

Abstract
Esophageal squamous cell carcinoma (ESCC) remains one of the most aggressive cancers with poor prognosis regardless of a several reports that indicate a better therapeutic efficacy using some new chemotherapeutic agents. Recent drug development has contributed to an improved specificity to suppress mTOR activity by which many types of malignancies can be explosively progressed. Temsirolimus (CCI-779, TricelTM) is one of recently synthesized analogs of rapamycin and has provided better outcomes for patients with renal cell carcinoma. In this study, we experimentally evaluated an efficacy of targeting mTOR by temsirolimus for ESCC treatment, with an assessment of its survival advantage using an advanced ESCC animal model.
First, we confirmed that the expression of phosphorylated mTOR was increased in 46 of 58 clinical ESCC tumor tissues (79.3%) and appeared to get strengthened with tumor progression. All of ESCC cell lines used in this study revealed an increase of mTOR phosphorylation, accompanied with the upregulation of hypoxia inducible factor-I α (HIF-1α), one of the critical effectors regulated by mTOR. Temsirolimus treatment apparently suppressed the activation of mTOR and its downstream effectors, resulting in the reduced ability of ESCC cell proliferation. Finally, the weekly administration of temsirolimus significantly diminished the size of subcutaneous tumors (vehicle, 3261.6 ± 722.0; temsirolimus, 599.2 ± 122.9; p = 0.007) in nude mice and effectively prolonged orthotopic esophageal cancer-bearing mice (median survival periods: control, 31 d; temsirolimus, 43 d; p = 0.0024).
These data suggests that targeting mTOR by temsirolimus may become a therapeutic alternative for esophageal cancer, with a contribution to a better outcome.
Keywords: temsirolimus, esophageal cancer, mTOR, prolonged survival, molecular-targeted therapy
Introduction
Esophageal cancer is an aggressive malignancy with poor prognosis.1 Pathologically, it mainly consists of adenocarcinoma and squamous cell carcinoma, the former often occurs in Western countries, whereas the latter predominates in Asia.2 The surest current therapy for esophageal cancer is surgical treatment. However, we often find both types of esophageal cancer at an advanced stage, resulting in the consideration of a combination therapy with both surgical and non-surgical treatments. Squamous cell carcinoma of the esophagus is relatively susceptible to many kinds of new medicine for chemotherapy that have been developed and introduced in clinical practice, but the mortality rate has not been improved.3 Recently, drug development strategy has focused on targeting particular molecules that are supposedly critical for cancer progression. Several molecules in growth factor receptor pathways are preferentially employed for specific targeting since those molecules are well recognized as being aberrantly regulated in cancers. For example, epidermal growth factor receptor (EGFR) and its downstream pathway are often upregulated due to gene amplification or mutation,4 and therefore targeting EGFR is a major therapeutic strategy for cancer treatment. However, in some cases these drugs only show a minimal effectiveness due to the aberrant regulation of downstream molecules located beneath the receptor tyrosine kinase pathways such as Ras-Raf-MAPK and phosphatidylinositol 3′-kinase (PI3K)-Akt.5-8 Among these downstream molecules, mammalian target of rapamycin (mTOR) is one of the major effectors regulated by the PI3K-Akt signaling pathway and plays a central role in this stimulated growth and survival signaling.9,10 Therefore, several compounds that selectively inhibit mTOR activity have been developed for clinical use.11,12 Temsirolimus (CCI-779, TricelTM), an analog of rapamycin, was recently synthesized to specifically inhibit mTOR and has provided better outcomes for patients with renal cell carcinoma.13 It was also reported that temsirolimus showed an antitumor effect on other types of cancer including breast cancer,14 glioblastoma,15 neuroendocrine carcinomas16 and mantle cell lymphoma.17 Based upon this evidence, we questioned whether temsirolimus treatment could be a good alternative strategy for ESCC. In this study, we evaluated the antiproliferative and antitumor effects of temsirolimus on ESCC in vitro and in vivo, with an assessment of its survival advantage in an advanced ESCC animal model.
Results
The activities of mTOR and its downstream molecules are upregulated in esophageal squamous cell carcinoma
First, the expression status of mTOR and phosphorylated mTOR in surgically resected tissues of esophageal squamous cell carcinoma was examined by immunohistochemistry. The expression of mTOR was equally detected both in normal esophageal epithelia and in cancer tissues (Fig. 1A, upper panels). The expression of phosphorylated mTOR was also detected in the cytoplasm of cancer tissues while its intensity in normal esophageal epithelia was very faint (Fig. 1A, left lower panel). Of note, the phosphorylated mTOR was highly expressed at the edge of the tumors (Fig. 1A, right lower panel). Progressive cancer cases tended to have a poor prognosis, and the intensity of p-mTOR expression became high in aggressive cases (Fig. 1B).
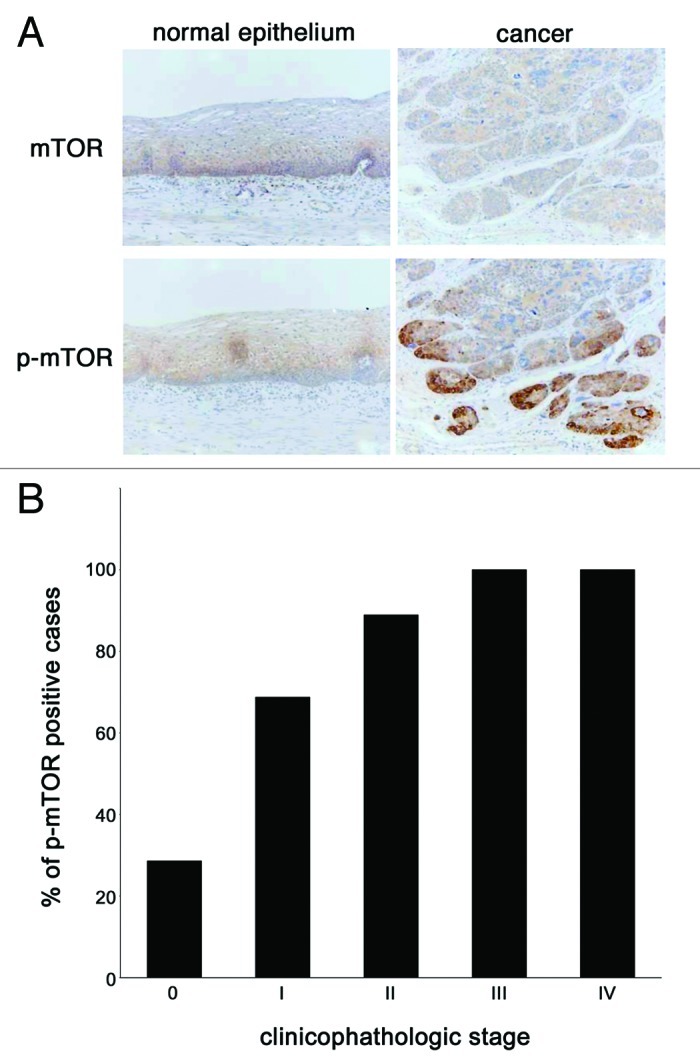
Figure 1. The expression and activation of mTOR is increased in esophageal cancer tissues. (A) Esophageal tissue samples were procured via surgery from the Okayama University Hospital and immunohistochemical analysis of mTOR and phosphorylated mTOR was done to compare its expression status between in cancer tissues (right panels) and in adjacent normal epithelia (left panels). (B) Fifty-eight cases of esophageal cancer whose tissues were used for this immunohistochemistory were categorized according to their pathological staging, which described briefly in “Materials and Methods” section, and the positive staining rate of phosphorylated mTOR in each stage was shown in histogram.
Immunohistochemistry was also performed to evaluate the expressions of mTOR and phosphorylated mTOR in cultured cells. As shown in Figure 2A, the expression of mTOR was detected in esophageal cancer cells (TE-1, TE-8 and TE-10) as well as in KOB9N and KOB12N cells, both of which were primary esophageal epithelial cells that were isolated from surgically resected human esophageal tissues.18 KOB9N and KOB9C cells, which were isolated from the surgically resected esophageal epithelia and tumor tissues of a single esophageal cancer patient, respectively, were used to compare the expression status of mTOR and phosphorylated mTOR to each other (Fig. 2A). Interestingly, both showed a similar expression of mTOR, while the intensity of phosphorylated mTOR in the cancer cells (KOB9C) was definitely stronger than in the normal epithelial cells (KOB9N). Similarly, all of the TE-1, TE-8 and TE-10 cells showed a higher intensity of phosphorylated mTOR than the normal esophageal epithelial cells (KOB9N and KOB12N) (Fig. 2A). These results indicated that mTOR appeared to exist ubiquitously regardless of tissue type, whereas cancer tissues/cells definitely increased the mTOR activity.
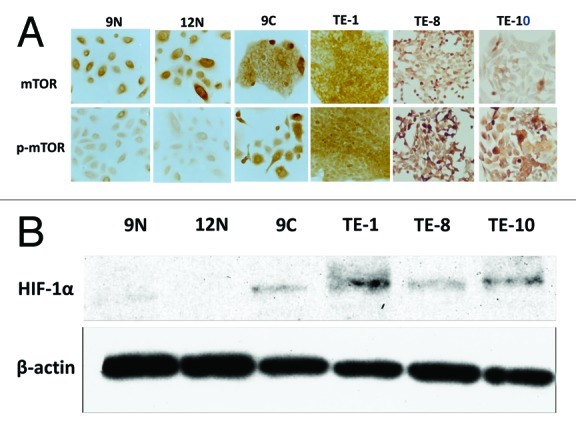
Figure 2. The activation of mTOR and its downstream effector is increased in esophageal cancer cell lines. (A) The expression status of mTOR (upper panels) and phosphorylated mTOR (lower panels) was determined in human primary normal esophageal epithelial cells (KOB9N and KOB12N) and in human esophageal cancer cell lines (KOB9C, TE-1, TE-8 and TE-10). (B) The expression of hypoxia inducible factor-1α (HIF-1α), a downstream effector of mTOR, was compared between in human primary normal esophageal epithelial cells (KOB9N and KOB12N) and in human esophageal cancer cell lines (KOB9C, TE-1, TE-8 and TE-10) by western blot.
The activation of mTOR is known to positively regulate protein translation and cell proliferation by upregulating downstream molecules such as hypoxia inducible factor-1 α-subunit (HIF-1α),21 which plays a critical role in cancer proliferation and angiogenesis. The expression status of HIF-1α in primary esophageal epithelial cells and in esophageal cancer cells was determined by western blot. Strikingly, all of the cancer cells had an apparent increase of HIF-1α expression, while the normal esophageal cells revealed no detectable level of expression (Fig. 2B). This suggests that the upregulation of downstream molecules such as HIF-1α paralleled that of mTOR, leading to the idea that targeting mTOR activity can be a potential therapeutic strategy for esophageal squamous cell carcinoma.
A selective mTOR inhibitor, temsirolimus, reveals an antiproliferative effect in esophageal cancer cells by inhibiting the mTOR pathway
Using temsirolimus, an analog of rapamycin that has recently been synthesized as a selective mTOR inhibitor, we aimed to inhibit mTOR activity to evaluate its possible anticancer effects in esophageal cancer cells. After the cancer cells (TE-1, TE-8 and TE-10) were treated with temsirolimus at different concentrations (0–1000nM), the expressions of mTOR and S6, a major downstream molecule, were examined by western blot. Temsirolimus treatment did not affect the expressions of total mTOR or S6 in esophageal cancer cells, but it significantly reduced the expressions of phosphorylated mTOR and phosphorylated S6 around a concentration of 1nM and the effect was observed in a dose-dependent manner, regardless of cell line (Fig. 3). Interestingly, the expression and activity of 4E-BP1, another downstream molecule of the mTOR pathway, did not change after temsirolimus treatment (Fig. 3).
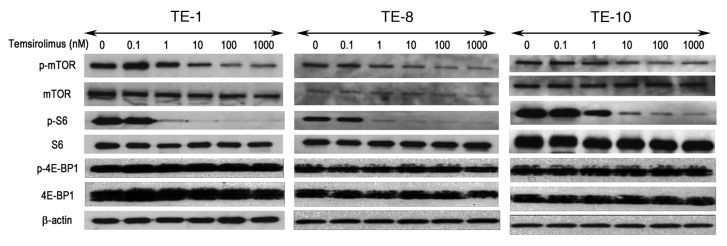
Figure 3. The mTOR inhibitor temsirolimus inhibits the activation of mTOR and its downstream molecules in esophageal cancer cells. Three esophageal cancer cell lines (TE-1, TE-8, and TE-10) were treated with different concentrations of temsirolimus (0–1000nM) and western blot was performed with appropriate antibodies to detect the expression of mTOR and its downstream effectors. β-actin was served as an internal control.
Next, we assessed the effect of mTOR inhibition from temsirolimus on cell proliferation in esophageal cancer cells. When the cancer cells were treated with temsirolimus at different concentrations (0–1000nM), their growth was dose-dependently suppressed (Fig. 4). These results indicated that the inhibition of the mTOR pathway by temsirolimus negatively affected the growth of esophageal cancer cells, and that targeting mTOR can be a potential alternative for esophageal cancer treatment.
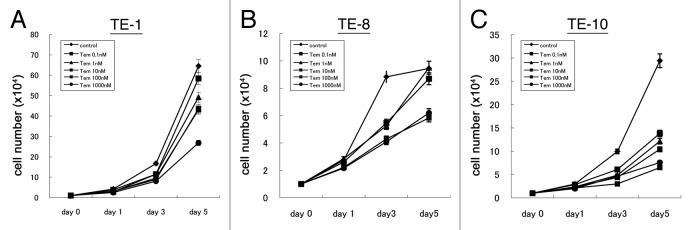
Figure 4. Temsirolimus suppresses cell proliferation of esophageal cancer cells. Three esophageal cancer cell lines (A, TE-1; B, TE-8; C, TE-10) were treated with different concentrations of temsirolimus (0–1000 nM) and the cell number at the indicated time point (day0, 1, 3, and 5) was counted to draw these growth curve.
Temsirolimus provides a survival advantage to tumor-bearing mice by retarding tumor growth
To clarify the potential effectiveness of temisrolimus for esophageal cancer treatment, we applied this selective mTOR inhibitor to animal experiments. An intravenous administration of temsirolimus was given to mice with subcutaneous tumors of TE-8 cells according to the schedule described in the Materials and Methods section and the tumor volume was measured once a week. The growth of the subcutaneous tumors was significantly reduced by temsirolimus treatment in a dose-dependent manner (Fig. 5A). On day 28, the mice treated with 10 mg/kg of temsirolimus had an approximately 6-fold less tumor volume than the control (vehicle only) mice (vehicle, 3261.6 ± 722.0; temsirolimus, 599.2 ± 122.9; p = 0.007). During the observation period the body weight of each mouse was tracked as a surrogate marker of drug toxicity and we observed no significant differences in body weight between the groups (data not shown).
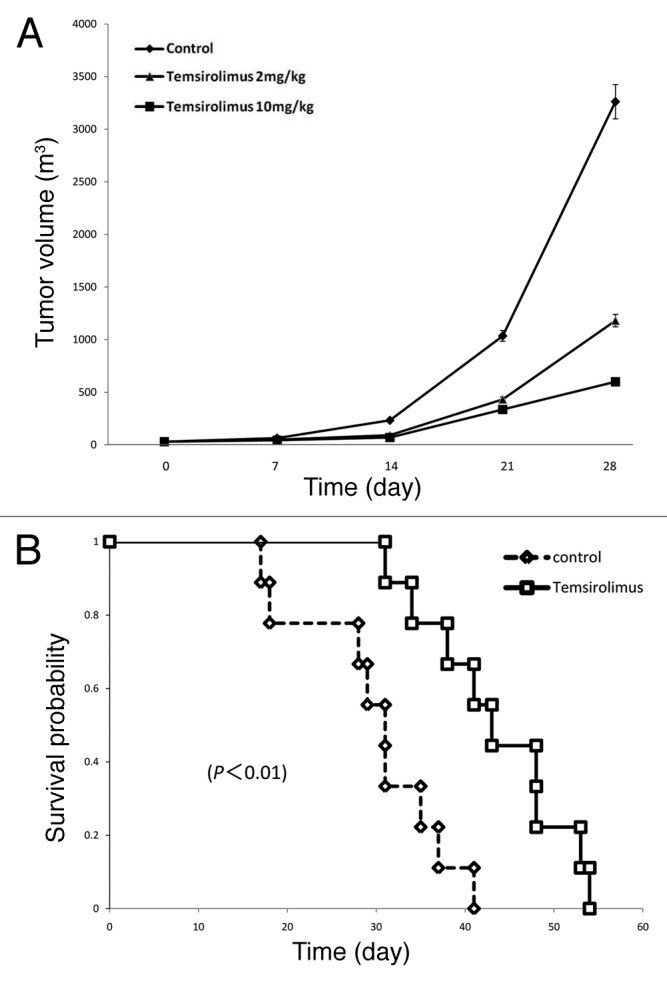
Figure 5. Administration of temsirolimus reduces tumor growth and prolongs the survival of tumor-bearing mice.(A) Subcutaneous tumor-bearing mice were subgrouped into three groups for intraperitoneal administration of either 2 different doses of temsirolimus (2mg/kg and 10mg/kg) or phosphate buffered saline (PBS) as a control group at the scheduled dates, as described in “Materials and Methods” section. Tumor volume [(length × width2)/2] was measured to draw a histogram. (B) Orthotopic esophageal cancer-bearing mice were divided into two groups for intraperitoneal administration of either temsirolimus or PBS (control) at the scheduled dates, as described in “Materials and Methods” section. Their survival was tracked to draw Kaplan-Meier survival curves and log-rank test was performed for the statistical comparison between two groups.
Furthermore, we applied a similar treatment with temsirolimus to an orthotopic esophageal cancer model that was recently established.19 As shown previously, this orthotopic mouse model shows a quick outcome from a lack of food intake due to esophageal stricture by the orthotopic tumor. Thus, we hypothesized that the inhibition of orthotopic tumor growth by temsirolimus would prolong the survival of this mouse model if the esophageal stricture could be retarded and normal food intake maintained. An intraperitoneal administration of temsirolimus or PBS as a vehicle was given to the mice with an orthotopic esophageal tumor once a week, and their survival was tracked (Fig. 5B). The mice with temsirolimus treatment significantly extended their survival compared with the control mice (median survival periods: control, 31 d; temsirolimus, 43 d; p = 0.0024).
These results support our idea that the inhibition of mTOR could provide a survival advantage for advanced esophageal cancer patients, and therefore, we propose that temsirolimus might be a potential alternative strategy for esophageal cancer treatment.
Discussion
Recent cancer therapy studies have been paying close attention to the key molecules of the signaling pathways that are important for cell migration, proliferation, progression and the invasion of tumors. mTOR is one of the molecules associated with basic biological processes such as the signal transduction of cell proliferation, migration, angiogenesis, and synthesis of tumorigenic proteins. In addition to its role as a basic controller in organisms, mTOR is also known to be involved in tumor progression.22 Aberrant PI3K-dependent signaling and protein translation may contribute to the development and progression of human cancers. Activation mutations of growth factor receptors and PI3K, as well as the amplification and/or overexpression of PI3K and Akt have been reported in different types of malignancies.23 If cancer cell growth and survival is dependent on the PI3K-Akt pathway, it is possible that this dependency in cancer cells would result in increased sensitivity to mTOR inhibitors.
In in vitro studies, it was revealed that mTOR is activated in esophageal squamous cell carcinoma cell lines and that mTOR expression is inhibited by rapamycin.21 There is an immunohistochemical study in which activated mTOR was detected in one-quarter of esophageal squamous cell carcinoma tissues.24 Our data in this study showed that the expression of phosphorylated mTOR was significantly higher in cancer tissues than in normal esophageal epithelia. In addition, the expression tended to be concentrated at the invasive front of tumors. This may suggest that intense tumor proliferation and progression may largely rely on a strong activation of mTOR signaling in esophageal cancer. Thus, it makes sense to apply mTOR inhibition to esophageal cancer treatment, and we believe that at least a certain population of esophageal cancer patients may potentially benefit from this therapy.
Hou G. et al. reported that the mTOR-p70S6K pathway is activated in esophageal squamous cell carcinoma and that rapamycin and siRNA rapidly inhibited the expression of mTOR and the phosphorylation of its major downstream effectors, p70S6K and 4E-BP1.25 They also reported that the inhibition of mTOR induced G0/G1 cell cycle arrest and apoptosis of esophageal squamous cell carcinoma cells. In our study, temsirolimus, an analog of rapamycin, suppressed the phosphorylation of mTOR and S6 at a concentration of 1nM in all of the tested esophageal cancer cell lines. Interestingly, 4E-BP1, another downstream molecule beneath mTOR, was not affected by this treatment in our experiments (Fig. 3). Moreover, our observation by cell cycle analysis and TUNEL assay did not show any apparent cell death or the induction of apoptosis from temsirolimus treatment (data not shown). In general, the inhibition of mTOR downregulates the translation of specific mRNAs that are required for cell cycle progression from the G1 phase to the S phase.26,27 Taken together, we speculate that temsirolimus induced the inhibition of cell proliferation with cell accumulation in the G0/G1 phase rather than leading to apoptosis and therefore the intrinsic effect of temsirolimus in human esophageal cancer is cytostatic.
In our study, treatment with a nanomolar level of temsirolimus was enough to suppress the phosphorylation of mTOR and its substrates S6. Shor B. et al. proved that a low micromolar concentration of temsirolimus completely suppressed the proliferation of a broad panel of tumor cells including lung cancer, colon cancer, breast cancer, and human embryonic kidney cell lines.28 They hypothesized that the response of tumor cells to the commonly used nanomolar concentrations of rapamycin may be limited by the feedback loop, whereas the suppression of S6 signaling by rapamycin stimulates the IRS-PI3K pathway to promote AKT activity. Our data did not support their opinion, but further experiments will be needed in the future.
Using our established primary human esophageal epithelial cells and cancer cells,18 we compared the expression and activation of mTOR and HIF-1α, a downstream molecule affected by the mTOR pathway. Our data that these proteins showed less activity in normal esophageal epithelial cells led us to investigate the tolerance of these normal esophageal cells to an mTOR inhibitor, specifically temsirolimus. This may provide some knowledge to help determine the most effective dose of this drug for cancer therapy. Although we used esophageal squamous cell carcinoma cells in this study, we also recently reported that a dual tyrosine kinase inhibitor for focal adhesion kinase (FAK) and insulin-like growth factor-I receptor (IGF-IR) exhibits anticancer effects in esophageal adenocarcinoma in vitro and in vivo.29 We also confirmed that this dual tyrosine kinase inhibitor suppressed mTOR activity in esophageal cancer cells.30 It would be intriguing to evaluate the effectiveness of temsirolimus in esophageal adenocarcinoma in further basic and clinical studies.
In conclusion, we showed the importance of the mTOR signaling pathway on the regulation of cell proliferation in the esophagus and temsirolimus could inhibit the activity of mTOR and its downstream molecule S6. Temsirolimus may be useful for esophageal cancer treatment as a novel therapeutic instrument.
Materials and Methods
Clinical tissues
Fifty-eight esophageal squamous cell carcinoma tissues were used in this study. These tissues were surgically resected from individual patients who had surgery at the Okayama University Hospital between 2000 and 2002. Informed consent was fully given by each patient involved in this study. These tissues were subject to immunohistochemical analysis.
Cell lines
Human primary esophageal epithelial cells KOB9N and 12N, both of which were isolated from normal esophageal epithelia that were surgically resected from two independent patients, were maintained as monolayer cultures in KSFM supplemented with EGF, BPE, 100 units/ml of penicillin G and 100 μg/ml of streptomycin.18 Human esophageal cancer cells, KOB9C cells, were also isolated from an esophageal squamous cell carcinoma tissue that was resected from the same patient as the KOB9N cells. Established esophageal squamous cell carcinoma cell lines (TE-1, TE-8 and TE-10) were also used in this study and all of these cancer cells were cultured in a medium consisting of RPMI1640 supplemented with 10% heat-inactivated fetal calf serum (FCS), 100 units/ml of penicillin G and 100 μg/ml of streptomycin. Those normal and cancer cells were maintained in a humidified 5% CO2 atmosphere at 37°C.
Antibodies and reagents
The following antibodies used in this study were purchased from Cell Signaling Technology, Inc.; Phospho-mTOR (Ser2448) (Cat.No.2971), mTOR (Cat.No.2972), Phospho-p70 S6 Kinase (Thr389) (Cat.No.9205), p70 S6 Kinase (Cat.No.9202), Phospho-S6 Ribosomal Protein (Ser235/236) (Cat.No. 2211) and Hydroxy-HIF-1α (Pro564) (D43B5) (Cat.No.3434). β-actin (Cat.No.A1978) was obtained from Sigma-Aldrich.
The selective mTOR inhibitor temsirolimus, commercialized as ToriselTM (Cat.No.45714) by Wyeth K.K., was purchased from OZ International Inc. Temsirolimus was dissolved in attached diluents and diluted to the working concentrations (0–1000nM) with culture media before use in vitro. When temsirolimus was used for animal experiments, it was dissolved in attached diluents, and diluted to a final concentration of either 2 mg/kg or 10 mg/kg with 0.9% sodium chloride.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections mounted on silanized slides were deparaffinized in xylene for 20 min and rehydrated through a graded ethanol series. Endogenous peroxidase was blocked by incubating the sections in 3.0% H2O2 in methanol for 15 min. Antigen retrieval on the paraffin sections was performed by heating two times in a 10 mM citrate buffer solution (pH 6.0) in a microwave. After blocking nonspecific reactivity with rabbit serum for 10 min at room temperature, sections were incubated overnight at 4°C with the primary antibodies, followed by immuno-bridging with Avidin DH-biotinylated horseradish peroxide complex (Histofine SAB PO kit; Cat.No. 424031; Nichirei). Immunostaining was developed using a 3,3′-diaminobenzidine tetrahydrochloride (DAB)/hydrogen peroxidase solution (Histofine DAB substrate kit; Cat.No.415192; Dako Japan), and sections were counterstained with Mayer’s hematoxylin. Culture cells that were seeded into chamber slides were fixed in 4% paraformaldehyde for 10 min at room temperature, and then were made permeable by treating them with Triton X-100 for 2 min at room temperature before the subsequent incubation step with the antibodies.
Western blotting
Cells were collected by trypsinization 48 h after treatment with temsirolimus and washed twice in cold PBS. Whole cell lysates were extracted as follows. Cell pellets were dissolved at 4°C for 30 min in a protein lysis buffer containing 50 mM TRIS-HCl (pH 7.5), 150 mM NaCl, 0.5% Triton X-100, and protease inhibitors (0.2 mM phenylmethylsulfonyl fluoride, 0.2 mM 4-(2-aminoethyl) benzenesulfonylfluoride, 10 μg/ml leupeptin, 10 μg/ml pepstatin, and 1 μg/ml aprotinin). Cell lysates were centrifuged at 15,000 rpm and the supernatants were employed to determine the protein concentration using the Bio-Rad protein determination method (Bio-Rad). Equal amounts (15 μg) of proteins were first electrophoresed under reducing conditions on 12% (w/v) polyacrylamide gels, and then were electrophoretically transferred to Hybond- polyvinylidene difluoride transfer membranes (Amersham) and incubated with the primary antibodies, followed by incubation with a peroxidase-linked secondary antibody. An ECL Western Blotting System (Cat.No.RPN2109; Amersham) was used for signal detection.
Cell proliferation assay
To determine the growth inhibitory effect of temsirolimus, esophageal cancer cells were seeded into 24-well culture plates at a density of 1.0 × 105 per well and incubated for 24 h in culture medium. The medium was then replaced with a fresh one containing 10% FBS with different concentrations of temsirolimus (0, 0.1, 1, 10, 100 or 1000nM) (day 0) and was refreshed every 48 h. On days 1, 3, and 5, the cells were trypsinized and were counted using the Trypan blue exclusion method.
Animal experiments
For animal studies, six week-old male BALB/cA nude mice were purchased from Clea Japan and were maintained in a barrier facility in accordance with the Institutional Animal Care and Use Committee regulations of Okayama University. A cell suspension of 3 × 106 TE-8 cells mixed with Matrigel (BD Biosciences) was inoculated subcutaneously into those nude mice (day 0). From day 7, the tumor-bearing mice were randomized into three groups and an intravenous administration of either 2 mg/kg or 10 mg/kg of temsirolimus or phosphate buffered saline (PBS) as a vehicle was given to each group. The treatment was repeated once a week and continued for four weeks. During the treatment, tumor volume [(length × width2)/2] was measured with a digital caliper every week and was tracked up to day 28.
To prepare an orthotopic esophageal cancer model, we followed a procedure that we recently reported on.19 Briefly, a cell suspension of 5 × 106 TE-8 cells mixed with Matrigel (Cat.No.356234) was injected via the lumen into the esophagus of an anesthetized mouse (day 0) using a needle and barrel. The orthotopic tumor-bearing mice were randomized into 2 groups and from day 7 the intraperitoneal administration of either 10 mg/kg of temsirolimus or PBS as a vehicle was given to each group. The treatment was repeated once a week and was continued until the mice died. The survival period of each mouse was tracked for comparison between the two groups. The doses of temsirolimus used in the animal studies were based on our previous study using lung cancer cells.20
Statistical analysis
Overall survival was calculated using the Kaplan-Meier method and compared by the Wilcoxon test. A P-value less than 0.05 denoted the presence of a statistically significant difference.
Disclosure of Potential Conflicts of Interest
The authors have no conflicts of interest to declare.
Acknowledgments
We are grateful to Mr. Toru Tanida and Tae Yamanishi (Okayama University) for their technical assistance and to Drs. Minoru Haisa (Okayama Citizens’ Hospital), Junji Matsuoka, Kazuhiro Noma, Shunsuke Tanabe (Kawasaki Medical School) for useful discussions.
Glossary
Abbreviations:
- mTOR
mammalian target of rapamycin
- EGFR
epidermal growth factor receptor
- HIF-1α
hypoxia inducible factor-1 α-subunit
- PI3K
phosphatidylinositol 3′-kinase
- FCS
fetal calf serum
- PBS
phosphate buffered saline
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/23294
References
- 1.Horner MJ, Ries RAG, Krapcho M, Neyman N, Aminou R, et al. SEER Cancer Statistics Review, 1975-2006, National Cancer Institute. Bethesda, MD. Available from URL: http://seer.cancer.gov/csr/1975_2006/, based on November 2008 SEER data submission, posted to the SEER web site, 2009.
- 2.Trivers KF, Sabatino SA, Stewart SL. Trends in esophageal cancer incidence by histology, United States, 1998-2003. Int J Cancer. 2008;123:1422–8. doi: 10.1002/ijc.23691. [DOI] [PubMed] [Google Scholar]
- 3.Neuner G, Patel A, Suntharalingam M. Chemoradiotherapy for esophageal cancer. Gastrointest Cancer Res. 2009;3:57–65. [PMC free article] [PubMed] [Google Scholar]
- 4.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–85. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Settleman J, Kurie JM. Drugging the bad “AKT-TOR” to overcome TKI-resistant lung cancer. Cancer Cell. 2007;12:6–8. doi: 10.1016/j.ccr.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 6.Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–72. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- 7.Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol. 2009;6:352–66. doi: 10.1038/nrclinonc.2009.62. [DOI] [PubMed] [Google Scholar]
- 8.Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011;6:707–15. doi: 10.1097/JTO.0b013e31820a3a6b. [DOI] [PubMed] [Google Scholar]
- 9.Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000;103:253–62. doi: 10.1016/S0092-8674(00)00117-3. [DOI] [PubMed] [Google Scholar]
- 10.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 11.Shor B, Gibbons JJ, Abraham RT, Yu K. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8:3831–7. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 12.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7:209–19. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 13.Hudes GR, Berkenblit A, Feingold J, Atkins MB, Rini BI, Dutcher J. Clinical trial experience with temsirolimus in patients with advanced renal cell carcinoma. Semin Oncol. 2009;36(Suppl 3):S26–36. doi: 10.1053/j.seminoncol.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Chan S, Scheulen ME, Johnston S, Mross K, Cardoso F, Dittrich C, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–22. doi: 10.1200/JCO.2005.66.130. [DOI] [PubMed] [Google Scholar]
- 15.Chang SM, Wen P, Cloughesy T, Greenberg H, Schiff D, Conrad C, et al. North American Brain Tumor Consortium and the National Cancer Institute Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Invest New Drugs. 2005;23:357–61. doi: 10.1007/s10637-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 16.Duran I, Kortmansky J, Singh D, Hirte H, Kocha W, Goss G, et al. A phase II clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br J Cancer. 2006;95:1148–54. doi: 10.1038/sj.bjc.6603419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, et al. Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27:3822–9. doi: 10.1200/JCO.2008.20.7977. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi M, Naomoto Y, Nobuhisa T, Okawa T, Takaoka M, Shirakawa Y, et al. Heparanase regulates esophageal keratinocyte differentiation through nuclear translocation and heparan sulfate cleavage. Differentiation. 2006;74:235–43. doi: 10.1111/j.1432-0436.2006.00072.x. [DOI] [PubMed] [Google Scholar]
- 19.Ohara T, Takaoka M, Sakurama K, Nagaishi K, Takeda H, Shirakawa Y, et al. The establishment of a new mouse model with orthotopic esophageal cancer showing the esophageal stricture. Cancer Lett. 2010;293:207–12. doi: 10.1016/j.canlet.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Ohara T, Takaoka M, Toyooka S, Tomono Y, Nishikawa T, Shirakawa Y, et al. Inhibition of mTOR by temsirolimus contributes to prolonged survival of mice with pleural dissemination of non-small-cell lung cancer cells. Cancer Sci. 2011;102:1344–9. doi: 10.1111/j.1349-7006.2011.01967.x. [DOI] [PubMed] [Google Scholar]
- 21.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8:851–64. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 22.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 24.Boone J, Ten Kate FJ, Offerhaus GJ, van Diest PJ, Rinkes IH, van Hillegersberg R. mTOR in squamous cell carcinoma of the oesophagus: a potential target for molecular therapy? J Clin Pathol. 2008;61:909–13. doi: 10.1136/jcp.2008.055772. [DOI] [PubMed] [Google Scholar]
- 25.Hou G, Xue L, Lu Z, Fan T, Tian F, Xue Y. An activated mTOR/p70S6K signaling pathway in esophageal squamous cell carcinoma cell lines and inhibition of the pathway by rapamycin and siRNA against mTOR. Cancer Lett. 2007;253:236–48. doi: 10.1016/j.canlet.2007.01.026. [DOI] [PubMed] [Google Scholar]
- 26.Le Tourneau C, Faivre S, Serova M, Raymond E. mTORC1 inhibitors: is temsirolimus in renal cancer telling us how they really work? Br J Cancer. 2008;99:1197–203. doi: 10.1038/sj.bjc.6604636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapoor A, Figlin RA. Targeted inhibition of mammalian target of rapamycin for the treatment of advanced renal cell carcinoma. Cancer. 2009;115:3618–30. doi: 10.1002/cncr.24409. [DOI] [PubMed] [Google Scholar]
- 28.Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ, et al. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 2008;68:2934–43. doi: 10.1158/0008-5472.CAN-07-6487. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe N, Takaoka M, Sakurama K, Tomono Y, Hatakeyama S, Ohmori O, et al. Dual tyrosine kinase inhibitor for focal adhesion kinase and insulin-like growth factor-I receptor exhibits anticancer effect in esophageal adenocarcinoma in vitro and in vivo. Clin Cancer Res. 2008;14:4631–9. doi: 10.1158/1078-0432.CCR-07-4755. [DOI] [PubMed] [Google Scholar]
- 30.Wang ZG, Fukazawa T, Nishikawa T, Watanabe N, Sakurama K, Motoki T, et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncol Rep. 2008;20:1473–7. [PubMed] [Google Scholar]