

Abstract
Knockout mice have been informative in the discovery of unexpected biological functions of aquaporins. Knockout mice have confirmed the predicted roles of aquaporins in transepithelial fluid transport, as in the urinary concentrating mechanism and glandular fluid secretion. A less obvious, though predictable role of aquaporins is in tissue swelling under stress, as in the brain in stroke, tumor and infection. Phenotype analysis of aquaporin knockout mice has revealed several unexpected cellular roles of aquaporins whose mechanisms are being elucidated. Aquaporins facilitate cell migration, as seen in aquaporin-dependent tumor angiogenesis and tumor metastasis, by a mechanism that may involve facilitated water transport in lamellipodia of migrating cells. The ‘aquaglyceroporins’, aquaporins that transport both glycerol and water, regulate glycerol content in epidermis, fat and other tissues, and lead to a multiplicity of interesting consequences of gene disruption including dry skin, resistance to skin carcinogenesis, impaired cell proliferation and altered fat metabolism. An even more surprising role of a mammalian aquaporin is in neural signal transduction in the central nervous system. The many roles of aquaporins might be exploited for clinical benefit by modulation of aquaporin expression/function – as diuretics, and in the treatment of brain swelling, glaucoma, epilepsy, obesity and cancer.
1 Introduction
The mammalian aquaporins (AQPs) are a family of at least 12 related proteins expressed in epithelial, endothelial and other tissues, many of which are involved in fluid transport such as kidney tubules, while others are not such as skin and fat cells. Functional measurements indicate that AQPs 1, 2, 4, 5 and 8 are primarily water-selective, whereas aquaporins 3, 7 and 9 (the ‘aquaglyceroporins’) also transport glycerol and perhaps other small solutes. Tissue distribution and regulation studies have provided indirect evidence for roles of AQPs in a variety of physiological processes. In the case of AQP2, nephrogenic diabetes insipidus in subjects with AQP2 mutation provides direct evidence for AQP2 involvement in the urinary concentrating mechanism (Deen et al. 1994). However, the unavailability to date of aquaporin inhibitors suitable for use in vivo has precluded direct investigation of their function.
This review will focus on physiologically important functions of the mammalian AQPs discovered from phenotype analysis of AQP knockout mice and follow-up cell/tissue studies. One of the paradigms that has emerged from phenotype studies of knockout mice is that tissue-specific AQP expression does not mandate AQP involvement in a physiologically important process, as in the case of lung AQPs (reviewed in Verkman 2007), AQPs in the intestine (reviewed in Verkman and Thiagarajah 2006), AQP4 in skeletal muscle (Yang et al. 2000), AQP5 in sweat gland (Song et al. 2002), and AQP8 in multiple tissues (Yang et al. 2005). However, data from knockout mice implicate important roles of AQPs in kidney, brain, eye, skin, fat and exocrine glands, suggesting their involvement in major organ functions and disease processes including urinary concentrating, brain swelling, epilepsy, glaucoma, cancer and obesity.
2 AQPs and the Urinary Concentrating Mechanism
A major and anticipated role of AQPs in kidney is in water transport across kidney tubules and vasa recta for the formation of concentrated urine. Figure 1a shows the distribution of the major renal AQPs. Deletion of AQP1 and/or AQP3 in mice results in marked polyuria, as seen in 24 h urine collections (Fig. 1B) (Ma et al. 1998, 2000b). A qualitatively similar urinary concentrating defect was found in rare humans with defective AQP1 (King et al. 2001). Figure 1c summarizes urine osmolalities in mice before and after 36 h water deprivation. Urinary osmolality in AQP1 null mice is low and does not increase with water deprivation, resulting in severe dehydration. AQP3 null mice are able to generate partially concentrated urine in response to water deprivation, whereas AQP4 null mice manifest only a mild defect in maximum urinary concentrating ability.
Fig. 1.

Impaired urinary concentrating ability in AQP null mice. (a) Sites of AQP expression in kidney. (b) Twenty-four hour urine collections showing polyuria in mice lacking AQP1 and AQP3, individually and together. (c) Urine osmolalities before and after 36 h water deprivation (S.E.). (d) Transepithelial osmotic water permeability (Pf) in microperfused proximal tubule, thin descending limb of Henle and inner medullary collecting from mice of indicated genotype (S.E.). Data summarized from Ma et al. (1997, 1998, 2000b) and Schnermann et al. (1998)
AQP1 deletion produces polyuria and unresponsiveness to vasopressin or water deprivation by two distinct mechanisms – impaired near-isosmolar water reabsorption in proximal tubule and reduced medullary hypertonicity resulting from impaired countercurrent multiple and exchange. Transepithelial osmotic water permeability (Pf) in the isolated microperfused S2 segment of proximal tubule was reduced by ~fivefold in AQP1 knockout mice (Fig. 1d) (Schnermann et al. 1998), indicating that most water transport in proximal tubule is transcellular and AQP1-dependent. Free-flow micropuncture with end-proximal tubule fluid sampling showed ~50% reduced fluid absorption, with little effect on single nephron glomerular filtration rate (Schnermann et al. 1998), supporting a three-compartment model in which mild luminal hypotonicity drives osmotic water movement through highly water permeable cell membranes. AQP1 also provides the major route for transepithelial water permeability in thin descending limb of Henle (TDLH) and outer medullary descending vasa recta (OMDVR) (Chou et al. 1999; Pallone et al. 2000). These results support the conclusions that AQP1 is the principal water channel in TDLH and OMDVR and causes osmotic equilibration in these segments, and plays a key role in the renal countercurrent concentrating mechanism. AQP1 deletion thus impairs urinary concentrating ability by impairing near-isosmolar fluid absorption by proximal tubule and by interfering with the normally hypertonic medullary interstitium generated by countercurrent multiplication and exchange. The precise degree of impairment in urinary concentrating ability is also influenced by secondary renal mechanisms, such as tubuloglomerular feedback, as well as altered expression of other genes in AQP1 null mice. AQP7 is expressed in a small distal segment (S3 segment) of the proximal tubule, though its deletion in mice is not associated with significant impairment in urinary concentrating ability, but rather with an impairment of glycerol clearance, whose significance remains unclear (Sohara et al. 2005).
AQP3 and AQP4 are expressed at the basolateral membrane of collecting duct epithelium, with relatively greater expression of AQP3 in cortical and outer medullary collecting duct and AQP4 in inner medullary collecting duct. In contrast to AQP1 null mice, countercurrent multiplication and exchange mechanisms in AQP3/AQP4 null mice are basically intact. The polyuria in AQP3 null mice results in large part from the more than threefold reduction in Pf of cortical collecting duct basolateral membrane (Ma et al. 2000b). In addition, AQP2 expression is reduced in AQP3 null mice, which appears to be a maladaptive renal response seen in various forms of acquired polyuria. AQP4 null mice manifest only a mild impairment in maximal urinary concentrating ability (Ma et al. 1997), despite fourfold reduced water permeability in microperfused inner medullary collecting duct (Fig. 1d) (Chou et al. 1998). Several mouse models of AQP2 gene deletion and mutation (reviewed in Verkman 2008) support the conclusion that AQP2 is the major vasopressin-regulated water channel whose apical membrane targeting in collecting duct during antidiuresis is crucial for the formation of concentrated urine.
3 AQPs in Active, Near-Isosmolar Fluid Secretion
As mentioned above, impaired fluid absorption in kidney proximal tubule in AQP1 deficiency indicates the need for high cell membrane water permeability for rapid, near-isosmolar fluid transport. The involvement of AQPs in fluid absorption or secretion in various exocrine glands (salivary, submucosal, sweat, lacrimal), absorptive epithelia (lung, airways), and secretory epithelia (choroid plexus and ciliary body) has been investigated using knockout mouse models. The general conclusion is that AQPs facilitate active, transepithelial secretion when sufficiently rapid, such that AQP deletion causes reduced secreted fluid volume and increased ion/solute content of secreted fluid. Because active, near-isosmolar fluid transport involves transepithelial water transport driven by relatively small osmotic gradients produced by salt pumping, AQP deficiency reduces transepithelial water flow. This paradigm has been confirmed from defective fluid secretion in AQP5 knockout mice in salivary gland (Ma et al. 1999; Krane et al. 2001) and airway submucosal gland (Song and Verkman 2001). Defective fluid secretion has also been found in AQP1 knockout mice in ciliary epithelium (Zhang et al. 2002), which produces aqueous fluid in the eye and in choroid plexus (Oshio et al. 2005), which produces cerebrospinal fluid. However, AQPs appear not to be needed when fluid absorption or secretion rate (per unit epithelial surface area) is low, as in lung (Bai et al. 1999; Ma et al. 2000a; Song et al. 2000a), airways (Song and Verkman 2001), lacrimal gland (Moore et al. 2000) and sweat gland (Song et al. 2002), where AQP-independent water permeability is high enough to support slow fluid transport. In the case of the peritoneal and pleural cavities, though AQP1 facilitates osmotically driven water transport, AQP1 deficiency in mice does not impair the relatively slow absorption of excess fluid in these cavities (Yang et al. 1999; Song et al. 2000b). Together, these results support the anticipated role of AQPs in active fluid absorption and secretion, but indicate that the requirement for AQP-facilitated water transport depends on the rate of water transport.
4 AQP4 in Brain Swelling and Neural Signal Transduction
AQP4 is expressed in the brain and spinal cord at sites of fluid transport at blood–brain and brain–cerebrospinal fluid (CSF) interfaces. AQP4 expression is polarized to astrocytic foot processes in contact with blood vessels and in the dense astrocyte cell processes that form the glia limitans, which lines the CSF-bathed pial and ependymal surfaces in the subarachnoid space and the ventricles (Nielsen et al. 1997). Evidence that AQP4 provides the major route for water transport across astrocyte cell membranes came from osmotic water permeability measurements showing sevenfold reduced water transport in astrocytes cultured from AQP4 null mice (Solenov et al. 2004). Also, accumulation of brain water was found to be greatly slowed in AQP4 null mice in response to serum hypoosmolality, as monitored continuously in vivo by a non-invasive near-infrared optical method (Thiagarajah et al. 2005), or by brain wet-to-dry weight ratio measurements done at different times after water intoxication (Papadopoulos and Verkman 2005). Interestingly, AQP4 has been identified as a structural component of membrane ‘orthogonal arrays of proteins’ (OAPs), which are cobblestone-appearing structures, seen by freeze-fracture electron microscopy. OAPs were found in stably transfected CHO cells expressing functional AQP4 (Yang et al. 1996) and were absent in brain, kidney and skeletal muscle in AQP4 null mice (Verbavatz et al. 1997). Subsequently freeze-label electron microscopy studies confirmed the presence of AQP4 in OAPs (Rash et al. 1998). However, the functional significance of AQP4 assembly in OAPs is not known.
Evidence for involvement of AQP4 in brain water balance came from experiments showing reduced brain swelling and improved survival in AQP4 null vs. wildtype mice after water intoxication (Fig. 2a), reduced hemispheric swelling after focal cerebral ischemia (Fig. 2b) and improved survival after bacterial meningitis (Fig. 2c) (Manley et al. 2000; Papadopoulos and Verkman 2005). Reduced brain swelling was also reported in alpha-syntrophin null mice, which secondarily manifest disrupted brain AQP4 expression (Amiry-Moghaddam et al. 2003a). According to the Klatzo classification of brain edema, these are primarily models of cytotoxic (cell swelling) edema in which excess water moves from the vasculature into the brain parenchyma through an intact blood–brain barrier. The forces driving water flow to form cytotoxic edema are osmotic, generated in water intoxication by reduced plasma osmolality and in ischemia by impaired Na+/K+ ATPase pump function with consequent Na+ and water flow from the intravascular and extracellular compartments into the intracellular compartment. AQP4 also plays a key role in elimination of excess brain water. When the blood–brain barrier becomes disrupted (brain tumor, brain abscess, focal freeze injury), water moves from the vasculature into the extracellular space of the brain in an AQP4-independent manner down a hydrostatic gradient to form, what was termed by Klatzo vasogenic edema. Excess water is eliminated primarily through the glia limiting membrane into the cere-brospinal fluid. We found greater brain water accumulation and intracranial pressure in AQP4 null vs. wildtype mice with brain tumor, brain abscess, focal cortical freeze injury and after infusion of normal saline directly into the brain extracellular space (Fig. 2d) (Papadopoulos et al. 2004; Bloch et al. 2005), supporting the conclusion that in vasogenic edema fluid is eliminated by an AQP4-dependent route. In a kaolin model of hydrocephalus AQP4 null mice develop marked hydrocephalus, probably due to reduced transependymal water clearance (Fig. 2e) (Bloch et al. 2006). AQP4 thus is a major determinant in fluid movement into and out of the brain.
Fig. 2.
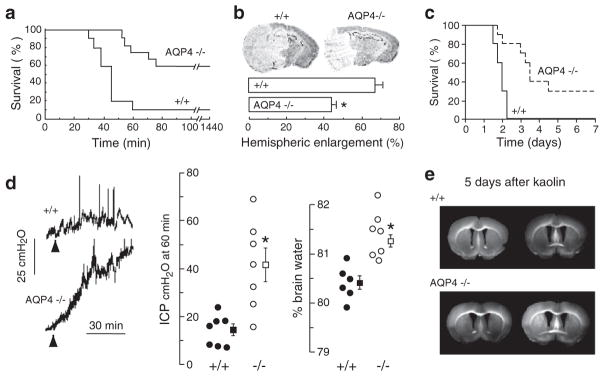
AQP4 deletion reduces brain water accumulation in cytotoxic edema, but slows removal of excess brain water in vasogenic edema and hydrocephalus. (a) Water intoxication model of cytotoxic edema. Mouse survival after acute water intoxication produced by intraperitoneal water injection. (b) (top) Ischemic stroke model of cytotoxic edema. Brain sections of mice at 24 h after ischemic stroke produced by permanent middle cerebral artery occlusion. (bottom) Average hemispheric enlargement expressed as a percentage determined by image analysis of brain sections (S.E., *P < 0.01). (c) Mouse survival in a bacterial model of meningitis produced by cisternal injection of S. pneumoniae. (d) Reduced elevation in intracranial pressure (ICP, S.E., *P < 0.01) and brain water content (S.E., *P < 0.001) following continuous intraparenchymal infusion of artificial cerebrospinal fluid at 0.5μL min−1. (e) Accelerated progression of hydrocephalus in AQP4 null mice. Coronal sections wildtype and AQP4 null mouse brain at 5 days after kaolin injection. Data from Manley et al. (2000), Papadopoulos et al. (2004), and Bloch et al. (2006)
AQP4 also appears to play an unexpected role in neural signal transduction. AQP4 is expressed in supportive cells adjacent to electrically excitable cells, as in glia vs. neurons in brain and spinal cord, Müller vs. bipolar cells in retina, and hair vs. supportive cells in the inner ear. We found evidence for impaired auditory and visual signal tranduction in AQP4 null mice, seen as increased auditory brainstem response thresholds (Li and Verkman 2001; Mhatre et al. 2002) and reduced electroretinographic potentials (Li et al. 2002). In brain, seizure susceptibility in response to the convulsant (GABA antagonist) pentylenetetrazol was remarkably increased in AQP4 null mice (Binder et al. 2004a); at 40mg kg−1 pentylenetetrazol, all wildtype mice exhibited seizure activity, whereas six out of seven AQP4 null mice did not exhibit seizure activity. In freely moving mice (Fig. 3a, top), electrically-induced seizures following hippocampal stimulation showed greater threshold and remarkably longer seizure duration in AQP4 null mice compared to wildtype mice (Fig. 3a, bottom) (Binder et al. 2006). In support of these findings, using a hyperthermia model of seizure induction, alpha-syntrophin deficient mice developed more severe seizures than wildtype mice (Amiry-Moghaddam et al. 2003b).
Fig. 3.
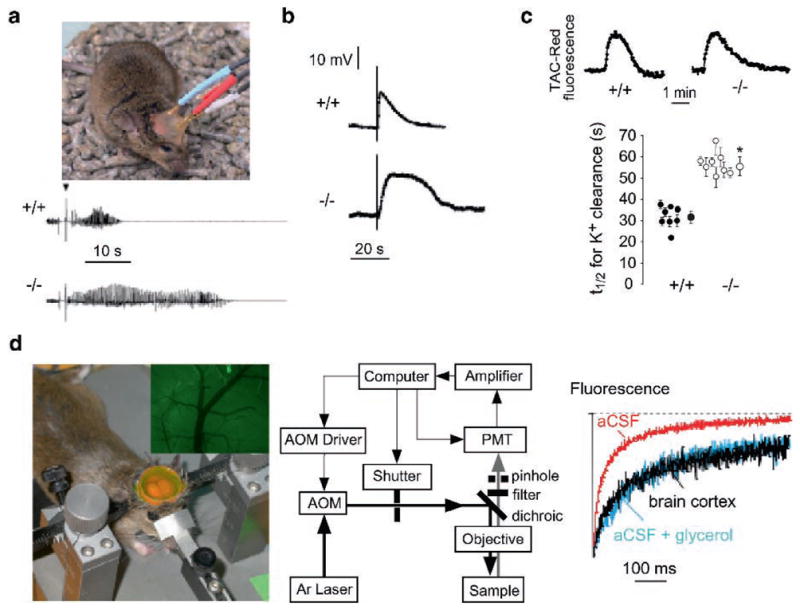
Involvement of AQP4 in brain neuroexcitation. (a) Increased seizure duration in AQP4 null mice. (top) Bipolar electrodes implanted in the right hippocampus were connected to a stimulator and electroencephalograph recording system. (bottom) Representative electroencephalographic recordings in freely moving mice following electrically induced generalized seizures. (b) Delayed K+ clearance in brain following electrically induced seizure-like neuroexcitation. Measurements done using K+-sensitive microelectrodes inserted into brain cortex in living mice. (c) Slowed K+ clearance in brain ECS during cortical spreading depression measured using TAC-Red, a K+-sensitive fluorescent probe. (top) Representative data. (bottom) Half-times (t1/2) for K+ reuptake. (d) Expanded brain ECS in AQP4 null mice measured by cortical surface photobleaching. (left) Mouse brain surface exposed to FITC-dextran with dura intact following craniectomy and fluorescence imaging of cortical surface after loading with FITC-dextran (inset). (middle) Photobleaching apparatus. A laser beam is modulated by an acousto-optic modulator and directed onto the surface of the cortex using a dichroic mirror and objective lens. (right) In vivo fluorescence recovery in cortex of wildtype mouse shown in comparison to aCSF and 30% glycerol in a CSF. Taken from Binder et al. (2004b, 2006) and Padmawar et al. (2005)
The mechanisms for altered neuroexcitation in AQP4 deficiency remain unknown. Delayed K+ uptake from brain extracellular space (ECS) in AQP4 deficiency has been suggested, which may account for the prolonged seizure phenotype. Direct measurements of [K+] in brain cortex in living mice using K+-sensitive microelectrodes showed significant slowing of K+ clearance following electrical stimulation (Fig. 3b). Using a K+-sensitive fluorescent dye, altered K+ wave dynamics was found in a cortical spreading depression model of neuroexcitation, again with delayed K+ clearance (Fig. 3c). It was proposed from immunocolocalization and immunopercipitation studies that AQP4 is closely associated with the inwardly rectifying K+ channel Kir4.1 in astroglia and Müller cells, suggesting that reduced K+ channel function in AQP4 deficiency might account for the delay in K+ clearance. However, recent patch-clamp studies in astroglia (Zhang and Verkman 2008) and Müller cells (Ruiz-Ederra et al. 2007) provide evidence against this hypothesis. Another possible explanation involves ECS expansion in AQP4 deficiency, which may account in part for reduced seizure susceptibility and prolonged seizure duration in AQP4 deficiency. An expanded ECS would provide a larger aqueous volume to dilute K+ released into the ECS during neuroexcitation, thereby slowing changes in ECS K+ concentration. Evidence for an expanded ECS in AQP4 deficiency came from cortical surface photobleaching measurements of the diffusion of fluorescently-labeled macromolecules (Binder et al. 2004b). The ECS in mouse brain was stained with fluorescein-dextran by exposure of the intact dura after craniectomy (Fig. 3d, left), and diffusion detected by fluorescence recovery after photobleaching (Fig. 3d, middle). Diffusion was slowed ~threefold in brain ECS relative to saline solutions (Fig. 3d, right), and significantly accelerated in AQP4 null mice, indicating an expanded ECS. ECS expansion in AQP4 deficiency was also shown utilizing a microfiberoptic epifluorescence photobleaching method to measure diffusion in deep brain structures (Zador et al. 2008). It remains unclear, however, whether ECS expansion in AQP4 deficiency could account quantitatively for the altered ECS K+ dynamics, as well as the mechanisms involved in chronic ECS expansion in AQP4 deficiency.
5 AQPs and Eye Physiology
The eye expresses several AQPs at putative sites of fluid transport (Fig. 4a). The expression of MIP (major intrinsic protein, also referred to as AQP0) in lens fiber has been known for many years. Mutations in AQP0 in humans are associated with congenital cataracts (Berry et al. 2000). AQP1 is expressed in corneal endothelia and at sites of aqueous fluid production (ciliary epithelium) and outflow (trabecular meshwork). AQP3 is expressed in the conjunctival epithelium. AQP4 is expressed in Müller cells in retina, which support the electrically excitable hair cells, as well as in optic nerve. AQP4 is also expressed with AQP1 in non-pigmented ciliary epithelium. AQP5 is expressed in corneal epithelia and lacrimal gland. The expression pattern of AQPs provides indirect evidence for their involvement in intraocular pressure (IOP) regulation (AQP1 and AQP4), corneal and lens transparency (AQP0, AQP1 and AQP5), visual signal transduction (AQP4), tear film homeostasis (AQP3 and AQP5), conjunctival barrier function (AQP3) and tear formation by lacrimal glands (AQP5).
Fig. 4.

Ocular phenotype in AQP deficient mice. (a) Sites of AQP expression in the eye. (b) (top) Stained plastic sections of corneas of mice indicated genotype showing epithelium (upper surface), stroma and endothelium. (bottom) Photographs of wildtype and AQP1 null mice at 40 min after a 10 min exposure of the corneal surface to distilled water showing corneal opacification in the AQP1 null mouse (white arrow). (c) (top) Retinal morphology in an ischemia-reperfusion model of retinal injury. Hematoxylin and eosin-stained retinal sections before and at 12 and 96 h after 60 min retinal ischemia in wildtype and AQP4 null mice. Note retinal swelling at 12 h and degeneration at 96 h. (bottom) Functional analysis by electroretinography. Representative electroretinograms before and at 1, 2 and 4 days after 45 min retinal ischemia in wildtype and AQP4 null mice. From Thiagarajah and Verkman (2002) and Da and Verkman (2004)
These possibilities have been examined systematically by phenotype analysis of AQP knockout mice. Interestingly, compared to wildtype mice that have a corneal thickness of 123μm, corneal thickness was reduced in AQP1 null mice (101μm) and increased in AQP5 null mice (144μm) (Thiagarajah and Verkman 2002). Thickness measurements were made in fixed eyes (Fig. 4b, top), as well as by brightfield scanning confocal microscopy in vivo. After exposure of the external corneal surface to hypotonic saline the rate of corneal swelling was reduced ~twofold by AQP5 deletion. After exposure of the corneal endothelial surface to hypotonic saline by anterior chamber perfusion, the rate of corneal swelling was reduced ~fourfold by AQP1 deletion. Remarkably, though baseline corneal transparency was not impaired by AQP1 deletion, the recovery of corneal transparency and thickness after hypotonic swelling (10 min exposure of corneal surface to distilled water) was remarkably delayed in AQP1 null mice, with ~75% recovery at 7 min in wildtype mice compared to 5% recovery in AQP1 null mice (Fig. 4b, bottom). These data provide evidence for AQP1 involvement in active extrusion of fluid from the corneal stroma across the corneal endothelium. Water permeability has also been measured in conjunctiva, where it is significantly slowed in AQP3 knockout mice (Levin and Verkman 2004). Together with permeability data for cornea, a mathematical model of tear film osmolarity was developed, providing insights into the pathophysiology of dry eye disorders and possible roles of ocular surface AQPs.
The principal determinants of IOP are the rate of aqueous fluid production by the ciliary epithelium and the rate of fluid drainage (outflow) in the canal of Schlemm. As mentioned above, aqueous fluid production involves passive, near-isosmolar fluid secretion driven by active salt transport across the ciliary epithelium. Aqueous fluid drainage primarily involves pressure-driven bulk fluid flow in the canal of Schlemm. Using a modified micro-needle method to measure IOP, a small though significant reduction in IOP was found in mice lacking AQP1, with reduced aqueous fluid secretion, though unimpaired aqueous fluid drainage (Zhang et al. 2002). Whether larger differences in IOP will be found with AQP deficiency in models of glaucoma remains to be determined.
Like the AQP1-expressing corneal endothelium covering the corneal inner surface, the anterior surface of the lens is covered by an AQP1-expressing epithelium. Motivated by the marked impairment of AQP1 deletion on corneal transparency in an experimental model of corneal swelling, we tested the possibility that a lens AQP1 might serve a similar function (Ruiz-Ederra et al. 2006). Osmotic water permeability across lens epithelium was ~threefold reduced in lenses from AQP1 null mice. Though AQP1 deletion did not alter baseline lens morphology or transparency, lens water content was significantly greater by ~4% in AQP1 null mice. In vitro and in vivo models were used to test for possible involvement of AQP1 in cataract formation. In vitro, following incubation of lenses in high glucose solutions, loss of lens transparency was greatly accelerated in AQP1-null lenses as measured by optical contrast analysis of transmitted grid images. In vivo, cataract formation was significantly accelerated in AQP1 null mice in a model of acetaminophen toxicity. The data suggested that AQP1 facilitates the maintenance of lens transparency and opposes cataract formation.
AQP4 is expressed in Müller cells in retina, where as mentioned above it appears to be involved in light signal transduction. We found evidence also for AQP4 involvement in retinal swelling (Da and Verkman 2004). Motivated by the protection against cytotoxic brain edema conferred by AQP4 gene deletion, the possibility was tested that AQP4 deletion in mice protects the retina in a transient ischemia-reperfusion model produced by 45–60 min IOP elevation to 120 mmHg. Retinal structure and cell number were remarkably preserved in AQP4 null mice, particularly in inner nuclear and plexiform layers of retina where Müller cells are concentrated (Fig. 4C). Retinal function and cell survival were also significantly improved in AQP4 deficient mice. By electroretinography, b-wave amplitude was reduced by ~75% at 1–4 days after ischemia in wildtype mice vs. less than 50% in AQP4 null mice (Fig. 4C). AQP4 deletion in mice is thus neuroprotective in a transient ischemia model of retinal injury, though the precise neuroprotective mechanism(s) remain to be established.
6 AQPs in Angiogenesis and Cell Migration
Phenotype analysis of AQP1 null mice led to discovery of AQP involvement in cell migration. Based on the expression of AQP1 in tumor microvessels (Endo et al. 1999), we tested the possible involvement of AQP1 in tumor angiogenesis (Saadoun et al. 2005a). We found that AQP1 deletion in mice reduces tumor growth following subcutaneous injection of melanoma cells (Fig. 5a), which was associated with increased tumor necrosis and reduced blood vessel formation within the tumor bed. In experiments designed to elucidate the mechanism of defective tumor angiogenesis in AQP1 deficiency, we found that cultured aortic endothelial cells from AQP1 null mice migrate more slowly towards a chemotactic stimulus compared with AQP1-expressing endothelial cells (Fig. 5b). Figure 5c shows increased movement of cells expressing AQP1. Interestingly, in migrating cells, AQP1 becomes polarized to the front end of cells (Fig. 5d) and is associated with increased turnover of cell membrane protrusions, suggesting an important role for AQPs at the leading edge of migrating cells.
Fig. 5.
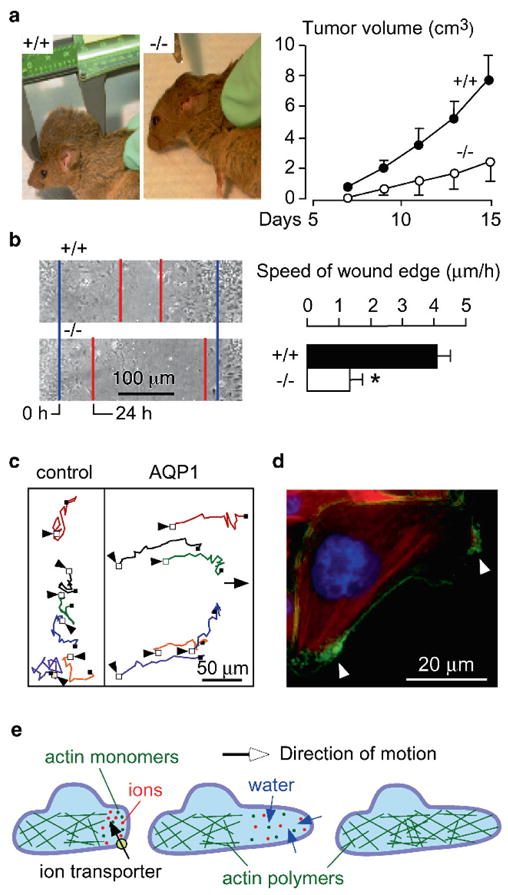
Impaired tumor growth and endothelial cell migration in AQP1 null mice. (a) (left) Tumor in a wildtype vs. AQP1 null mouse, 2 weeks after subcutaneous injection of 106 B16F10 melanoma cells. (right) Tumor growth data (ten mice per group, S.E., P < 0.001). (b) (left) Wound healing of cultured endothelial cells (initial wound edge blue, after 24 h red). (right) Wound edge speed (n = 4 per group, S.E., *P < 0.01). (c) Tracks of six migrating CHO cells expressing AQP1 vs. six non-AQP expressing control CHO cells, tracked for 4 h. Initial cell positions indicated by arrows. (d) AQP1 protein (green) polarization to lamellipodia (arrows) in a migrating CHO cell. (e) Proposed mechanism of AQP-facilitated endothelial cell migration: Actin de-polymerization and ion movements increase osmolality at the anterior end of the cell. Water entry increases local hydrostatic pressure, producing cell membrane expansion to form a protrusion. Actin re-polymerizes stabilizing the protrusion. Adapted from Saadoun et al. (2005a)
Follow-up experiments showed that AQPs facilitate cell migration independent of AQP and cell types. AQP4 facilitates astrocyte cell migration (Saadoun et al. 2005b; Auguste et al. 2007), AQP3 facilitates migration of corneal epithelial cells (Levin and Verkman 2006) and epidermal cells (Hara-Chikuma and Verkman 2008a), and AQP1 facilitates the migration of cultured renal proximal tubule cells (Hara-Chikuma and Verkman 2006), B16F10 melanoma and 4T1 breast cancer cells (Hu and Verkman 2006). In addition to defective angiogenesis in AQP1 deficiency, other consequences of AQP-facilitated cell migration include AQP involvement in tumor spread, glial scar formation, and wound healing. We have obtained evidence for each of these processes: (a) AQP1 expression in tumor cells increases their migration across endothelial barriers, local invasiveness and metastatic potential (Hu and Verkman 2006); (b) AQP4 deletion in glial cells reduces their migration toward a wound in vivo (Auguste et al. 2007) and the rate of glial scar formation (Saadoun et al. 2005b); and (c) AQP3 deletion impairs closure of cutaneous wounds (Hara-Chikuma et al. 2005) and corneal wounds (Levin and Verkman 2006). Possible involvement of AQP-dependent cell migration in other processes, such as organ regeneration and leukocyte chemotaxis, remain to be tested.
The mechanisms by which AQPs enhance cell migration are under investigation. The enhanced cell migration found for multiple structurally different AQPs, independent of their modulation method (transfection, knock-out, RNA inhibition), suggests that AQP-facilitated transmembrane water transport is the responsible mechanism. One mechanism by which AQPs may accelerate cell migration is by facilitating rapid changes in cell volume, which accompany changes in cell shape as cells squeeze through the irregularly shaped extracellular space. Water flow across the cell membrane may also allow migrating cells to generate hydrostatic forces that ‘push apart’ adjacent stationary cells. This mechanism, however, does not account for the polarization of AQPs to the front end of migrating cells or for AQP enhancement of lamellipodial dynamics. These observations support a role for water movement into and out of the leading edge during cell migration, as was proposed previously (Condeelis 1993). According to this hypothesis, actin de-polymerization and ion influx increase cytoplasmic osmolality at the front end of the migrating cell (Fig. 5e). These localized changes in cytoplasmic osmolality drive water influx through the plasma membrane. Consistent with the idea that water flows into and out of migrating cells are reports that migration can be inhibited or accelerated by changing the osmolality of the extracellular medium. We proposed that water influx thus causes expansion of the adjacent plasma membrane by increased local hydrostatic pressure, which is followed by rapid actin re-polymerization to stabilize the cell membrane protrusion. Recent evidence shows that regional hydrostatic pressure changes within cells do not equilibrate throughout the cytoplasm on scales of 10μm and 10 s (Charras et al. 2005) and could thus contribute to the formation of localized cell membrane protrusions. Direct measurements of water flow across the leading edge of migrating cells are needed for validation of these ideas.
7 Physiological Roles for Glycerol Transport by Aquaglyceroporins
For many years the physiological significance of glycerol transport by a subset of the AQPs, the aquaglyceroporins, was unclear. Phenotype studies of aquaglyceroporin knockout mice have addressed this question, producing a number of remarkable findings. There is now strong evidence for involvement of AQP3 in epidermal biology and cell proliferation and of AQP7 in adipocyte metabolism. A recent report on the phenotype of AQP9 null mice showed a subtle phenotype suggestive of impaired hepatic glycerol uptake (Rojek et al. 2007), though the mechanism remains to be established as does its proposed significance to diabetes.
7.1 AQP3 and Skin Function
The most superficial layer of skin is the stratum corneum (SC), which consists of terminally differentiated keratinocytes (corneocytes) that originate from actively proliferating keratinocytes in lower epidermis and contain a lamellar lipid layer (Fig. 6a). Hydration of the SC is an important determinant of skin appearance and physical properties and depends on a number of factors including the external humidity and its structure, lipid/protein composition, barrier properties and concentration of water-retaining osmolytes.
Fig. 6.
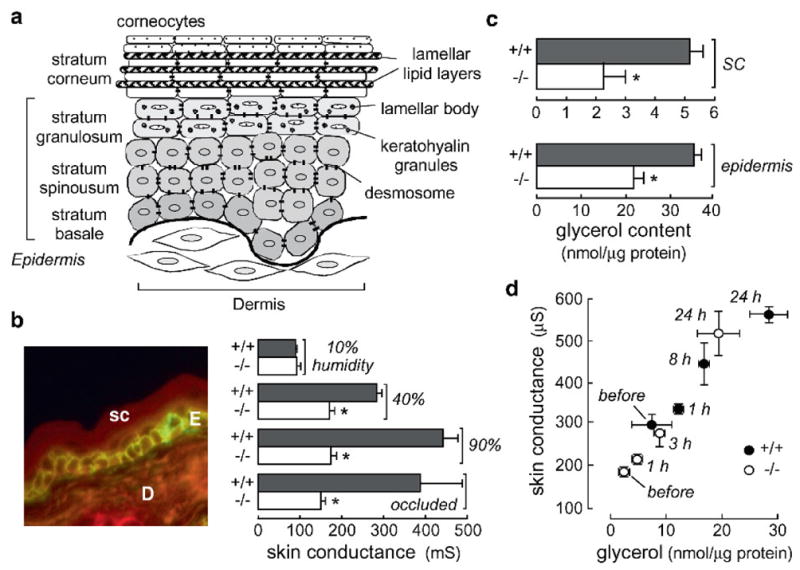
Reduced skin hydration in AQP3 deficiency. (a) Schematic showing stratum corneum and epidermal layers. (b) (left) Immunofluorescence showing AQP3 in mouse epidermal cells. E epidermis; D dermis; sc stratum corneum. (right) High-frequency superficial skin surface conductance in dorsal skin of hairless wildtype and AQP3 null mice (S.E., 20 mice per group). Skin conductance measured after 24 h exposure to relative humidity of 10, 40, or 90%; ‘occluded’ indicates a plastic occlusion dressing that prevents evaporative water loss (S.E., five mice per group). (c) Glycerol content measured in SC and epidermis (S.E., *P < 0.01). (d) Correlation between SC glycerol content and skin conductance for wildtype (filled circles) and AQP3 null (open circles) mice in a 90% humidity atmosphere for indicated times. Mice were given glycerol orally ad libitum as their only fluid source. From Hara et al. (2002), Ma et al. (2002) and Hara and Verkman (2003)
Phenotype analysis of AQP3 deficient mice has provided compelling evidence for a role of AQP3 in epidermal biology. AQP3 is expressed strongly in the basal layer of kerinocytes in mammalian skin (Fig. 6b, left). Figure 6b (right) shows reduced SC hydration in AQP3 null mice as measured by high frequency skin conductance (Ma et al. 2002), which is a linear index of SC water content. Exposure of mice to high humidity or occlusion increased SC hydration in wildtype, but not AQP3 null mice, indicating that water transport through AQP3 is not a rate-limiting factor in transepidermal water loss. If reduced SC hydration is related to a balance between evaporative water loss from the SC and water replacement through AQP3-containing basal keratinocytes, then preventing water loss by high humidity or occlusion should have corrected the defect in SC hydration in the AQP3 null mice. Further skin phenotype analysis indicated delayed barrier recovery after SC removal by tape-stripping in AQP3 null mice, as well as decreased skin elasticity and delayed wound healing (Hara et al. 2002).
Many types of experiments were done to investigate the mechanism by which AQP3 deficiency produces the pleotropic defects in skin function. A systematic analysis of the ultrastructure and composition of the epidermis and stratum corneum in AQP3 deficient mice revealed reduced glycerol content in SC and epidermis (Fig. 6c), with normal glycerol in dermis and serum; suggesting reduced glycerol transport from blood into the epidermis in AQP3 deficiency through the basal keratinocytes. No significant differences in wildtype vs. AQP3 deficient mice were found in SC structure, cell turnover, lipid profile, protein content and the concentrations of amino acids, ions and other small solutes (Hara et al. 2002). From these observations it was postulated that reduced epidermal and SC glycerol content was responsible for the abnormal skin phenotype in AQP3 null mice. Because glycerol is a ‘natural moisturizing factor’, reduced SC glycerol is predicted to reduce SC hydration and skin elasticity; because of the biosynthetic role of glycerol in the epidermis, reduced epidermal glycerol is predicted to delay barrier recovery function and wound healing. In support of this hypothesis, it was found that glycerol replacement by topical or systemic routes corrected each of the phenotype abnormalities in AQP3 null mice (Hara and Verkman 2003). SC glycerol content and water content, as assessed by skin conductance, correlated well for mice placed in a 90% humidity atmosphere and given oral glycerol (Fig. 6d). Further, glycerol transport from blood into the epidermis and SC was found to be reduced in AQP3 deficiency, suggesting impaired glycerol transport into the epidermis and SC through the relatively glycerol impermeable basal keratinocyte layer resulting in reduced steady-state epidermal and SC glycerol content. These findings indicated an important role for AQP3 and glycerol in epidermal function, providing a rational scientific basis for the longstanding practice of including glycerol in cosmetic and skin medicinal preparations.
7.2 AQP3 and Cell Proliferation
We recently discovered a remarkable phenotype in AQP3 null mice – resistance to the development of skin tumors (Hara-Chikuma and Verkman 2008b). Our motivation for studying AQP3 and skin tumors was the strong expression of AQP3 in basal cells in human skin squamous cell carcinomas. Figure 7a shows one of many such examples. Also, we recently discovered proliferation defects in AQP3 knockout mice in normally AQP3-expressing cells in cornea (Levin and Verkman 2006), resulting in delayed healing of corneal wounds, in colon (Thiagarajah et al. 2007), producing severe colitis in experimental models, and skin (Hara-Chikuma and Verkman 2008a), slowing cutaneous wound healing.
Fig. 7.
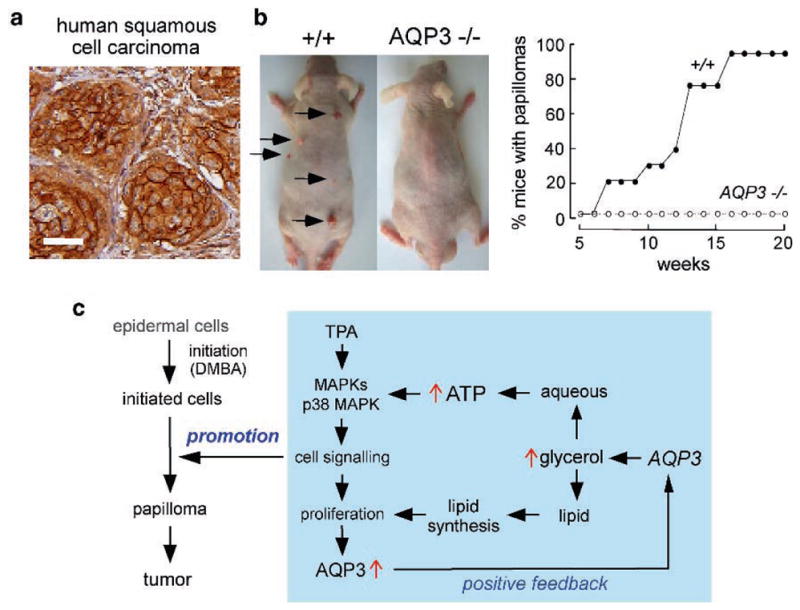
AQP3 expression in human squamous cell carcinoma and protection against cutaneous papillomas in AQP3-null mice. (a) AQP3 immunostaining in human skin squamous cell carcinoma. Bar, 50μm. (b) (left) Dorsal skin of mice was treated with a single application of DMBA, followed by twice-weekly applications of TPA for 20 weeks. Representative photographs showing multiple papillomas in wildtype mouse but no papillomas in AQP3 null mouse. (right) Percentage of mice with papillomas. (c) Proposed cellular mechanism of AQP3-facilitated tumorigenesis. Adapted from Hara-Chikuma and Verkman (2008b)
An established multistage carcinogenesis model was used to study skin tumor formation, which involves skin exposure to a tumor initiator and phorbol ester promoter. The remarkable phenotype finding, as shown in Fig. 7b was the absence of cutaneous papillomas in AQP3 knockout mice, whereas multiple tumors were produced in wild-type mice (Hara-Chikuma and Verkman 2008b). Experiments to establish the cellular mechanisms responsible for the impaired tumorigenesis phenotype showed impaired promoter-induced cell proliferation in AQP3-null or knock-down keratinocytes. AQP3-deficient keratinocytes had reduced content of glycerol, its metabolite glycerol-3-phosphate, and ATP, without impairment of mitochondrial function. Glycerol supplementation or AQP3 adenoviral infection (but not AQP1 adenoviral infection) corrected the defects in keratinocyte proliferation and reduced ATP generation. Further studies revealed correlations between cell proliferation and ATP and glycerol content. As diagrammed in Fig. 7c, we propose that AQP3-facilitated glycerol transport is an important determinant of epidermal cell proliferation and tumorigenesis by a mechanism in which cellular glycerol is a key regulator of cellular ATP energy. The mechanism also shows glycerol biosynthetic incorporation into lipids, ATP-facilitated MAP kinase signaling and positive feedback in which cell proliferation increases AQP3 expression. It is not known whether this mechanism applies to non-epidermal, AQP3-expressing cells and to non-skin cancers. Our findings also raise potential concerns in the use of recently marketed cosmetics containing ingredients that increase epidermal AQP3 expression whose goal is to improve skin moisture and appearance.
7.3 AQP7 and Fat Metabolism
We discovered a remarkable phenotype in AQP7 null mice (Hara-Chikuma et al. 2005). Although wildtype and AQP7 mice grew at similar rates as assessed by mouse weight, AQP7 null mice had remarkably greater fat mass compared to wild-type mice as seen grossly (Fig. 8a, top). Fat mass from multiple sites was significantly elevated in both male and female AQP7 null mice at age 16 weeks. Epididymal fat mass was comparable in wildtype and AQP7 null mice until age 4 weeks, but became different as the mice aged (Fig. 8b). Histologically, adipocytes at 16 weeks were remarkably larger in AQP7 null mice than in wildtype mice (Fig. 8a, bottom), suggesting that the greater fat mass in the AQP7 null mice is a consequence of adipocyte hypertrophy. Adipocyte size was similar in young wildtype and AQP7 deficient mice.
Fig. 8.
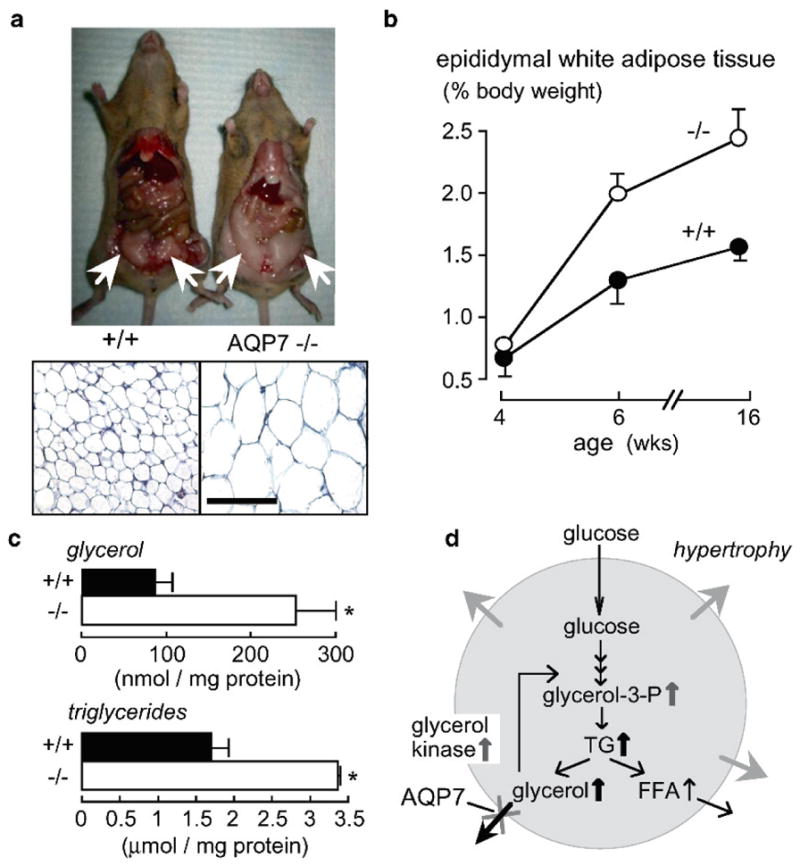
Progressive fat accumulation and adipocyte hypertrophy in AQP7 deficiency. (a) (top) Photographs of mice showing increased gonadal fat in AQP7 null mice at age 16 weeks (white arrows). (bottom) Histology of gonadal fat (stained with hematoxylin and eosin). Bar, 100μm. (b) Age-dependent epididymal fat mass (S.E., six mice per group). (c) Glycerol and triglyceride content in adipocytes from mice of age 16 weeks (S.E., *P < 0.01). (d) Proposed mechanism for adipocyte hypertrophy in AQP7 deficiency. See text for explanations. From Hara-Chikuma et al. (2005)
The concentrations of glycerol and triglycerides in serum were unaffected by AQP7 deletion, though adipocyte glycerol and triglyceride concentrations were significantly elevated in AQP7 null mice (Fig. 8c). To investigate the mechanism for the progressive adipocyte hypertrophy in AQP7 deficiency, measurements were made of adipocyte plasma membrane glycerol permeability, glycerol release, lipolysis and lipogenesis. Plasma membrane glycerol permeability was measured from the initial uptake of 14C-glycerol into isolated adipocytes from the younger wildtype and AQP7 null mice, where adipocyte size is comparable. 14C-glycerol uptake was reduced by ~threefold in AQP7 null mice compared to wildtype mice. Glycerol release was also significantly reduced in the AQP7 null mice, whereas lipolysis, as measured by free fatty acid release from isolated adipocytes, was similar in wild-type and AQP7 deficient mice. Also, lipogenesis was similar in the wildtype and knockout mice, as assayed from the incorporation of 14C-glucose into triglycerides. From these results we proposed a simple mechanism for progressive TG accumulation in AQP7 deficient adipocytes (Fig. 8d). Reduced plasma membrane glycerol permeability in AQP7 deficiency results in an increase in steady-state glycerol concentration in adipocyte cytoplasm. Increased adipocyte glycerol concentration would then increase glycerol 3-phosphate and hence triglyceride (TG) biosynthesis. Similar conclusions about fat metabolism in AQP7 deficiency were reported independently (Hibuse et al. 2005), though with some relatively minor differences in phenotype findings compared to our results. From these data, it was speculated that AQP7 plays an important role in the pathogenesis of human obesity (reviewed in Funahashi et al. 2006), though whether this is the case remains to be determined. Interestingly, a weak association of AQP7 polymorphisms with the risks of obesity and diabetes was reported (Prudente et al. 2007).
8 Summary and Perspective
The phenotype data on AQP-deficient mice suggests that AQP-facilitated water permeability is important: (a) when water movement is driven across a barrier by a continuous osmotic gradient (as in kidney collecting duct); (b) for active, near-isosmolar fluid absorption/secretion (as in kidney proximal tubule and salivary gland); (c) for neural signal transduction (as in brain and inner ear); and (d) for cell migration (as in tumor angiogenesis). Aquaglyceroporin-facilitated glyercol transport is important in skin hydration and cell proliferation (AQP3) and adipocyte metabolism (AQP7). It is likely that additional new roles of AQPs will be discovered. Much work remains to be done in following up the mouse phenotype observations, in discovering new roles of AQPs in mammalian physiology and in developing clinical therapies based on the new insights emerging from the mouse phenotype analyses. Additional data are needed to establish a firm mechanistic basis for AQP involvement in migration, neural signal transduction and in clearance of excess brain water in vasogenic edema, and in the precise role of the aquaglyceroporins in metabolism and cell proliferation. Small-molecule modulators of AQP expression/function could have clinical applications in the therapy of congestive heart failure and hypertension, cytotoxic and vasogenic brain edema, epilepsy, obesity, cancer, glaucoma and other conditions.
Acknowledgments
Supported by grants DK35124, EB00415, EY13574, HL59198, DK72517 and HL73856 from the National Institutes of Health and Research Development Program and Drug Discovery grants from the Cystic Fibrosis Foundation
References
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A. 2003a;100:2106–2111. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci U S A. 2003b;100:13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS. Greatly impaired migration of implanted aquaporin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Bai C, Fukuda N, Song Y, Ma T, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-1 and aquaporin-4 knockout mice. J Clin Invest. 1999;103:555–561. doi: 10.1172/JCI4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry V, Francis P, Kaushal S, Moore A, Bhattacharya S. Missense mutations in MIP underlie autosomal dominant ‘polymorphic’ and lamellar cataracts linked to 12q. Nat Genet. 2000;25:15–17. doi: 10.1038/75538. [DOI] [PubMed] [Google Scholar]
- Binder DK, Oshio K, Ma T, Verkman AS, Manley GT. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport. 2004a;15:259–262. doi: 10.1097/00001756-200402090-00009. [DOI] [PubMed] [Google Scholar]
- Binder DK, Papadopoulos MC, Haggie PM, Verkman AS. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci. 2004b;24:8049–8056. doi: 10.1523/JNEUROSCI.2294-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Yao X, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Bloch O, Papadopoulos MC, Manley GT, Verkman AS. Aquaporin-4 gene deletion in mice increases focal edema associated with brain abscess. J Neurochem. 2005;95:254–262. doi: 10.1111/j.1471-4159.2005.03362.x. [DOI] [PubMed] [Google Scholar]
- Bloch O, Manley GT, Verkman AS. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4 deficient mice. J Cereb Blood Flow Metab. 2006;26:1527–1537. doi: 10.1038/sj.jcbfm.9600306. [DOI] [PubMed] [Google Scholar]
- Charras GT, Yarrow JC, Horton MA, Mahadevan L, Mitchison TJ. Non-equilibration of hydrostatic pressure in blebbing cells. Nature. 2005;435:365–336. doi: 10.1038/nature03550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou CL, Ma T, Yang B, Knepper MA, Verkman AS. Fourfold reduction of water permeability in inner medullary collecting duct of aquaporin-4 knockout mice. Am J Physiol. 1998;274:C549–C554. doi: 10.1152/ajpcell.1998.274.2.C549. [DOI] [PubMed] [Google Scholar]
- Chou CL, Knepper MA, Hoek AN, Brown D, Yang B, Ma T, Verkman AS. Reduced water permeability and altered ultrastructure in thin descending limb of Henle in aquaporin-1 null mice. J Clin Invest. 1999;103:491–496. doi: 10.1172/JCI5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condeelis J. Life at the leading edge: the formation of cell protrusions. Annu Rev Cell Biol. 1993;9:411–444. doi: 10.1146/annurev.cb.09.110193.002211. [DOI] [PubMed] [Google Scholar]
- Da T, Verkman AS. Aquaporin-4 gene disruption in mice protects against impaired retinal function and cell death after ischemia. Invest Ophthalmol Vis Sci. 2004;45:4477–4483. doi: 10.1167/iovs.04-0940. [DOI] [PubMed] [Google Scholar]
- Deen PM, Verdijk MA, Knoers NV, Wieringa B, Monnens LA, van Os CH, van Oost BA. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994;264:92–95. doi: 10.1126/science.8140421. [DOI] [PubMed] [Google Scholar]
- Endo M, Jain RK, Witwer B, Brown D. Water channel (aquaporin 1) expression and distribution in mammary carcinomas and glioblastomas. Microvasc Res. 1999;58:89–98. doi: 10.1006/mvre.1999.2158. [DOI] [PubMed] [Google Scholar]
- Funahashi T, Nagasawa A, Hibuse T, Maeda N. Impact of glycerol gateway molecule in adipocytes. Cell Mol Biol (Noisy-le-grand) 2006;52:40–45. [PubMed] [Google Scholar]
- Hara M, Verkman AS. Glycerol replacement corrects defective skin hydration, elasticity, and barrier function in aquaporin-3-deficient mice. Proc Natl Acad Sci U S A. 2003;100:7360–7365. doi: 10.1073/pnas.1230416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Ma T, Verkman AS. Selectively reduced glycerol in skin of aquaporin-3-deficient mice may account for impaired skin hydration, elasticity, and barrier recovery. J Biol Chem. 2002;277:46616–46621. doi: 10.1074/jbc.M209003200. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M, Verkman AS. Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J Am Soc Nephrol. 2006;17:39–45. doi: 10.1681/ASN.2005080846. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M, Verkman AS. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J Mol Med. 2008a;86:221–231. doi: 10.1007/s00109-007-0272-4. [DOI] [PubMed] [Google Scholar]
- Hara-Chikuma M, Verkman AS. Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol Cell Biol. 2008b;28:328–332. doi: 10.1128/MCB.01482-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara-Chikuma M, Sohara E, Rai T, Ikawa M, Okabe M, Sasaki S, Uchida S, Verkman AS. Progressive adipocyte hypertrophy in aquaporin-7 deficient mice: Adipocyte glycerol permeability as a novel regulator of fat accumulation. J Biol Chem. 2005;280:15493–15496. doi: 10.1074/jbc.C500028200. [DOI] [PubMed] [Google Scholar]
- Hibuse T, Maeda N, Funahashi T, Yamamoto K, Nagasawa A, Mizunoya W, Kishida K, Inoue K, Kuriyama H, Nakamura T, Fushiki T, Kihara S, Shimomura I. Aquaporin 7 deficiency is associated with development of obesity through activation of adipose glycerol kinase. Proc Natl Acad Sci U S A. 2005;102:10993–10998. doi: 10.1073/pnas.0503291102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Verkman AS. Increased migration and metastatic potential of tumor cells expressing aquaporin water channels. FASEB J. 2006;20:1892–1894. doi: 10.1096/fj.06-5930fje. [DOI] [PubMed] [Google Scholar]
- King LS, Choi M, Fernandez PC, Cartron JP, Agre P. Defective urinary-concentrating ability due to a complete deficiency of aquaporin-1. N Engl J Med. 2001;345:175–179. doi: 10.1056/NEJM200107193450304. [DOI] [PubMed] [Google Scholar]
- Krane CM, Melvin JE, Nguyen HV, Richardson L, Towne JE, Doetschman T, Menon AG. Salivary acinar cells from aquaporin 5-deficient mice have decreased membrane water permeability and altered cell volume regulation. J Biol Chem. 2001;276:23413–23420. doi: 10.1074/jbc.M008760200. [DOI] [PubMed] [Google Scholar]
- Levin MH, Verkman AS. Aquaporin-dependent water permeation at the mouse ocular surface: in vivo microfluorimetric measurements in cornea and conjunctiva. Invest Ophthalmol Vis Sci. 2004;45:4423–4432. doi: 10.1167/iovs.04-0816. [DOI] [PubMed] [Google Scholar]
- Levin MH, Verkman AS. Aquaporin-3-dependent cell migration and proliferation during corneal re-epithelialization. Invest Ophthalmol Vis Sci. 2006;47:4365–4372. doi: 10.1167/iovs.06-0335. [DOI] [PubMed] [Google Scholar]
- Li J, Verkman AS. Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem. 2001;276:31233–31237. doi: 10.1074/jbc.M104368200. [DOI] [PubMed] [Google Scholar]
- Li J, Patil RV, Verkman AS. Mildly abnormal retinal function in transgenic mice without Müller cell aquaporin-4 water channels. Invest Ophthalmol Vis Sci. 2002;43:573–579. [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Severely impaired urinary concentrating ability in transgenic mice lacking aquaporin-1 water channels. J Biol Chem. 1998;273:4296–4299. doi: 10.1074/jbc.273.8.4296. [DOI] [PubMed] [Google Scholar]
- Ma T, Song Y, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Defective secretion of saliva in transgenic mice lacking aquaporin-5 water channels. J Biol Chem. 1999;274:20071–20074. doi: 10.1074/jbc.274.29.20071. [DOI] [PubMed] [Google Scholar]
- Ma T, Fukuda N, Song Y, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-5 knockout mice. J Clin Invest. 2000a;105:93–100. doi: 10.1172/JCI8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Song Y, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc Natl Acad Sci U S A. 2000b;97:4386–4391. doi: 10.1073/pnas.080499597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Hara M, Sougrat R, Verbavatz JM, Verkman AS. Impaired stratum corneum hydration in mice lacking epidermal water channel aquaporin-3. J Biol Chem. 2002;277:17147–17153. doi: 10.1074/jbc.M200925200. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Mhatre AN, Stern RE, Li J, Lalwani AK. Aquaporin 4 expression in the mammalian inner ear and its role in hearing. Biochem Biophys Res Commun. 2002;297:987–996. doi: 10.1016/s0006-291x(02)02296-9. [DOI] [PubMed] [Google Scholar]
- Moore M, Ma T, Yang B, Verkman AS. Tear secretion by lacrimal glands in transgenic mice lacking water channels AQP1, AQP3, AQP4 and AQP5. Exp Eye Res. 2000;70:557–562. doi: 10.1006/exer.1999.0814. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshio K, Watanabe H, Song Y, Verkman AS, Manley GT. Reduced cerebrospinal fluid production and intracranial pressure in mice lacking choroid plexus water channel aquaporin-1. FASEB J. 2005;19:76–78. doi: 10.1096/fj.04-1711fje. [DOI] [PubMed] [Google Scholar]
- Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nature Meth. 2005;2:825–827. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]
- Pallone TL, Edwards A, Ma T, Silldorff EP, Verkman AS. Requirement of aquaporin-1 for NaCl-driven water transport across descending vasa recta. J Clin Invest. 2000;105:215–222. doi: 10.1172/JCI8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem. 2005;280:13906–13912. doi: 10.1074/jbc.M413627200. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch. 2008;456:693–700. doi: 10.1007/s00424-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudente S, Flex E, Morini E, Turchi F, Capponi D, De Cosmo S, Tassi V, Guida V, Avogaro A, Folli F, Maiani F, Frittitta L, Dallapiccola B, Trischitta V. A functional variant of the adipocyte glycerol channel aquaporin 7 gene is associated with obesity and related metabolic abnormalities. Diabetes. 2007;56:1468–1474. doi: 10.2337/db06-1389. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci U S A. 1998;95:11981–11986. doi: 10.1073/pnas.95.20.11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojek AM, Skowronski MT, Füchtbauer EM, Füchtbauer AC, Fenton RA, Agre P, Frøkiaer J, Nielsen S. Defective glycerol metabolism in aquaporin 9 (AQP9) knockout mice. Proc Natl Acad Sci U S A. 2007;104:3609–3614. doi: 10.1073/pnas.0610894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Verkman AS. Accelerated cataract formation and reduced lens epithelial water permeability in aquaporin-1deficient mice. Invest Opthalmol Vis Sci. 2006;47:3960–3967. doi: 10.1167/iovs.06-0229. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Zhang H, Verkman AS. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 K+ channels in retinal Müller cells. J Biol Chem. 2007;282:21866–21872. doi: 10.1074/jbc.M703236200. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted of aquaporin-1 gene disruption. Nature. 2005a;434:786–792. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005b;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Schnermann J, Chou CL, Ma T, Traynor T, Knepper MA, Verkman AS. Defective proximal tubular fluid reabsorption in transgenic aquaporin-1 null mice. Proc Natl Acad Sci U S A. 1998;95:9660–9664. doi: 10.1073/pnas.95.16.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohara E, Rai T, Miyazaki J, Verkman AS, Sasaki S, Uchida S. Defective water and glycerol transport in the proximal tubules of AQP7 knockout mice. Am J Physiol. 2005;289:F1195–F1200. doi: 10.1152/ajprenal.00133.2005. [DOI] [PubMed] [Google Scholar]
- Solenov E, Watanabe H, Manley GT, Verkman AS. Sevenfold-reduced osmotic water permeability in primary astrocyte cultures from AQP-4-deficient mice, measured by a fluorescence quenching method. Am J Physiol. 2004;286:C426–C432. doi: 10.1152/ajpcell.00298.2003. [DOI] [PubMed] [Google Scholar]
- Song Y, Verkman AS. Aquaporin-5 dependent fluid secretion in airway submucosal glands. J Biol Chem. 2001 doi: 10.1074/jbc.M107257200. [DOI] [PubMed] [Google Scholar]
- Song Y, Fukuda N, Bai C, Ma T, Matthay MA, Verkman AS. Role of aquaporins in alveolar fluid clearance in neonatal and adult lung, and in oedema formation following acute lung injury: studies in transgenic aquaporin null mice. J Physiol. 2000a;525(Pt 3):771–779. doi: 10.1111/j.1469-7793.2000.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Yang B, Matthay MA, Ma T, Verkman AS. Role of aquaporin water channels in pleural fluid dynamics. Am J Physiol. 2000b;279:C1744–C1750. doi: 10.1152/ajpcell.2000.279.6.C1744. [DOI] [PubMed] [Google Scholar]
- Song Y, Sonawane N, Verkman AS. Localization of aquaporin-5 in sweat glands and functional analysis using knockout mice. J Physiol. 2002;541:561–568. doi: 10.1113/jphysiol.2001.020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajah JR, Verkman AS. Aquaporin deletion in mice reduces corneal water permeability and delays restoration of transparency after swelling. J Biol Chem. 2002;277:19139–19144. doi: 10.1074/jbc.M202071200. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Papadopoulos MC, Verkman AS. Non-invasive early detection of brain edema in mice by near-infrared light scattering. J Neurosci Res. 2005;80:293–299. doi: 10.1002/jnr.20439. [DOI] [PubMed] [Google Scholar]
- Thiagarajah JR, Zhao D, Verkman AS. Impaired enterocyte proliferation in aquaporin-3 deficiency in mouse models of colitis. Gut. 2007;56:1529–1535. doi: 10.1136/gut.2006.104620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbavatz JM, Ma T, Gobin R, Verkman AS. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J Cell Sci. 1997;110:2855–2860. doi: 10.1242/jcs.110.22.2855. [DOI] [PubMed] [Google Scholar]
- Verkman AS. Role of aquaporins in lung fluid physiology. Resp Physiol Neurobiol. 2007;159:324–330. doi: 10.1016/j.resp.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS. Dissecting the role of aquaporins in renal pathophysiology using transgenic mice. Semin Nephrol. 2008;28:217–226. doi: 10.1016/j.semnephrol.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS, Thiagarajah JR. Physiology of water transport in the gastrointestinal tract. In: Johnson LR, Barrett K, Ghishan F, Manchant J, Said H, Wood J, editors. Physiology of the gastrointestinal tract. Vol. 4. New York: Academic Press; 2006. pp. 1827–1845. [Google Scholar]
- Yang B, Brown D, Verkman AS. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem. 1996;271:4577–4580. [PubMed] [Google Scholar]
- Yang B, Folkesson HG, Yang J, Matthay MA, Ma T, Verkman AS. Reduced osmotic water permeability of the peritoneal barrier in aquaporin-1 knockout mice. Am J Physiol. 1999;276:C76–C81. doi: 10.1152/ajpcell.1999.276.1.C76. [DOI] [PubMed] [Google Scholar]
- Yang B, Verbavatz JM, Song Y, Vetrivel L, Manley G, Kao WM, Ma T, Verkman AS. Skeletal muscle function and water permeability in aquaporin-4 deficient mice. Am J Physiol. 2000;278:C1108–C1115. doi: 10.1152/ajpcell.2000.278.6.C1108. [DOI] [PubMed] [Google Scholar]
- Yang B, Song Y, Zhao D, Verkman AS. Phenotype analysis of aquaporin-8 null mice. Am J Physiol. 2005;288:C1161–C1170. doi: 10.1152/ajpcell.00564.2004. [DOI] [PubMed] [Google Scholar]
- Zador Z, Magzoub M, Jin S, Manley GT, Papadopoulos MC, Verkman AS. Microfiberoptic fluorescence photobleaching reveals size-dependent macromolecule diffusion in extracellular space deep in brain. FASEB J. 2008;22:326–332. doi: 10.1096/fj.07-9468com. [DOI] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci. 2008;37:1–10. doi: 10.1016/j.mcn.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Vetrivel L, Verkman AS. Aquaporin deletion in mice reduces intraocular pressure and aqueous fluid production. J Gen Physiol. 2002;119:561–569. doi: 10.1085/jgp.20028597. [DOI] [PMC free article] [PubMed] [Google Scholar]