

Abstract
Objective
To characterize the shape of the trajectories of Alzheimer’s Disease (AD) biomarkers as a function of MMSE.
Design
Longitudinal registries from the Mayo Clinic and the Alzheimer’s Disease Neuroimaging Initiative (ADNI).
Patients
Two different samples (n=343 and n=598) were created that spanned the cognitive spectrum from normal to AD dementia. Subgroup analyses were performed in members of both cohorts (n=243 and n=328) who were amyloid positive at baseline.
Main Outcome Measures
The shape of biomarker trajectories as a function of MMSE, adjusted for age, was modeled and described as baseline (cross-sectional) and within-subject longitudinal effects. Biomarkers evaluated were cerebro spinal fluid (CSF) Aβ42 and tau; amyloid and fluoro deoxyglucose position emission tomography (PET) imaging, and structural magnetic resonance imaging (MRI).
Results
Baseline biomarker values generally worsened (i.e., non-zero slope) with lower baseline MMSE. Baseline hippocampal volume, amyloid PET and FDG PET values plateaued (i.e., non-linear slope) with lower MMSE in one or more analyses. Longitudinally, within-subject rates of biomarker change were associated with worsening MMSE. Non-constant within-subject rates (deceleration) of biomarker change were found in only one model.
Conclusions
Biomarker trajectory shapes by MMSE were complex and were affected by interactions with age and APOE status. Non-linearity was found in several baseline effects models. Non-constant within-subject rates of biomarker change were found in only one model, likely due to limited within-subject longitudinal follow up. Creating reliable models that describe the full trajectories of AD biomarkers will require significant additional longitudinal data in individual participants.
Keywords: Alzheimer’s disease biomarkers, Magnetic Resonance Imaging, cerebro spinal fluid, amyloid PET imaging, FDG PET imaging
BACKGROUND
The five most well established biomarkers of Alzheimer’s disease (AD) at this time can be divided into two major categories: 1) measures of brain Aβ amyloid deposition; these are cerebro spinal fluid (CSF) Aβ421–8 and position emission tomography (PET) amyloid imaging9–15 and, 2) measures of neuronal injury and degeneration; these are CSF tau (total and phosphorylated tau)1,2,4,5,16–18, fluoro deoxyglucose (FDG) PET19,20, and structural magnetic resonance imaging (MRI)21–26. Some of the authors recently proposed a hypothetical model describing the temporal evolution of these five biomarkers over the entire adult lifespan of an individual who develops AD dementia27. This model is based on the assumption that different AD biomarkers do not change in an identical fashion over time, but rather in an ordered and sequential manner, and likewise approach a pathological level in an ordered manner27–32. This was proposed as a hypothetical model with validation awaiting additional data.
This hypothetical model can be divided into two conceptual components, first is the order in which each biomarker significantly departs from normal (which was addressed in an earlier manuscript33). The second conceptual component, which is the subject of this paper, is the shape of the trajectory of each biomarker curve as the disease progresses. The trajectory shape can be envisioned from a plot of each biomarker where the horizontal axis represents clinical disease severity and the vertical axis represents the degree of abnormality of each biomarker, from most normal to most abnormal. In our model, we hypothesized that biomarker trajectories have a sigmoid shape27. For reasons described later, we did not directly test for sigmoid shaped trajectories in this paper. Rather, we evaluated the shape of biomarker curves by modeling the baseline and longitudinal within-subject rate of change in five AD biomarkers as a function of MMSE, adjusted for age, in two large cohorts separately for APOE ε4 non-carriers and carriers. Then, as illustrated in Figure 1, we tested for evidence of non-zero, non-linear, non-constant and interaction terms in baseline values and within-subject rates of biomarker change based on longitudinal values.
Figure 1.
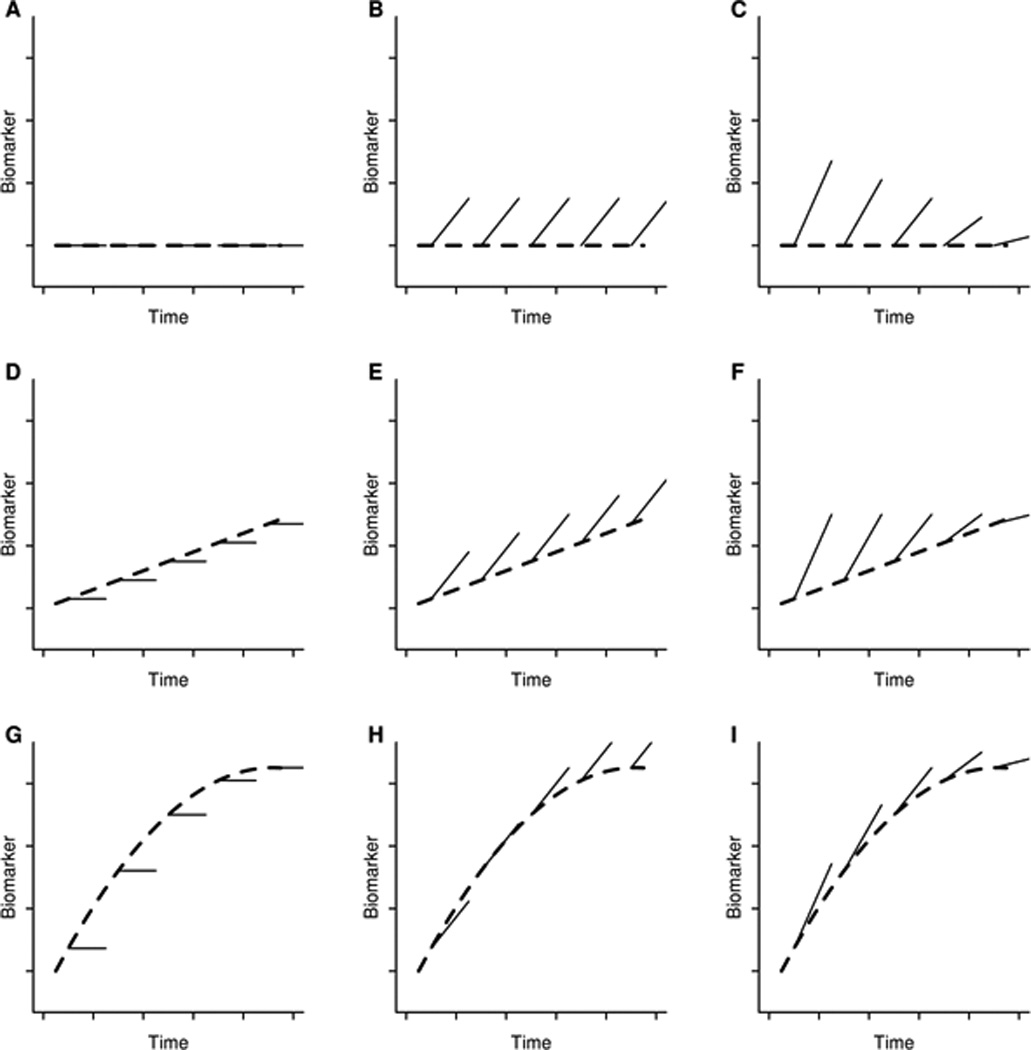
Prototypical linear mixed effects models based on different baseline shapes and longitudinal change. The dashed line in each panel characterizes the mean value of the biomarker at baseline as a function of disease severity while the solid lines characterize the mean within-subject rate of change. The dashed lines show either no baseline effect (flat line, Panels A, B, and C), a linear baseline effect (Panels D, E, and F), or a non-linear baseline effect which in this case is reaching an asymptote or saturation point (Panels G, H, and I). The solid lines show either no within-subject changes with increasing disease severity (flat lines, Panels A, D, and G), constant within-subject changes over time (parallel increasing lines, Panels B, E, and H), or non-constant within-subject changes which in this case are greater early in the disease and less when the disease becomes more severe (Panels C, F, and I).
METHODS
Participants and Diagnostic Evaluation
Two separate cohorts were created by pooling data from two Mayo Clinic studies and the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Participants at Mayo were recruited from the Mayo Clinic study of aging (MCSA), an epidemiologic cohort study of normal aging and mild cognitive impairment (MCI) in individuals aged 70–90 years in Rochester, Olmsted County, Minnesota34, and the Mayo Alzheimer’s disease research center (ADRC). For all participants, written informed consent was obtained for participation as approved by the local Institutional Review Boards.
At baseline, all participants met diagnostic criteria for cognitively normal (CN), MCI, or AD dementia35. Clinical disease severity was scored with the Mini Mental State Exam (MMSE)36. For Mayo Clinic participants, a 38-point test, the Short Test of Mental Status (STMS)37, was converted to MMSE scores using an algorithm developed at our center38. STMS values transformed to MMSE scores are reported simply as MMSE throughout the manuscript.
While we wished to maximize sample size we also recognized that all participants within a cohort must have every biomarker test to perform valid within-cohort comparisons. None of the Mayo Clinic participants had CSF samples taken, while many more Mayo than ADNI participants had amyloid PET studies available. We therefore created two cohorts, whom we refer to as the CSF/MRI cohort and the PET/MRI cohort. The CSF/MRI cohort included only ADNI participants and was used to evaluate trajectories of hippocampal volume, CSF Aβ42, and CSF t-tau. The PET/MRI cohort included both ADNI and Mayo participants and was used to evaluate trajectories of hippocampal volume, amyloid PET with Pittsburgh Compound B (PIB), and FDG-PET. Only visits with all biomarkers available were used in analysis.
CSF Methods
CSF was analyzed using a multiplex xMAP Luminex platform (Luminex Corp, Austin, TX) with Innogenetics (INNO-BIA AlzBio3, Ghent, Belgium) immunoassay kit-based reagents5,39 (http://www.adni-info.org/index.php).
MRI Methods
ADNI participants received 1.5T MRI scans and Mayo participants were scanned at either 1.5T or 3T. We used a standard 3D magnetization prepared rapid acquisition gradient echo (MPRAGE) imaging sequence and standardized data post processing described in Jack, et al 200840. Hippocampal and total intracranial volumes (TIV) were measured at Mayo Clinic; the hippocampus using FreeSurfer software (version 4.5.0)41 and TIV using an in-house algorithm42. We evaluated the statistical agreement between FreeSurfer hippocampal volumes obtained at 1.5T versus 3T among 91 ADNI participants (32 CN, 39 MCI, 20 AD) who underwent MRI exams at both field strengths at the same visit. Lin’s concordance correlation coefficient (CCC), which measures agreement about the identity line43, was excellent (CCC = 0.98, p < 0.001).
PET Amyloid and FDG imaging Methods
PET Amyloid and FDG imaging methods were similar for Mayo and ADNI. Amyloid imaging was performed with11C Pittsburgh Compound B (PIB)44. Quantitative image analysis for both PIB and FDG was done using our in-house fully automated image processing pipeline described in45. PIB-PET ROIs were based on anatomically defined regions while FDG-PET ROIs were not. A global cortical PIB PET retention summary ratio was formed by combining the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus values and dividing the median value across all voxels in these cortical regions of interest (ROIs) by the median across all voxels in the cerebellum46,47. FDG PET scans were analyzed in a similar manner using medial parietal, angular gyrus and inferior temporal cortical ROIs as described in Landau, et al 201048 normalized to pons uptake.
Statistical Methods
We used linear mixed effect models49 to investigate the shape of the biomarker trajectories. For each biomarker, the model always included terms for baseline age, baseline MMSE, change-in-age from baseline, and change-in-MMSE from baseline. Using this model parameterization, the baseline MMSE term allowed us to assess the baseline, or cohort-level, relationship between biomarker and MMSE while the change in MMSE term allowed us to assess the within subject rates of biomarker change with worsening MMSE. The age terms were included as a necessary adjustment to impose the correct ordering of a subject’s visits over time.
As a first step in model fitting, baseline age and baseline MMSE were fit as a restricted cubic spline with knots at the 10th, 50th, and 90th percentiles. If the p-value from a likelihood ratio test comparing the spline fit to the linear fit was less than p=0.10, the non-linear effects were retained. Otherwise, they were kept as linear terms. We next fit a full model for each biomarker with all two-way interactions between baseline MMSE, baseline age, change in MMSE, and change in age to test for significant interactions but only retained interactions with p <0.10 in the final models. Interactions with baseline age and baseline MMSE or change in MMSE allowed us to assess if the biomarker-MMSE relationship depends on age. An interaction term with baseline MMSE and change in MMSE allowed us to assess if the within subject rates of biomarker change depend on the level of disease severity (i.e. are non-constant over disease severity). Nine possible prototype models based on different baseline shapes and longitudinal change are shown in Figure 1.
Random intercepts and slopes for change in age and change in MMSE were included when possible. All models were adjusted for sex. Hippocampal volume models were additionally adjusted for total intracranial volume (TIV). Because of the known effect of APOE on rates of cognitive change50; separate models were fit within APOE ε4 non carriers and carriers for each biomarker.
For each participant, all available time points where all biomarker tests were performed were used in the models. Because of differences in when each biomarker was collected, this reduced the amount of follow-up used for some biomarkers (MRI and FDG), but it was necessary to use only those visits where all biomarkers were available within a cohort so that all biomarkers would be evaluated on equal footing.
RESULTS
The CSF/MRI cohort (111 CN, 154 MCI, 78 AD) was composed entirely of ADNI participants; all 343 had baseline data and 262 had longitudinal data (Table 1). The PET/MRI cohort (429 CN, 129 MCI, 40 AD) was composed of Mayo and ADNI participants; all 598 had baseline data and 182 had longitudinal data (Table 1).
Table 1.
Descriptive characteristics of all participants
| Characteristic | All | CN | MCI | AD |
|---|---|---|---|---|
| CSF/MRI Cohort | ||||
| Number of participants | 343 | 111 | 154 | 78 |
| Age, years | 77 (73, 81) | 76 (72, 78) | 77 (73, 81) | 78 (73, 82) |
| Female gender, no. (%) | 134 (39.1) | 56 (50.5) | 45 (29.2) | 33 (42.3) |
| Education, years | 16 (14, 18) | 16 (14, 18) | 16 (14, 18) | 16 (12, 18) |
| APOE ε4 carriers, no. (%) | 160 (46.6) | 24 (21.6) | 81 (52.6) | 55 (70.5) |
| MMSE | 27 (25, 29) | 29 (29, 30) | 27 (25, 28) | 24 (22, 25) |
| Hippocampal volume, cm3 | 6.3 (5.5, 7.3) | 7.3 (6.9, 7.7) | 6.1 (5.4, 6.9) | 5.4 (4.9, 6.1) |
| Aβ1–42 | 152 (132, 223) | 220 (154, 245) | 144 (130, 183) | 142 (120, 155) |
| T-tau | 81 (57, 120) | 61 (50, 84) | 87 (64, 131) | 116 (69, 141) |
| Number with follow-up, (%) | 262 (76.4) | 88 (79.3) | 120 (77.9) | 54 (69.2) |
| 1 follow-up visit | 175 (66.8) | 56 (63.6) | 76 (63.3) | 43 (79.6) |
| 2 follow-up visits | 83 (31.7) | 30 (34.1) | 42 (35.0) | 11 (20.4) |
| 3 follow-up visits | 4 (1.5) | 2 (2.3) | 2 (1.7) | 0 (0) |
| Years of follow-up | 1.1 (1.1, 2.1) | 1.1 (1.1, 2.1) | 1.1 (1.1, 2.1) | 1.1 (1.0, 1.2) |
| PET/MRI Cohort | ||||
| Number of participants | 598 | 429 | 129 | 40 |
| ADNI participants, no. (%) | 81 (13.5) | 21 (4.9) | 44 (34.1) | 16 (40.0) |
| Age, years | 79 (76, 83) | 79 (76, 83) | 81 (75, 83) | 80 (76, 84) |
| Female gender, no. (%) | 257 (43.0) | 199 (46.4) | 44 (34.1) | 14 (35.0) |
| Education, years | 14 (12, 16) | 14 (12, 16) | 14 (12, 17) | 14 (12, 18) |
| APOE ε4 carriers, no. (%) | 205 (34.3) | 113 (26.3) | 61 (47.3) | 31 (77.5) |
| MMSE | 28 (27, 29) | 28 (27, 29) | 27 (24, 28) | 23 (21, 24) |
| Hippocampal volume, cm3 | 6.9 (6.3, 7.5) | 7.2 (6.6, 7.7) | 6.4 (5.7, 6.9) | 5.4 (4.5, 6.3) |
| PIB Ratio | 1.44 (1.33, 1.98) | 1.39 (1.32, 1.69) | 1.88 (1.39, 2.26) | 2.21 (1.90, 2.66) |
| FDG Ratio | 1.36 (1.25, 1.46) | 1.39 (1.29, 1.48) | 1.27 (1.18, 1.39) | 1.09 (1.01, 1.21) |
| Number with follow-up, (%) | 182 (30.4) | 110 (25.6) | 58 (45.0) | 14 (35.0) |
| 1 follow-up visit | 151 (83.0) | 99 (90.0) | 40 (69.0) | 12 (85.7) |
| 2 follow-up visits | 29 (15.9) | 11 (10.0) | 17 (29.3) | 1 (7.1) |
| 3 follow-up visits | 2 (1.1) | 0 (0) | 1 (1.7) | 1 (7.1) |
| Years of follow-up | 1.3 (1.1, 1.5) | 1.3 (1.2, 1.5) | 1.3 (1.1, 2.1) | 1.0 (1.0, 1.1) |
Median (Inter-quartile) range shown unless otherwise noted.
Abbreviations: APOE, apolippoprotein E; MMSE, Mini Mental State Exam
The data are summarized in Figures 2 and 3. Each biomarker value is plotted in its native units, with the vertical axis oriented so that increasing values correspond to worsening. Similarly, the x-axis is oriented so that left-to-right movement corresponds to worsening cognition. Each figure contains three columns with the left-most column showing the biomarker values for individual participants in a random subset of the same n=100 participants; a subset was used to reduce overlapping values and allow individual trajectories to be discerned. The middle and right-most columns illustrate the shape of the curve based on the baseline effects (dotted), and the within-subject rates of biomarker change based on longitudinal data (solid) vs. MMSE for APOE ε4 non carriers (middle column) and carriers (right column) from the model estimates. Within-subject rates of change are shown as an average change in biomarker for a three-point MMSE worsening. Non-zero terms indicate biomarker values that change with worsening MMSE. Non-linear terms indicate baseline effects that are non-linear (most often saturating or plateauing) with advancing MMSE. Interaction terms indicate the relationship between biomarker and MMSE depends on baseline age. Non-constant terms indicate the within-subject rate of change depends on baseline MMSE, or disease severity. Significant non-linear, interaction, and non-constant terms imply a non-zero relationship with biomarker and MMSE. Significant terms from the models summarized in Figures 2 and 3 are displayed in table form in Table 2.
Figure 2. CSF/MRI Cohort.
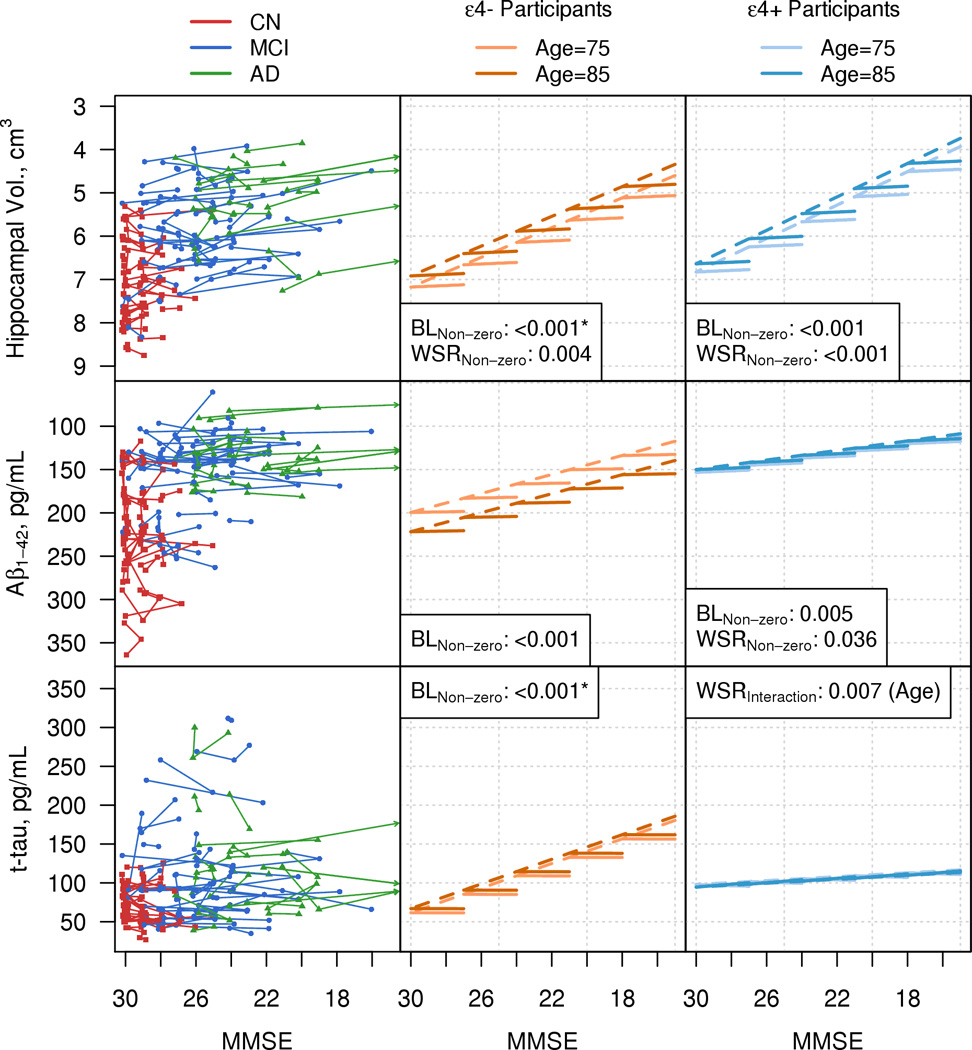
Individual trajectory plots by MMSE for hippocampal volume, CSF Aβ1–42, and CSF t-tau are plotted in the left column. Because of the large number of subjects, the left hand column illustrates a random subset of the MRI-CSF cohort. Plots in the middle and right hand columns are based on modeling the entire cohort. CN participants are represented with red squares, MCI participants with blue circles, and AD participants with green triangles. Arrows indicate trajectories that extend beyond the plotting region. The center and right columns are model summary plots in the MRI-CSF cohort for ε4 non carriers (center) and ε4 carriers (right). The dashed lines represent the baseline relationship with biomarker and MMSE estimated from the linear mixed effect models. The solid lines represent the within-subject rate of change in biomarker with a decrease in MMSE of 3 points. Light orange (ε4 non carriers) and light blue (ε4 carriers) lines represent the effects for a subject with a baseline age of 75 and dark orange (ε4 non carriers) and dark blue (ε4 carriers) lines represent the effects for a subject with a baseline age of 85. P-values are shown for all effects in the model with a p-value <0.10. BL indicates baseline biomarker and MMSE effects. These may be non-zero, non-linear, or interact with baseline age. WSR indicates within-subject rates of change in biomarker with worsening MMSE. These may be non-zero, interact with baseline age, or be non-constant such that the rate of change differs by baseline MMSE. Asterisks indicate p-values reported when the change in age and the change in MMSE are zero.
Figure 3. PET/MRI Cohort.
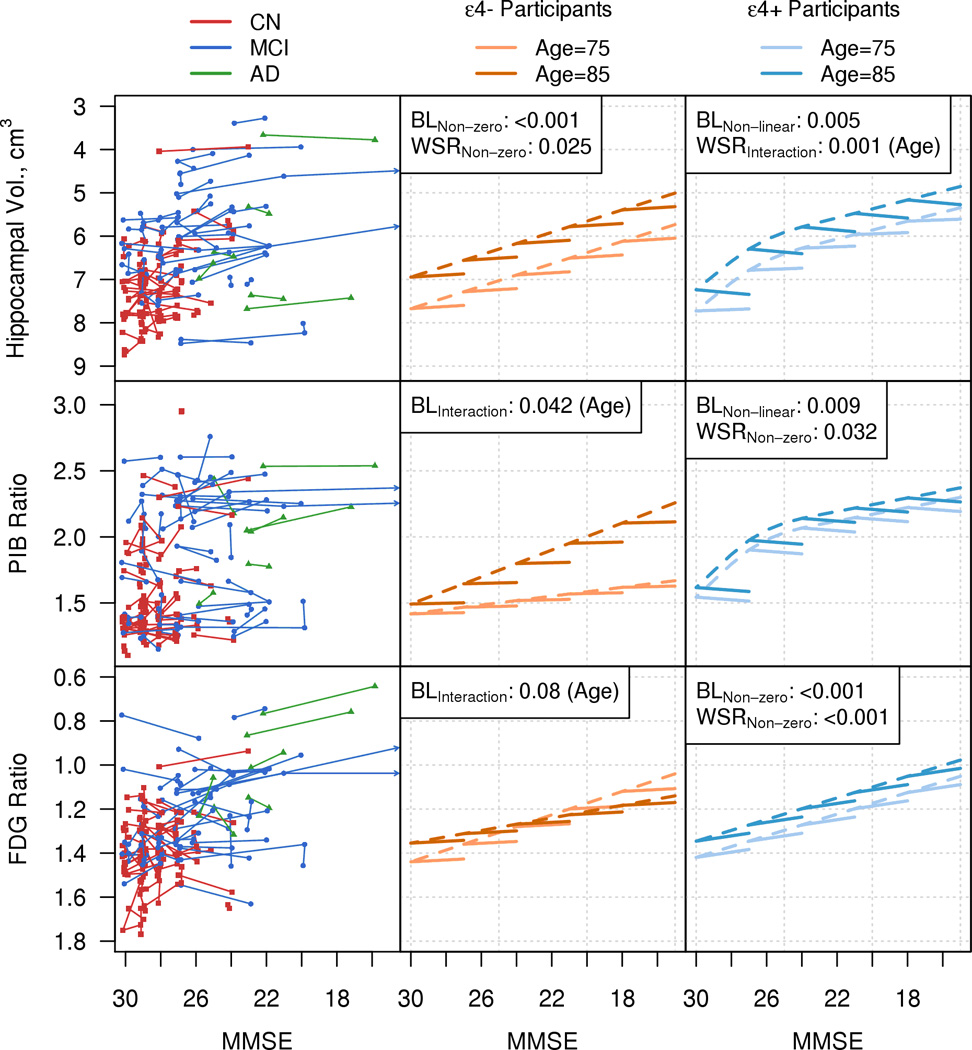
Individual trajectory plots by MMSE for hippocampal volume, PIB Ratio, and FDG Ratio in a random subset of the MRI-PET cohort. Model summary plots by MMSE for hippocampal volume, PIB Ratio, and FDG Ratio by APOE ε4 genotype. The organization of Fig. 3 is analogous to that of Fig 2.
Table 2.
Summary of all baseline and within-subject effects that were significant (p<0.10) in the linear mixed effects models
| Baseline MMSE Effects | Within-Subject MMSE Effects | |||||
|---|---|---|---|---|---|---|
| Non- Zero |
Non- Linear |
Inter- action |
Non- Zero |
Inter- action |
Non- Constant |
|
| ε4- Participants | ||||||
| Hippocampal vol. (CSF/MRI cohort) | x | x | ||||
| Aβ1–42 | x | |||||
| T-tau | x | |||||
| Hippocampal vol. (PET/MRI cohort) | x | x | ||||
| PIB | x | x | ||||
| FDG | x | x | ||||
| ε4+ Participants | ||||||
| Hippocampal vol. (CSF/MRI cohort) | x | x | ||||
| Aβ1–42 | x | x | ||||
| T-tau | x | x | ||||
| Hippocampal vol. (PET/MRI cohort) | x | x | x | x | ||
| PIB | x | x | x | |||
| FDG | x | x | ||||
Non-Zero: MMSE is associated with biomarker
Non-Linear: MMSE is non-linearly associated with biomarker
Interaction: MMSE association with biomarker depends on baseline age
Non-Constant: MMSE association with biomarker depends on baseline MMSE
Xs in the table indicate those effects that were significant (<0.10) in the linear mixed effects models
Figure 2 illustrates biomarkers vs. MMSE in the CSF/MRI cohort. Baseline effects: all biomarkers worsen (i.e., display non-zero slope) with lower MMSE scores at baseline (p≤0.005) except for t-tau in APOE ε4 positive participants (Table 2). A cross-sectional age effect in the CSF Aβ model was unexpected in that CSF Aβ values decrease/worsen as ages increase from 70 to 78 and then increase for older ages .Longitudinal effects: within-subject rates in hippocampal volume worsen (i.e. non-zero slope) as MMSE worsens in both ε4 negative and positive participants (p=0.004, p<0.001) (Table 2). Within-subject rates of Aβ42 worsen (i.e. non-zero slope) as MMSE worsens in ε4 positive participants only (p=0.036). The within-subject t-tau rate is changing as MMSE worsens (i.e., non-zero slope) for ε4 positive participants only (p=0.007) but the rate of change depends on baseline age.
Figure 3 biomarkers vs. MMSE in the PET/MRI cohort. Baseline effects: Hippocampal volumes in ε4 negative participants and FDG in ε4 positive participants worsen (i.e., non-zero slope) with lower MMSE scores (p<0.001) (Table 2). The relationship with biomarker and baseline MMSE for both PIB and FDG PET in ε4 negative participants depend on baseline age (p=0.042, p=0.08). Baseline hippocampal volume and amyloid PET values worsened in ε4 positive participants with lower MMSE but the effect plateaued at lower MMSE values (i.e. non-linear slope) (p=0.005, p=0.009). Longitudinal effects: within-subject rates of change in hippocampal volume worsen (i.e. non-zero slope) as MMSE worsens (p=0.025) in ε4 negative participants (Table 2). In ε4 positive participants, within-subject rates of change in hippocampal volume also worsen as MMSE worsens, but this effect depends on baseline age (p=0.001). Within-subject rates of change in PIB PET decrease (i.e., non-zero slope) as MMSE worsens in ε4 positive participants (0.032) but not in ε4 negative participants. Within-subject rates of FDG worsen as MMSE worsens (i.e. non-zero slope) in ε4 positive participants (p<0.001) but not in ε4 negative participants.
We performed a subgroup analysis restricted to participants who had evidence of amyloid deposition either by amyloid PET or CSF Aβ42 and thus are likely on the AD pathophysiological pathway. We selected PIB > 1.4 as the cut-point. This reflects a lenient cut-point but one that still likely eliminated individuals clearly not on the AD pathophysiological pathway. Using data from an analysis of 41subjects who had PIB and CSF on the same visit51 we used linear regression to identify the CSF cut-point that corresponds to PIB 1.4 as being 209 pg/ml. This resulted in an “amyloid positive” CSF cohort of 243 participants and a PET cohort of 328 participants (Table 3). Figure 3 illustrates biomarkers vs. MMSE in the amyloid positive participants. Because the range of amyloid values was truncated by design in this subgroup analysis, we did not create trajectory plots for PIB PET or CSF Aβ42 and limited the plots to MRI, CSF tau and FDG.
Table 3.
Descriptive characteristics of amyloid positive participants (PIB > 1.4 or CSF Aβ1–42 < 209 pg/ml)
| Characteristic | All | CN | aMCI | AD |
|---|---|---|---|---|
| CSF/MRI Cohort | ||||
| Number | 243 | 50 | 121 | 72 |
| Age, years | 77 (73, 80) | 77 (74, 78) | 76 (73, 80) | 78 (74, 81) |
| Female gender, no. (%) | 92 (37.9) | 25 (50.0) | 38 (31.4) | 29 (40.3) |
| Education, years | 16 (14, 18) | 16 (14, 18) | 16 (14, 18) | 16 (12, 18) |
| APOE ε4 positive, no. (%) | 148 (60.9) | 19 (38.0) | 74 (61.2) | 55 (76.4) |
| MMSE | 26 (24, 29) | 30 (29, 30) | 27 (25, 28) | 24 (22, 25) |
| Hippocampal volume, cm3 | 6.1 (5.3, 7.0) | 7.2 (6.9, 7.7) | 6.1 (5.5, 6.7) | 5.4 (4.9, 6.0) |
| Aβ1–42 | 141 (127, 155) | 149 (135, 175) | 138 (127, 152) | 140 (119, 151) |
| t-tau | 93 (66, 135) | 74 (53, 96) | 99 (70, 143) | 117 (74, 145) |
| Number with follow-up, (%) | 191 (78.6) | 38 (76.0) | 100 (82.6) | 53 (73.6) |
| 1 follow-up visit | 128 (67.0) | 23 (60.5) | 63 (63.0) | 42 (79.2) |
| 2 follow-up visits | 61 (31.9) | 15 (39.5) | 35 (35.0) | 11 (20.8) |
| 3 follow-up visits | 2 (1.0) | 0 (0) | 2 (2.0) | 0 (0) |
| Years of follow-up | 1.1 (1.0, 2.0) | 1.1 (1.1, 2.0) | 1.1 (1.1, 2.1) | 1.1 (1.0, 1.2) |
| PET/MRI Cohort | ||||
| Number | 328 | 196 | 95 | 37 |
| ADNI subjects, no. (%) | 56 (17.1) | 10 (5.1) | 32 (33.7) | 14 (37.8) |
| Age, years | 80 (76, 83) | 80 (76, 83) | 81 (75, 83) | 80 (76, 84) |
| Female gender, no. (%) | 139 (42.4) | 88 (44.9) | 37 (38.9) | 14 (37.8) |
| Education, years | 14 (12, 16) | 14 (12, 16) | 14 (12, 16) | 15 (12, 18) |
| APOE ε4 positive, no. (%) | 159 (48.5) | 76 (38.8) | 53 (55.8) | 30 (81.1) |
| MMSE | 27 (26, 29) | 28 (27, 29) | 27 (25, 27) | 23 (21, 24) |
| Hippocampal volume, cm3 | 6.7 (6.0, 7.4) | 7.1 (6.5, 7.7) | 6.3 (5.7, 6.8) | 5.4 (4.6, 6.3) |
| PIB Ratio | 1.90 (1.58, 2.26) | 1.77 (1.49, 2.07) | 2.14 (1.69, 2.32) | 2.32 (1.98, 2.66) |
| FDG Ratio | 1.32 (1.21, 1.42) | 1.36 (1.27, 1.46) | 1.27 (1.18, 1.39) | 1.08 (1.00, 1.23) |
| Number with follow-up, (%) | 100 (30.5) | 44 (22.4) | 43 (45.3) | 13 (35.1) |
| 1 follow-up visit | 80 (80.0) | 39 (88.6) | 30 (69.8) | 11 (84.6) |
| 2 follow-up visits | 19 (19.0) | 5 (11.4) | 13 (30.2) | 1 (7.7) |
| 3 follow-up visits | 1 (1.0) | 0 (0) | 0 (0) | 1 (7.7) |
| Years of follow-up | 1.3 (1.1, 1.6) | 1.3 (1.2, 1.4) | 1.3 (1.1, 2.1) | 1.0 (1.0, 1.1) |
Median (Inter-quartile) range shown unless otherwise noted.
Abbreviations: APOE, apolippoprotein E; MMSE, Mini Mental State Exam
Significant effects in the amyloid positive cohorts are summarized in Table 4. While many findings were the same between the amyloid positive cohorts and the entire sample there were some differences. In the amyloid positive MRI/CSF cohort, baseline hippocampal volume increased non-linearly – i.e., plateaued at lower MMSE values in ε4 carriers. Within subject hippocampal volume was non constant (rates of atrophy decelerated at higher MMSE) in ε4 non carriers. In the amyloid positive MRI/PET cohort, baseline hippocampal volume and FDG PET increased non-linearly – i.e., plateaued at lower MMSE values in ε4 non-carriers. Within subject change was non-zero for FDG PET in ε4 non-carriers. There was also an interaction between baseline age and baseline MMSE in the hippocampal volume model for the ε4 non-carriers.
Table 4.
Summary of all baseline and within-subject effects that were significant (p<0.10) in the linear mixed effects models among the amyloid positive subgroup
| Baseline MMSE Effects | Within-Subject MMSE Effects | |||||
|---|---|---|---|---|---|---|
| Non- Zero |
Non- Linear |
Inter- action |
Non- Zero |
Inter- action |
Non- Constant |
|
| ε4- Participants | ||||||
| Hippocampal vol. (CSF/MRI cohort) | X | X | X | |||
| T-tau | X | |||||
| Hippocampal vol. (PET/MRI cohort) | X | X | X | X | ||
| FDG | X | X | X | X | ||
| ε4+ Participants | ||||||
| Hippocampal vol. (CSF/MRI cohort) | X | X | X | |||
| T-tau | X | X | ||||
| Hippocampal vol. (PET/MRI cohort) | X | X | X | X | ||
| FDG | X | X | ||||
Non-Zero: MMSE is associated with biomarker
Non-Linear: MMSE is non-linearly associated with biomarker
Interaction: MMSE association with biomarker depends on baseline age
Non-Constant: MMSE association with biomarker depends on baseline MMSE
Xs in the table indicate those effects that were significant (p<0.10) in the linear mixed effects models
DISCUSSION
Our major findings were: (1) overall biomarker trajectory shapes were complex and were affected by interactions with age and APOE status. 2) Baseline biomarker values generally worsened (i.e., non-zero slope) with lower baseline MMSE. 3) Baseline hippocampal volume, amyloid PET and FDG PET values plateaued (i.e., non-linear slope) with lower MMSE in one or more analyses. 4) Longitudinally, within-subject rates of biomarker change were associated with worsening MMSE. 5) Non-constant within-subject rates of biomarker change were found in only one model; the rate of hippocampal volume change decelerated with worsening MMSE in amyloid positive e4 negative participants. 6) Trajectories for a given biomarker were often different in ε4 carriers vs non carriers in the overall samples. This was less often so in the amyloid positive sub samples. 7) While most findings were the same between the amyloid positive cohorts and the entire sample there was a slightly greater tendency toward non-linear baseline effects in amyloid positive participants.
Our hypothetical biomarker model27 predicts that each biomarker follows a sigmoid shaped trajectory. The rationale for this prediction starts with the assumption that the rate of change of a biomarker denoting accumulating AD pathophysiology in the brain should be zero from birth through at least early adulthood. At some point, e.g., age 50s – 70s, AD biomarkers deflect from the normal baseline and begin to become abnormal, which by definition represents acceleration in rate. Based on prior evidence that some biomarker rates of change (i.e., amyloid PET, CSF Aβ42 and t-tau) do not accelerate in the dementia phase of the disease31,52, we presumed that biomarker rates do not continue to accelerate indefinitely, but instead begin to saturate or plateau at some point, which represents deceleration. An initial period of acceleration followed later by deceleration defines a trajectory that is approximately sigmoidal, with the midpoint of the curve defined as the initiation of deceleration. A second reason to suspect that biomarkers should follow a sigmoid shaped trajectory relates to sensitivity limits of any measurement technique at extremes. Floor and ceiling measurement sensitivity effects impart a sigmoid shape to a data distribution.
Sources outside the field of human biomarker studies suggest that amyloid and neurodegenerative biomarkers might follow a sigmoid-shaped function. Inglesson, et al 200428 found in human autopsy studies that amyloid accumulation plateaus with increasing disease duration. Amyloid deposition in transgenic AD mice follows a sigmoidal-shaped function with advancing age53. Tau fibrillization follows a sigmoid shaped function with time in vitro54. A cumulative damage model of neurodegenerative disease where the risk of cell death in the vulnerable population of cells changes over time predicts a sigmoid-shaped trajectory of neurodegenerative brain atrophy55,56.
Reports from analyses of ADNI data draw somewhat inconsistent conclusions about the shapes of biomarker trajectories. Caroli, et al 201057 analyzed cross-sectional ADNI data and found that mean baseline hippocampal volume, CSF Aβ42, and CSF tau data could be better modeled as a function of worsening cognition with sigmoid-shaped curves compared to linear fits. Lo, et al 201158 examined rates of change of biomarkers in ADNI and illustrated deceleration in CSF Aβ42 but acceleration in hippocampal atrophy rates with advancing disease. Schuff, et at 200959 found acceleration in atrophy rates in MCI and AD ADNI subjects. Sabuncu, et al 201160 examined brain atrophy rates in ADNI participants who had an AD-like CSF profile. They found that atrophy rates in a set of AD-signature ROIs exhibit early acceleration followed by deceleration which was consistent with a sigmoid shaped curve. Conversely, they found rates of hippocampal volume loss exhibited positive acceleration.
In the present study, we fit the models in such a way that would allow us to assess the shape of the biomarker trajectories without imposing a particular structure (i.e. a sigmoid shape) upon the data. Flexible restricted cubic splines allowed for non-linearity if there was evidence for it. Interaction terms allowed for biomarker – MMSE relationships to depend on covariates. This way of modeling let the data “speak for themselves” and was preferred in this study because of several important limitations in the nature of the data. (1) The right and left hand portions of a sigmoid curve are where the maximum inflection occurs and thus the portions of the function where data are most needed to detect acceleration and deceleration. Unfortunately, our data are sparse in these regions. In participants with abnormal biomarkers at baseline, we have no data that would allow us to characterize the initial deviation of biomarkers from their normal baseline. The right-hand tail is equally problematic in that many patients survive a decade or more after the clinical diagnosis of AD dementia is made, but most stop participating in clinical research studies once they become moderately demented. Indeed, the AD subjects in our samples were only mildly demented (median MMSE of 24 for the CSF/MRI cohort and 23 for the PET/MRI cohort). (2) The median follow-up time in our data was only about 1 year with a maximum of only 4 years. This is a small fraction of the total duration of the disease which may span 30 plus years. Examining such a small window of time in each subject makes it difficult to detect acceleration or deceleration in within-subject rates. (3) We lacked a linear clinical measure of disease progression. Every cognitive testing instrument has a non-linear response function with both floor and ceiling effects50,61. Because subjects spanning the cognitive continuum were combined to estimate biomarker trajectories, a single universal cognitive test was needed to index all subjects on a common axis. The MMSE was the best option that was available in all ADNI and Mayo subjects. However, the limited range of the MMSE in cognitively normal participants (roughly 30–27) in particular made estimation of trajectory shape early in the disease particularly problematic. In many of our CN participants MMSE did not change, or fluctuated randomly from one time point to the next.
Our results do not disagree with sigmoid shaped biomarker trajectories in that most biomarkers worsened as MMSE worsened in both baseline and longitudinal analyses which is consistent with the middle, roughly linear, portion of a sigmoid curve. While cross sectional data may be influenced by cohort effects, we did see some baseline effects that were consistent with a sigmoid-shaped trajectory (i.e. baseline effects that plateaued with worsening MMSE). However, we found non-constant within-subject rates in only one analysis. Several prior studies (including one of our own) have shown that rates of brain atrophy accelerate prior to incident dementia62–65. However, these earlier MRI studies had considerably more within-subject longitudinal data than we had in the present study. Our failure to detect acceleration or deceleration in within-subject MRI rates may well be due to limited longitudinal data because we only used those time points in individual participants where all biomarkers were available.
AD biomarkers are poised to become an essential component of a comprehensive assessment of the disease. In particular, AD biomarkers constitute a major (some would say only) window into the disease in its long pre-clinical phase. Designing clinical trials in early symptomatic and preclinical disease will depend on acquiring a thorough understanding of the longitudinal trajectory of AD biomarkers. In addition, the notion of biomarker trajectories is central to the staging proposed in the recent preclinical AD research criteria66. However, creating reliable models that accurately describe the full trajectory shapes of AD biomarkers will require significant additional longitudinal data in individual participants beginning prior to deviation of biomarkers from normality (age 50s) through the end stage of the disease and ultimately to autopsy. Ideally, this data would be acquired in well-defined epidemiological cohorts.
Figure 4. Amyloid Positive Cohorts.
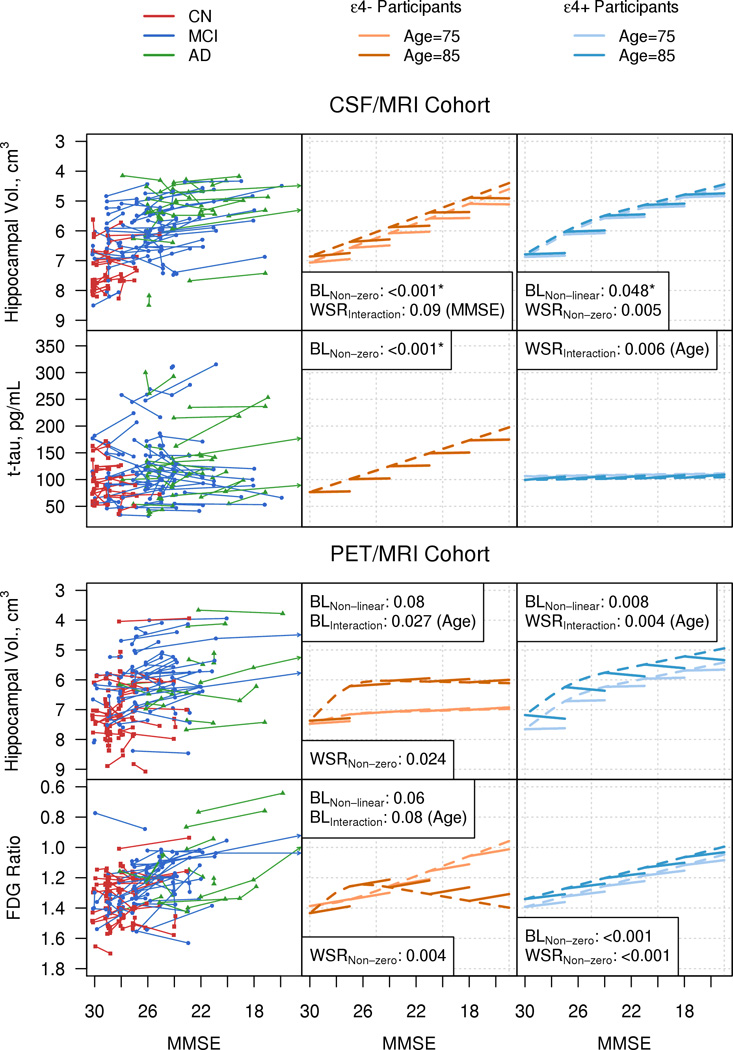
Individual trajectory plots by MMSE of hippocampal volume and CSF tau for the amyloid positive CSF/MRI cohort, and of hippocampal volume and FDG Ratio for the amyloid positive PET/MRI cohort. The organization of Fig. 4 is analogous to that of Figs 2 and 3.
ACKNOWLEDGMENTS
The National Institute on Aging (R01AG11378, P50 AG16574,U01 AG06786, and ADNI). The Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation, USA, and the Robert H. and Clarice Smith Alzheimer’s Disease Research Program of the Mayo Foundation, USA. Manuscript preparation by Samantha Wille. All authors had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.ucla.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.ucla.edu/wp-content/uploads/how_to_apply/ADNI_Authorship_List.pdf
REFERENCES
- 1.Peskind ER, Li G, Shofer J, et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol. 2006;63:936–939. doi: 10.1001/archneur.63.7.936. [DOI] [PubMed] [Google Scholar]
- 2.Bouwman FH, Schoonenboom NS, Verwey NA, et al. CSF biomarker levels in early and late onset Alzheimer's disease. Neurobiol Aging. 2009;30:1895–1901. doi: 10.1016/j.neurobiolaging.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 4.Fagan AM, Head D, Shah AR, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaw LM, Vanderstichele H, Knapik-Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Sokal I, Quinn JF, et al. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69:631–639. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- 7.Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 8.Visser PJ, Verhey F, Knol DL, et al. Prevalence and prognostic value of CSF markers of Alzheimer's disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol. 2009;8:619–627. doi: 10.1016/S1474-4422(09)70139-5. [DOI] [PubMed] [Google Scholar]
- 9.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 10.Mintun MA, Larossa GN, Sheline YI, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 11.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 12.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 13.Pike KE, Ellis KA, Villemagne VL, et al. Cognition and beta-amyloid in preclinical Alzheimer's disease: Data from the AIBL study. Neuropsychologia. 2011 doi: 10.1016/j.neuropsychologia.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rabinovici GD, Furst AJ, O'Neil JP, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007;68:1205–1212. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- 16.Hampel H, Frank R, Broich K, et al. Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9:560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 17.Blennow K. Biomarkers in Alzheimer's disease drug development. Nat Med. 2010;16:1218–1222. doi: 10.1038/nm.2221. [DOI] [PubMed] [Google Scholar]
- 18.Glodzik L, de Santi S, Tsui WH, et al. Phosphorylated tau 231, memory decline and medial temporal atrophy in normal elders. Neurobiol Aging. 2010 doi: 10.1016/j.neurobiolaging.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jagust WJ, Bandy D, Chen K, et al. The Alzheimer's Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6:221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowe VJ, Kemp BJ, Jack CR, Jr, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med. 2009;50:878–886. doi: 10.2967/jnumed.108.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dickerson BC, Stoub TR, Shah RC, et al. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology. 2011;76:1395–1402. doi: 10.1212/WNL.0b013e3182166e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vemuri P, Whitwell JL, Kantarci K, et al. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage. 2008;42:559–567. doi: 10.1016/j.neuroimage.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becker JA, Hedden T, Carmasin J, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol. 2010 doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CR, Jr, Wiste HJ, Vemuri P, et al. Brain beta-amyloid measure and magnetic resonance imaging atophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain. 2010;133:3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Desikan RS, Cabral HJ, Hess CP, et al. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer's disease. Brain. 2009;132:2048–2057. doi: 10.1093/brain/awp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hua X, Leow AD, Parikshak N, et al. Tensor-based morphometry as a neuroimaging biomarker for Alzheimer's disease: an MRI study of 676 AD, MCI, and normal subjects. Neuroimage. 2008;43:458–469. doi: 10.1016/j.neuroimage.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingelsson M, Fukumoto H, Newell KL, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 29.Mormino EC, Kluth JT, Madison CM, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villain N, Fouquet M, Baron JC, et al. Sequential relationships between grey matter and white matter atrophy and brain metabolic abnormalities in early Alzheimer's disease. Brain. 2010;133:3301–3314. doi: 10.1093/brain/awq203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jack CR, Jr, Vemuri P, Wiste HJ, et al. Evidence for Ordering of Alzheimer Disease Biomarkers. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30:58–69. doi: 10.1159/000115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petersen RC, Roberts RO, Knopman DS, et al. Mild cognitive impairment: ten years later. Arch Neurol. 2009;66:1447–1455. doi: 10.1001/archneurol.2009.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Folstein MF, Folstein SE, McHugh PR. "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 37.Kokmen E, Smith GE, Petersen RC, Tangalos E, Ivnik RC. The short test of mental status. Correlations with standardized psychometric testing. Arch Neurol. 1991;48:725–728. doi: 10.1001/archneur.1991.00530190071018. [DOI] [PubMed] [Google Scholar]
- 38.Tang-Wai DF, Knopman DS, Geda YE, et al. Comparison of the short test of mental status and the mini-mental state examination in mild cognitive impairment. Arch Neurol. 2003;60:1777–1781. doi: 10.1001/archneur.60.12.1777. [DOI] [PubMed] [Google Scholar]
- 39.Vanderstichele H, De Meyer G, Shapiro F, et al. Biomarkers For Early Diagnosis Of Alzheimer's Disease. Hauppauge;NY: Nova Science Publishers, Inc; 2008. [Google Scholar]
- 40.Jack CR, Jr, Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 42.Gunter JL, Bernstein MA, Borowski BJ, et al. Measurement of MRI scanner performance with the ADNI phantom. Med Phys. 2009;36:2193–21205. doi: 10.1118/1.3116776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin LI. A concordance correlation coefficient to evaluate reproducibility. Biometrics. 1989;45:255–268. [PubMed] [Google Scholar]
- 44.Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 45.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Price JC, Klunk WE, Lopresti BJ, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25:1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 47.McNamee RL, Yee SH, Price JC, et al. Consideration of optimal time window for Pittsburgh compound B PET summed uptake measurements. J Nucl Med. 2009;50:348–355. doi: 10.2967/jnumed.108.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fitzmaurice GM, Laird NM, Ware JH. Applied Longitudinal Analysis. Hoboken, N.J: Wiley-Interscience; 2004. [Google Scholar]
- 50.Caselli RJ, Dueck AC, Osborne D, et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. N Engl J Med. 2009;361:255–263. doi: 10.1056/NEJMoa0809437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weigand SD, Vemuri P, Wiste HJ, et al. Transforming cerebrospinal fluid Abeta42 measures into calculated Pittsburgh compound B units of brain Abeta amyloid. Alzheimers Dement. 2011;7:133–141. doi: 10.1016/j.jalz.2010.08.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vemuri P, Wiste HJ, Weigand SD, et al. Serial MRI and CSF Biomarkers in Normal Aging, MCI and AD. Neurology. 2010;75:143–151. doi: 10.1212/WNL.0b013e3181e7ca82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crowe A, Ballatore C, Hyde E, Trojanowski JQ, Lee VM. High throughput screening for small molecule inhibitors of heparin-induced tau fibril formation. Biochem Biophys Res Commun. 2007;358:1–6. doi: 10.1016/j.bbrc.2007.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lomasko T, Lumsden CJ. One-hit stochastic decline in a mechanochemical model of cytoskeleton-induced neuron death III: diffusion pulse death zones. J Theor Biol. 2009;256:104–116. doi: 10.1016/j.jtbi.2008.08.025. [DOI] [PubMed] [Google Scholar]
- 56.Clarke G, Lumsden CJ. Heterogeneous cellular environments modulate one-hit neuronal death kinetics. Brain Res Bull. 2005;65:59–67. doi: 10.1016/j.brainresbull.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Caroli A, Frisoni GB. The dynamics of Alzheimer's disease biomarkers in the Alzheimer's Disease Neuroimaging Initiative cohort. Neurobiol Aging. 2010;31:1263–1274. doi: 10.1016/j.neurobiolaging.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lo RY, Hubbard AE, Shaw LM, et al. Longitudinal Change of Biomarkers in Cognitive Decline. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schuff N, Woerner N, Boreta L, et al. MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009;132:1067–1077. doi: 10.1093/brain/awp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sabuncu MR, Desikan RS, Sepulcre J, et al. The Dynamics of Cortical and Hippocampal Atrophy in Alzheimer Disease. Arch Neurol. 2011;68:1040–1048. doi: 10.1001/archneurol.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mungas D, Reed BR, Kramer JH. Psychometrically matched measures of global cognition, memory, and executive function for assessment of cognitive decline in older persons. Neuropsychology. 2003;17:380–392. doi: 10.1037/0894-4105.17.3.380. [DOI] [PubMed] [Google Scholar]
- 62.Chan D, Janssen JC, Whitwell JL, et al. Change in rates of cerebral atrophy over time in early-onset Alzheimer's disease: longitudinal MRI study. Lancet. 2003;362:1121–1122. doi: 10.1016/S0140-6736(03)14469-8. [DOI] [PubMed] [Google Scholar]
- 63.Ridha BH, Barnes J, Bartlett JW, et al. Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet Neurol. 2006;5:828–834. doi: 10.1016/S1474-4422(06)70550-6. [DOI] [PubMed] [Google Scholar]
- 64.Carlson NE, Moore MM, Dame A, et al. Trajectories of brain loss in aging and the development of cognitive impairment. Neurology. 2008;70:828–833. doi: 10.1212/01.wnl.0000280577.43413.d9. [DOI] [PubMed] [Google Scholar]
- 65.Jack CR, Jr, Weigand SD, Shiung MM, et al. Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology. 2008;70:1740–1752. doi: 10.1212/01.wnl.0000281688.77598.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging and the Alzheimer Assocation Workgroup. Alzheimers Dement. 2011 doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]