correct coupling constants of the two diastereomers were derived by simulation,4b but could not be assigned to a specific configuration at the β,γ-bridging carbon. The absolute configuration at the chiral CHF carbon of 12b-2 is found to be (R) by X-ray crystallographic analysis of its ternary complex with DNA-pol β (Fig. 3, PDB ID: 4DO9), allowing assignment of the more upfield 19F resonance to this diastereomer (Table 1). It was possible to obtain the absolute configurations of the other three individual diastereomers (12b-1, 12a-1 and 12a-2) similarly (PDB IDs: 4DOA, 4DOC and 4DOB, respectively).
Figure 3.

Detailed view of the incoming nucleotide 12b-2, (R)-β,γ-CHF dGTP in the active site of the X-ray crystal structure of its ternary DNA pol β:DNA (PDB ID: 4DO9). The enzyme active site Arg183, Asp190 and Asp192 side chains are shown, along with the nucleotide-binding magnesium and a water molecule. The interatomic distance between the F and Nη2 of Arg183 is 3.11 Å.
Table 1. Relative 31P and 19F NMR chemical shifts and absolute configurations of β, γ-CHX dGTP diastereomer pairs.
| Compounds |
31P NMR Pα |
31P NMR Pβ |
19F NMR | A.C.a CHX |
|---|---|---|---|---|
| 12a-1 (CHCl) | Db | Uc | N/A | S |
| 12a-2 (CHCl) | U | D | N/A | R |
| 12b-1 (CHF) | D | U | D | S |
| 12b-2 (CHF) | U | D | U | R |
A.C. = absolute configuration;
D = downfield;
U = upfield. 12a-1/12a-2: Pα Δδ = 5.4 Hz, Pβ Δδ = 8.5 Hz (202 MHz, pH 10.2); 12b-1/12b-2: Pα Δδ = 2.4 Hz, Pβ Δδ = 5.3 Hz (162 MHz, pH 10.5); 19F Δδ = 22.6 Hz (376 MHz, pH 10.5). Δδ values are measured from the NMRs of the artificial mixtures.
The ability to detect discrete 31P resonances for non F-containing analogues such as 12a-1/12a-2 would render them more generally useful as stereoprobes. At pH ~10 and after removing traces of paramagnetic metal ions by passage through Chelex®-100, the individual diastereomeric Pα and Pβ resonances of 12a-1/12a-2 and 12b-1/12b-2 proved to be observable at both 162 and 202 MHz (0.30 Hz/point digital resolution). As shown in Fig. 4, a 2:1 mixture of 12a-1 and 12a-2 exhibits a 31P Δδ of 5.4 Hz for Pα at 202 MHz and assigns the more downfield signal to the (S) isomer, 12a-1. The Pβ resonances, which are separated by 8.5 Hz under same conditions, show the reverse relationship (Table 1).
Figure 4.

31P NMR (202 MHz, D2O) of Pα in 12a. a) Artificial mixture of 12a-1/12a-2, pH 10.2. 12a-1 was added in excess, demonstrating that the 12a-2 signal is more upfield (U in Table 1), Δδ 5.4 Hz (0.027 ppm). b) Individual diastereomer 12a-1, pH 10.6. c) Individual diastereomer 12a-2, pH 10.3.
In conclusion, the first examples (12) of individual β,γ-CXY dNTP diastereomers have been successfully prepared and their absolute configurations have been correlated with discrete features of their 31P and 19F NMR spectra. The synthetic strategy developed, based on a common chiral bisphosphonate synthon, should be adaptable to the synthesis of cognate nucleotide bisphosphonate diastereomers. The availability of the individual diastereomers of 12a and 12b now makes possible kinetic analysis of their individual binding and turnover interactions with pol β and other polymerases.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by National Institutes of Health Grant 5-U19-CA105010, and in part by Research Project Number Z01-ES050158 (S.H.W.), Intramural Research Program of the National Institutes of Health, National Institute of Environmental, Health and Sciences. We thank Ms. Inah Kang for assistance in preparing the manuscript.
Footnotes
SUPPORTING INFORMATION
Details of synthesis and characterization data for 2-7 and 10-12, NMR spectra of synthetic (6a, 7 and 12) and artificial (12) mixtures, Chelex®-100 effect on resolution of 12a 31P NMR, crystal structures of 12a-1, 12a-2 and 12b-1, crystallographic data for 12b-2, HPLC conditions (6a, 7, 11 and 12). This information is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).a) Myers TC, Nakamura K, Flesher JW. J. Am. Chem. Soc. 1963;85:3292. [Google Scholar]; b) Myers TC, Simon LN. J. Org. Chem. 1965;30:443. doi: 10.1021/jo01013a035. [DOI] [PubMed] [Google Scholar]; c) Myers TC, Nakamura K, Danielza AB. J. Org. Chem. 1965;30:1517. doi: 10.1021/jo01016a043. [DOI] [PubMed] [Google Scholar]
- (2).a) McKenna CE, Shen P-D. J. Org. Chem. 1981;46:4573. [Google Scholar]; b) Berkowitz DB, Bose M. J. Fluorine Chem. 2001;112:13. [Google Scholar]; c) Blackburn GM, Kent DE, Kolkmann F. J. Chem. Soc., Chem. Commun. 1981;1188 [Google Scholar]; d) Davisson VJ, Davis DR, Dixit VM, Poulter CD. J. Org. Chem. 1987;52:1794. [Google Scholar]
- (3).a) Alexandrova LA, Skoblov AY, Jasko MV, Victorova LS, Krayevsky AA. Nuc. Acids Res. 1998;26:778. doi: 10.1093/nar/26.3.778. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arabshahi L, Khan NN, Butler M, Noonan T, Brown NC, Wright GE. Biochemistry. 1990;29:6820. doi: 10.1021/bi00481a010. [DOI] [PubMed] [Google Scholar]; c) Hamilton CJ, Roberts SM, Shipitsin A. Chem. Commun. 1998;1087 [Google Scholar]; d) Krayevsky A, Arzumanov A, Shirokova E, Dyatkina N, Victorova L, Jasko M, Alexandrova L. Nucleosides Nucleotides. 1998;17:681. doi: 10.1080/07328319808005209. [DOI] [PubMed] [Google Scholar]; e) Martynov BI, Shirokova EA, Jasko MV, Victorova LS, Krayevsky AA. FEBS Lett. 1997;410:423. doi: 10.1016/s0014-5793(97)00577-2. [DOI] [PubMed] [Google Scholar]; f) Shipitsin AV, Victorova LS, Shirokova EA, Dyatkina NB, Goryunova LE, Beabealashvilli RS, Hamilton CJ, Roberts SM, Krayevsky A. J. Chem. Soc. Perkin Trans. 1. 1999;1039 doi: 10.1080/15257779908041639. [DOI] [PubMed] [Google Scholar]; g) Spelta V, Mekhalfia A, Rejman D, Thompson M, Blackburn GM, North RA. Br. J. Pharmacol. 2003;140:1027. doi: 10.1038/sj.bjp.0705531. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Kashemirov BA, Roze CN, McKenna CE. Phosphorus Sulfur Silicon Relat. Elem. 2002;177:2275. doi: 10.1080/10426507.2010.526675. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Boyle NA, Fagan P, Brooks JL, Prhavc M, Lambert J, Cook PD. Nucleosides Nucleotides Nucleic Acids. 2005;24:1651. doi: 10.1080/15257770500267055. [DOI] [PubMed] [Google Scholar]
- (4).a) McKenna CE, Kashemirov BA, Upton TG, Batra VK, Goodman MF, Pedersen LC, Beard WA, Wilson SH. J. Am. Chem. Soc. 2007;129:15412. doi: 10.1021/ja072127v. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Batra VK, Pedersen LC, Beard WA, Wilson SH, Kashemirov BA, Upton TG, Goodman MF, McKenna CE. J. Am. Chem. Soc. 2010;132:7617. doi: 10.1021/ja909370k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).This limitation as applied to β,γ-CHF ATP has long been recognized: Blackburn GM, Kent DE, Kolkmann F. J. Chem. Soc. Perkin Trans. 1. 1984;1119 These authors reported that the diastereomer resonances in this compound mixture could not be distinguished in 19F (94.08 MHz) and 31P (40.48 and 162 MHz) NMR.
- (6).a) Barnes DE, Lindahl T. Annu. Rev. Genet. 2004;38:445. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]; b) Beard WA, Prasad R, Wilson SH. Methods Enzymol. 2006;408:91. doi: 10.1016/S0076-6879(06)08007-4. [DOI] [PubMed] [Google Scholar]; c) Beard WA, Wilson SH. Chem. Rev. 2006;106:361. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- (7).Arndt JW, Gong WM, Zhong XJ, Showalter AK, Liu J, Dunlap CA, Lin Z, Paxson C, Tsai MD, Chan MK. Biochemistry. 2001;40:5368. doi: 10.1021/bi002176j. [DOI] [PubMed] [Google Scholar]
- (8).a) Upton TG, Kashemirov BA, McKenna CE, Goodman MF, Prakash GKS, Kultyshev R, Batra VK, Shock DD, Pedersen LC, Beard WA, Wilson SH. Org. Lett. 2009;11:1883. doi: 10.1021/ol701755k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sucato CA, Upton TG, Kashemirov BA, Batra VK, Martinek V, Xiang Y, Beard WA, Pedersen LC, Wilson SH, McKenna CE, Florian J, Warshel A, Goodman MF. Biochemistry. 2007;46:461. doi: 10.1021/bi061517b. [DOI] [PubMed] [Google Scholar]; c) Sucato CA, Upton TG, Kashemirov BA, Osuna J, Oertell K, Beard WA, Wilson SH, Florian J, Warshel A, McKenna CE, Goodman MF. Biochemistry. 2008;47:870. doi: 10.1021/bi7014162. [DOI] [PubMed] [Google Scholar]
- (9).a) Lin P, Pedersen LC, Batra VK, Beard WA, Wilson SH, Pedersen LG. Proc. Natl. Acad. Sci. USA. 2006;103:13294. doi: 10.1073/pnas.0606006103. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lin P, Batra VK, Pedersen LC, Beard WA, Wilson SH, Pedersen LG. Proc. Natl. Acad. Sci. USA. 2008;105:5670. doi: 10.1073/pnas.0801257105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).a) Bakhtina M, Roettger MP, Tsai MD. Biochemistry. 2009;48:3197. doi: 10.1021/bi802119f. [DOI] [PubMed] [Google Scholar]; b) Roettger MP, Bakhtina M, Tsai MD. Biochemistry. 2008;47:9718. doi: 10.1021/bi800689d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hardegger LA, Kuhn B, Spinnler B, Anselm L, Ecabert R, Stihle M, Gsell B, Thoma R, Diez J, Benz J, Plancher J-M, Hartmann G, Banner DW, Haap W, Diederich F. Angew. Chem. Int. Ed. 2011;50:314. doi: 10.1002/anie.201006781. [DOI] [PubMed] [Google Scholar]
- (12).a) Kowalska J, Zuberek J, Darzynkiewicz ZM, Lukaszewicz M, Darzynkiewicz E, Jemielity J. Collect. Symp. Ser. 2008;10:383. [Google Scholar]; b) Kowalska J, Lewdorowicz M, Zuberek J, Grudzien-Nogalska E, Bojarska E, Stepinski J, Rhoads RE, Darzynkiewicz E, Davis RE, Jemielity J. RNA. 2008;14:1119. doi: 10.1261/rna.990208. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kowalska J, Lukaszewicz M, Zuberek J, Darzynkiewicz E, Jemielity J. ChemBioChem. 2009;10:2469. doi: 10.1002/cbic.200900522. [DOI] [PubMed] [Google Scholar]; d) Lin JL, Shaw BR. Chem. Commun. 2000;2115 [Google Scholar]; e) Lin JL, Porter KW, Shaw BR. Nucleosides Nucleotides Nucleic Acids. 2001;20:1019. doi: 10.1081/NCN-100002482. [DOI] [PubMed] [Google Scholar]; f) Eckstein F, Goody RS. Biochemistry. 1976;15:1685. doi: 10.1021/bi00653a015. [DOI] [PubMed] [Google Scholar]
- (13).Chamberlain BT, Upton TG, Kashemirov BA, McKenna CE. J. Org. Chem. 2011;76:5132. doi: 10.1021/jo200045a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McKenna CE, Kashemirov BA, Peterson LW, Goodman MF. Biochim. Biophys. Acta, Proteins Proteomics. 2010;1804:1223. doi: 10.1016/j.bbapap.2010.01.005. [DOI] [PubMed] [Google Scholar]
- (15).McKenna CE, Khawli LA, Ahmad WY, Pham P, Bongartz JP. Phosphorus Sulfur Silicon Relat. Elem. 1988;37:1. [Google Scholar]
- (16).Marma MS, Khawli LA, Harutunian V, Kashemirov BA, McKenna CE. J. Fluorine Chem. 2005;126:1467. [Google Scholar]
- (17).a) Hutchinson DW, Semple G. J. Organomet. Chem. 1986;309:C7. [Google Scholar]; b) Hutchinson DW. J. Organomet. Chem. 1987;319:C39. [Google Scholar]
- (18).Nicholson DA, Cilley WA, Quimby OT. J. Org. Chem. 1970;35:3149. [Google Scholar]
- (19).a) Goldstein JA, McKenna C, Westheimer FH. J. Am. Chem. Soc. 1976;98:7327. [Google Scholar]; b) McKenna CE, Kashemirov BA, Roze CN. Bioorg. Chem. 2002;30:383. doi: 10.1016/s0045-2068(02)00521-7. [DOI] [PubMed] [Google Scholar]
- (20).Campbell DA. J. Org. Chem. 1992;57:6331. [Google Scholar]
- (21).a) McKenna CE, Higa MT, Cheung NH, McKenna MC. Tetrahedron Lett. 1977;155 [Google Scholar]; b) McKenna CE, Schmidhauser J. J. Chem Soc. Chem. Commun. 1979;739 [Google Scholar]
- (22).Moffatt JG, Khorana HG. J. Am. Chem. Soc. 1961;83:663. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.