

Abstract
The prognosis of patients suffering from tumors of the Ewing family (EFT) is still poor. Immunotherapy strategies are pursued and EFT-specific antigens have to be identified as targets for cytotoxic T-lymphocytes (CTL). Due to the lack of expression of cancer/testis antigens (CTA) in normal tissues, these antigens are partially able to induce immune responses in cancer patients. Therefore, they are promising targets for immunotherapy. EFT are characterized by chromosomal rearrangements involving members of the TET (translocated in liposarcoma, Ewing sarcoma breakpoint region 1, TATA box binding protein-associated factor 15) family of RNA binding proteins and members of the E-26 (ETS) family of transcription factors. The resulting onco-fusion proteins are highly specific for EFT and downstream targets of TET-ETS represent candidate tumor specific antigens. In order to identify new EFT-associated CTA, we analyzed microarray-data sets from EFT and normal tissues from the Gene Expression Omnibus (GEO) database. The impact of TET-ETS on expression of CTA was analyzed using GEO data sets from transgenic mesenchymal stem cells. One CTA with high specificity for EFT is lipase I (LIPI, membrane-associated phospholipase A1-β). CTL specific for LIPI-derived peptides LDYTDAKFV and NLLKHGASL were able to lyse HLA-A2 positive EFT cells in vitro which confirms the possible role of LIPI and other CTA for EFT-immunotherapy.
Keywords: Ewing sarcoma, gene expression, cancer/testis antigens, TET-ETS, LIPI
Introduction
Ewing sarcoma, also named as Ewing family tumors (EFT) are the second most common solid malignancies of bone and soft tissues in childhood and adolescence. EFT are characterized by expression of fusion proteins caused by chromosomal rearrangements, most often involving the RNA binding protein Ewing sarcoma breakpoint region 1 (EWSR1) and a member of the E-26 (ETS) family of transcription factors. EWSR1 is a member of the TET (translocated in liposarcoma, Ewing sarcoma breakpoint region 1, TATA box binding protein-associated factor 15) family of RNA binding proteins. Several different gene fusions between TET family members and a plethora of fusion partners have been described (for a review see ref. 1). Patients with localized EFT treated with a conventional multimodal therapy regime have a five-year-survival of approximately 65%, whereas patients with primary metastases have only a five-year-survival of 25%.2 The five-year overall survival for early recurrences (within less than two years) is only 7%.3 The risk of secondary neoplasms after high-dose chemotherapy is dramatically enhanced and 1–2% of patients suffer from it. New therapy strategies faced toward the poor prognoses of EFT that are pursued in many publications are, for example, different forms of immunotherapy. A class of EFT-specific antigens that could be used in immunotherapy is composed of so-called cancer/testis antigens (CTA) that are physiologically expressed only in germ cells, stem cells and during embryogenesis.4,5 CTA seem to have critical functions in oncogenesis and survival of malignant cells.5 Undifferentiated, highly neoplastic malignancies express very often a broader spectrum of different CTA. A high expression of CTA can be associated with a worse outcome.6 By using CTA as therapeutic targets, new ground is broken to fight the malignant phenotype of EFT because CTA are likely to be essential for the malignant and undifferentiated phenotype. For many CTA, approximately half of all known CTA listed in the CTA database of the Ludwig-Institute for Cancer Research,7 spontaneous humoral, cellular or inducible immune responses were described. For instance, X antigen family member 1 (XAGE1) that is also expressed in EFT, induces spontaneous cellular immune responses in patients after surgical therapy that was correlated with prolonged survival.8 A goal of the present study was to identify new EFT-specific CTA. Already identified CTA in other neoplastic tissues were checked in microarray data for their expression in EFT. Furthermore genes with a CTA-specific expression profile (overexpression restricted to testes and EFT) were identified as new CTA-candidates in EFT. The chimeric aberrant transcription factors of the TET-ETS family play a central role in malignant transformation and proliferation of EFT.1 Unfortunately, the fusion region of the most common EWSR1-Friend leukemia virus integration 1 (FLI1) onco-fusion proteins encode only weak HLA class I binding peptides for common HLA molecules.9 However, down-stream targets of TET-ETS in combination with a CTA-expression profile are interesting targets for immunotherapy. For that reason an induction of CTA in TET-ETS transgenic mesenchymal stem cells was investigated through analysis of public assessable microarray-data sets.
To evaluate the immunogenic and therapeutic potential of CTA in EFT, T-cells were generated with specificity for the known CTA lipase I alias membrane-associated phospholipase A1-β (LIPI) characteristically expressed in EFT10-12 and having probably important roles in pathophysiology in EFT via production of lysophosphatidic acid.13 Due to its putative role for oncogenesis and proliferation and its selective expression LIPI represents an excellent immunotherapeutic target.
Results
EFT-associated CTA
For identification of putative EFT-associated CTA we filtered microarray data (Table 1) for probe sets with high signal intensities in testis and at least one EFT sample. A total of 2625 probe sets showed at least 2.5 times higher signal intensities in testis than the 90. percentile of normal body atlas (NBA) samples. 953 of these probe sets showed also a minimal signal intensity of 100 in at least one of the EFT samples. In this analysis, cell lines were not included. The identified probe sets are shown in Table S1. Among these probe sets we identified known EFT associated CTA, e.g., enhancer of zeste homolog 2 (EZH2),10,14-16 XAGE1,17,18 and testis expressed 15 (TEX15).11 Approximately 1/3 of these probe sets showed overexpression in less than 5% of the EFT samples. Only 88 probe sets (9.2%) showed overexpression in more than 1/3 of the EFT samples. 75 of the identified 953 probe sets were specific for known CTA from the CTA database of the Ludwig Institute for Cancer Research.7 These probe sets were summarized in Table S2. In our earlier studies, we observed that expression of some CTA is not detectable in testis in microarray data sets. Therefore, we used the CTA database of the Ludwig Institute for Cancer Research7 and analyzed the expression of known CTA in EFT samples independent of microarray data from testis. The percentages of EFT samples in which expression of a known CTA was found are shown in Table S3. Among the known CTA from the database of the Ludwig Institute for Cancer Research, we found only one probe set (with specificity for LIPI) with high signal intensities in more than 80% of EFT samples. Nine additional probe sets showed high signal intensities in more than 50% of the EFT samples. A total of 14 probe sets showed overexpression in more than 1/3 of the EFT samples. From the probe sets in Tables S1 and S3 (probe sets that are present in both tables have been counted only once) 464 were statistically significant upregulated in EFT compared with NBA tissues (p < 0.01; Bonferroni corrected for 987 tests). As shown in Figure 1, EFT cell lines showed higher expression for several of these genes than native EFT samples. Moreover, embryonic stem cells, mesenchymal stem cells (MSC) and endothelial cells showed some expression of EFT-associated CTA. From the other tissues from NBA, weak expression of EFT-associated CTA was only detected in bone marrow and thymus. As expected, testis and isolated sperm showed high signal intensities for the majority of detected CTA (Fig. 1). The cluster analysis in Figure 1 clearly indicate the presence of two clusters of EFT-associated CTA, one cluster with expression in stem cells and one cluster with expression only in EFT and testis.
Table 1. Description of the analyzed microarray data sets.
| Experiments | Samples | Reference |
|---|---|---|
|
GSE8596 |
Ewing tumors (7 cell lines) |
42 |
|
GSE12102 |
Ewing tumors (37 tumors) |
43 |
|
GSE17679 |
Ewing tumors (11 cell lines) |
44 |
|
GSE34620 |
Ewing tumors (117 tumors) |
45 |
|
GSE34800 |
Ewing tumors (4 tumors) |
46 |
|
GSE36133 |
Ewing tumors (9 cell lines) |
47 |
|
GSE3526 |
Normal tissues (160 selected samples) |
48 |
|
GSE3526 |
HUVECS (2 samples) |
48 |
|
GSE9677 |
HUVECS (3 un-stimulated samples) |
49 |
|
GSE6257 |
Lymphatic endothelial cells (3 samples) |
50 |
|
GSE8590 |
Embryonic stem cells (6 samples) |
51 |
|
GSE22392 |
Embryonic stem cells (2 samples) |
52 |
|
GSE26862 |
Embryonic stem cells (3 samples) |
53 |
|
GSE3526 |
Testis (5 samples) |
48 |
|
GSE6969 |
Sperm (13 normal samples) |
54 |
|
GSE8665 |
MSC (14 samples; transgenic) |
42 |
|
GSE9520 |
MSC (6 samples) |
55 |
| GSE31215 | MSC (8 samples; transgenic) | 19 |
DNA microarray data sets from the sources above were down-loaded from the GEO database and used for analysis of known CTA and identification of putative new CTA in EFT.
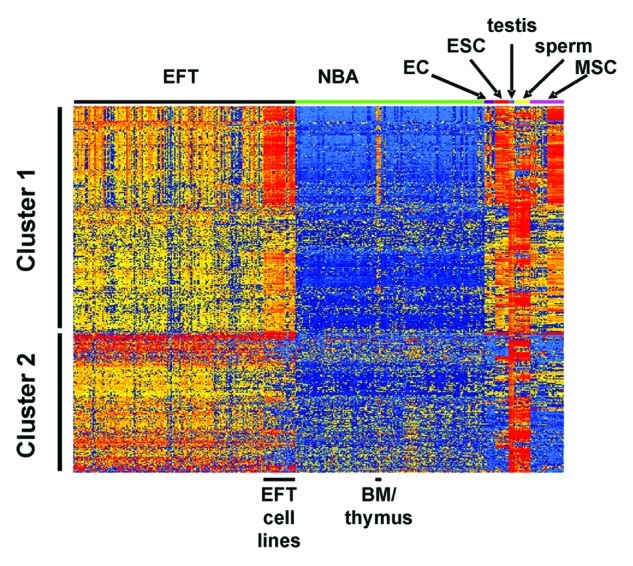
Figure 1. Expression pattern of EFT-associated CTA. EFT-associated CTA were identified as described in the text and used for cluster analysis (Manhattan distance, log2-transformed and median centered signal intensities). The positions of EFT samples and selected normal tissues (bone marrow (BM), endothelial cells (EC), embryonic stem cells (ESC), mesenchymal stem cells (MSC)) are indicated. The following GEO data sets were used: GSE8596, GSE12102, GSE17679, GSE34620, GSE34800, GSE36133, GSE3526, GSE9677, GSE6257, GSE8590, GSE22392, GSE26862, GSE6969, GSE8665, GSE9520 and GSE31215.
CTA expression in EWSR1-ETS transgenic MSC
We asked which of the identified CTA are induced by expression of EWSR1-ETS onco-fusion proteins in MSC, the suspected EFT cell of origin. For this end, we analyzed GEO data sets GSE8665 and GSE31215. Twenty-six probe sets from the total 464 probe sets showed at least in one of the analyzed experiments an upregulation by EWSR1-FLI1 or EWSR1-ERG (Table 2).
Table 2. TET-ETS regulated EFT-associated CTA.
| UNIQID | Gene symbol | Mean TET-ETS | Mean control |
|---|---|---|---|
| 203358_s_at |
EZH2 |
2659.83 |
1019.50 |
| 204170_s_at |
CKS2 |
13703.17 |
8938.17 |
| 206645_s_at |
NR0B1 |
1564.17 |
53.17 |
| 206743_s_at |
ASGR1 |
181.50 |
10.00 |
| 207666_x_at |
SSX3 |
127.83 |
7.67 |
| 208934_s_at |
LGALS8 |
4404.33 |
1244.50 |
| 209781_s_at |
KHDRBS3 |
1047.50 |
288.33 |
| 209967_s_at |
CREM |
1748.33 |
794.00 |
| 210187_at |
FKBP1A |
505.67 |
300.83 |
| 210497_x_at |
SSX2 /// LOC653088 |
219.17 |
31.33 |
| 210874_s_at |
NAT6 |
184.33 |
93.83 |
| 211425_x_at |
SSX4 /// SSX4B |
191.00 |
36.17 |
| 213644_at |
CCDC46 |
909.67 |
342.00 |
| 216471_x_at |
SSX2 /// LOC653088 |
240.33 |
22.50 |
| 219211_at |
USP18 |
596.00 |
247.33 |
| 222240_s_at |
ISYNA1 |
269.67 |
86.50 |
| 223557_s_at |
TMEFF2 |
4034.33 |
291.33 |
| 223700_at |
MND1 |
636.00 |
454.00 |
| 228988_at |
ZNF711 |
243.67 |
84.83 |
| 229622_at |
FLJ37034 |
3288.00 |
305.83 |
| 230511_at |
CREM |
305.83 |
155.17 |
| 232792_at |
RNF36 |
151.33 |
57.17 |
| 240950_s_at |
FLJ32658 |
1907.17 |
34.83 |
| 241117_at |
LOXHD1 |
90.50 |
5.17 |
| 242178_at |
LIPI |
191.00 |
19.50 |
| 242491_at | DKFZP686P18101 | 86.33 | 13.33 |
Regulation of the CTA from Figure 1 by TET-ETS onco-fusion proteins was analyzed in GEO data sets GSE8665 and GSE31215. Presented are the mean signal intensities of TET-ETS transgenic MSC and control MSC (from GSE8665 only the values 72h after induction of EWSR1-FLI1 or EWSR1-ERG expression were used).
Generation of LIPI-specific cytotoxic T-lymphocytes (CTL)
For the further experiments we selected LIPI because this CTA showed very high specificity for EFT (Fig. 2) and seems to be regulated by TET-ETS onco-fusion proteins at least in MSC from young donors19 (Table 2). Dendritic cells (DC) and T cell populations were characterized by flow cytometry. Mature DC expressed MHC class I and II, CD83, CD40, CD80 and CD86. Re-stimulated T-cells were a mixture of CD8-positive and CD4-positive cells and expressed CD25 and CD154 (data not shown). First we tested, whether LIPI specific T cells were generated by priming T cells with tumor lysate-pulsed DC. Peripheral blood mononuclear cells (PBMC) from 5 HLA-A2-positive donors were primed with SK-N-MC lysate-pulsed DC and were re-stimulated with SK-N-MC lysate-pulsed HLA-A2-positive lymphoblastoid B cells (LCL). Primed T cells were tested in interferon γ enzyme-linked immunospot assay (IFNγ ELISPOT). PBMC from one of the 5 donors led to significant EFT-specific IFNγ-release in ELISPOT (Fig. 3). For IFNγ-ELISPOT HLA-A2 negative SK-N-MC-cells and HLA-A2-positive A673-cells were used to investigate EFT-specific IFNγ-release. Furthermore HLA-A2-positive LCL were used as antigen presenting cells (APC) in ELISPOT and were pulsed with peptides derived from LIPI or from cyclin D1 (CCND1). These peptides were used because CCND1 is highly expressed in proliferating EFT cells10 and both peptides elicit immune responses in HLA-A2-positive donors.20 CTL without re-stimulation during ELISPOT were used as negative controls.
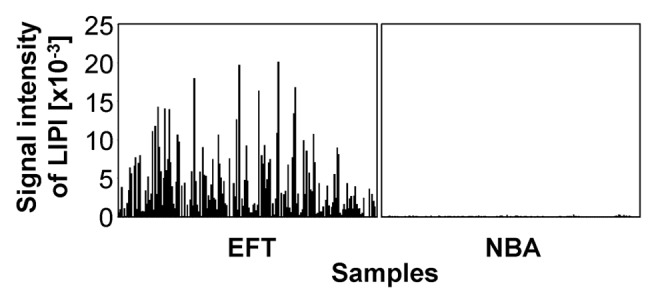
Figure 2. Expression pattern of the CTA LIPI. Presented are signal intensities of LIPI in the used microarray database. The following GEO data sets were used: GSE8596, GSE12102, GSE17679, GSE34620, GSE34800, GSE36133, GSE3526, GSE9677, GSE6257, GSE8590, GSE22392, GSE26862, GSE6969 and GSE8665.
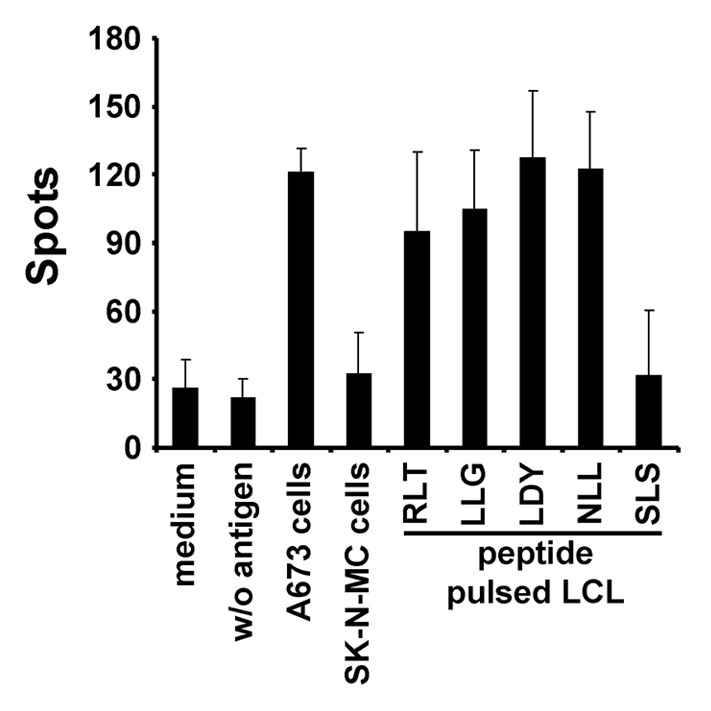
Figure 3. ELISPOT analysis of PBMC after priming with tumor lysate. PBMC of a HLA-A2-positive donor were primed with SK-N-MC lysate-pulsed DC and re-stimulated once with SK-N-MC lysate-pulsed HLA-A2-positive LCL. No significant release of IFNγ was observed in the presence of HLA-A2-negative SK-N-MC cells during ELISPOT assay and in the presence of LCL pulsed with LIPI peptide SLS. Significant IFNγ-release was observed for CCND1 peptides LLG and RLT and for LIPI peptides LDY and NLL. CTL re-stimulated during the ELISPOT assay with HLA-A2-positive A673 cells elicited also significantly increased levels of IFNγ.
No significant IFNγ-release was observed for HLA-A2-negative SK-N-MC-cells (p = 0.16) as APC. No significant results were received for LIPI peptide SLS (p = 0.26) presented by LCL. Significant IFNγ-release was gained for CCND1 peptides LLG (p = 0.02) and RLT (p = 0.02) and for LIPI peptides LDY (p = 0.03) and NLL (p = 0.03) as well as for HLA-A2-positive21-23 A673-cells (p = 0.01).
We asked whether it is possible to stimulate LIPI-specific T cells directly by peptides. T-cells from 8 donors were primed with LIPI-peptides. CCND1-peptides were used as positive control.2 In addition, T cells were primed with tumor lysate-pulsed DC. Re-stimulation was accomplished with HLA-A2-positive LCL and the same antigens. CTL that were primed with un-pulsed DC and re-stimulated with un-pulsed LCL served as negative control. In order to measure cell mediated cytotoxicity against HLA-A2-positive A673-cells, lactate dehydrogenase release assay (LDH assay) was performed as described below. Two of the 8 donors showed specific killing of A673 tumor cells after priming with LIPI peptides (Fig. 4). CTL from donor 1 exerted significant lysis of target cells after priming with A673 lysate (p = 0.02), LIPI peptides LDY (p = 0.01) and NLL (p = 0.03) as well as CCND1 peptide RLT (p = 0.03). No significant release was elicited by CTL stimulated with LIPI peptide SLS (p = 0.5), CCND1 peptide LLG (p = 0.08) or SK-N-MC lysate (p = 0.31). Similarly, CTL from donor 2 primed with LIPI peptide SLS showed no significant LDH-release (p = 0.46). Significant cell mediated cytotoxicity occurred for CTL from donor 2 after stimulation with A673 lysate (p = 0.04), SK-N-MC lysate (p = 0.005), LIPI peptides LDY (p = 0.004) and NLL (p = 0.003) as well as CCND1 peptides LLG (p = 0.003) and RLT (p = 0.01).
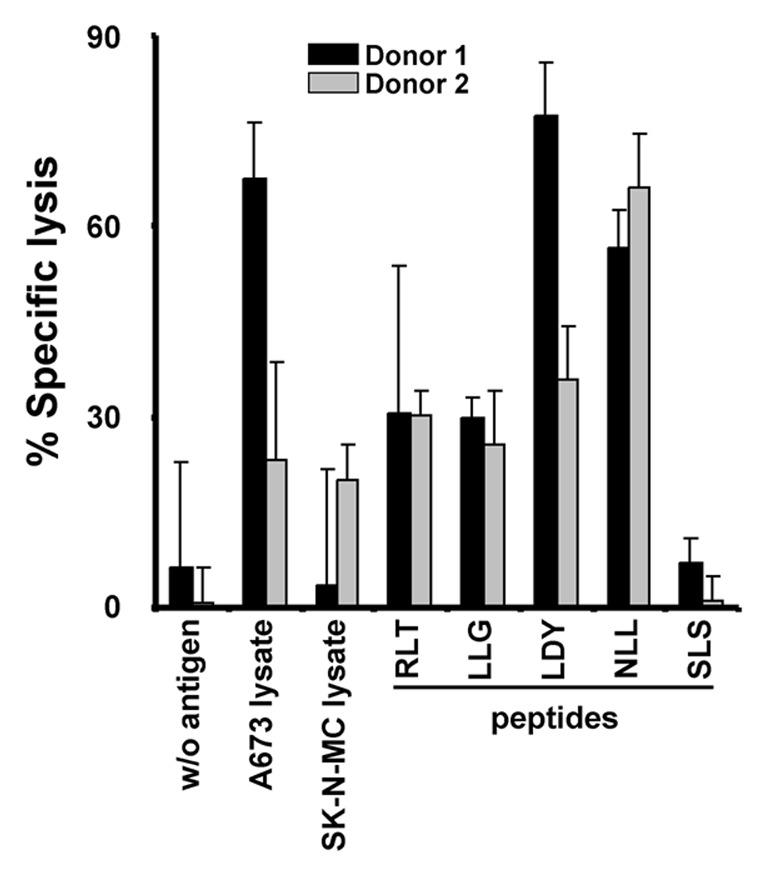
Figure 4. Lysis of HLA-A2-positive A673 cells by LIPI-stimulated CTL. T-cells from two HLA-A2-positive donors were stimulated with DC that were pulsed with SK-N-MC lysate, A673 lysate, CCND1 peptides RLT and LLG or LIPI peptides SLS, NLL and LDY. Cells were re-stimulated with HLA-A2-positive LCL and the same antigens. CTL that were primed with un-pulsed DC and re-stimulated with un-pulsed LCL served as negative controls. The cell mediated cytotoxicity of the CTL against living A673 tumor cells was analyzed in LDH assay.
Discussion
It is known that different CTA are often simultaneously expressed in one tumor tissue and that high expression of CTA is correlated with bad outcome.6 The high portion of CTA that could be detected in the investigated EFT samples in our microarray-analysis emphasizes the malignant phenotype of EFT. CTA are often involved in pathways that are critical for metastatic capacity, proliferation and maintenance of an undifferentiated phenotype with stem cell character.6 Expression of CTA during embryogenesis and in MSC was already described4 and was proved in the microarray-analysis of this paper. For some CTA moderate expression in MSC, embryonic stem cells and endothelial cells was found (Fig. 1). Selective expression of CTA during embryogenesis and in stem cells highlights the function of CTA for maintenance of stem cell character in malignancies. A loss of antigens by using CTA as therapeutic targets is unlikely to occur when CTA have critical functions to support the malignant character. Pathways of newly identified CTA-candidates in this paper are predominantly associated with resistance to chemotherapy or enhanced metastatic capability of EFT,24 e.g., new CTA-candidates are involved in transcription, cell cycle, apoptosis or the ubiquitin/proteasom pathway.
Many known CTA being expressed in other malignancies and registered in the CTA-database7 were detected here for the first time in EFT or were expressed in a high percentage of all EFT-samples (Tables S2 and S3). Only few of those have been described already to be expressed in EFT: aristaless related homeobox (ARX),10 LIPI,10-12 TEX15,11 and XAGE1.17,18 LIPI showed no expression in testes in the microarray analysis. Lack of detectability of CTA in testes in microarray analysis was also found by other authors.25 By reverse transcriptase-polymerase chain reaction LIPI is detectable in testis.11 Some of the identified CTA (LIPI, ARX, EZH2) are known to be highly specific in EFT.10
Sustained expression of TET-ETS onco-fusion proteins is crucial for the malignant phenotype of EFT. Several studies that aimed at inhibiting EWSR1-ETS function were able to point out this aspect.26 Genes featuring CTA-specific expression patterns and being downstream-targets of EWSR1-ETS are likely to be specific for EFT and constituting the malignant phenotype. Thus we investigated whether the newly identified CTA-candidates and CTA in EFT are downstream-targets of EWSR1-ETS. The EFT cell-of-origin may be derived from a cell with neural- and mesenchymal stem cell characteristics.10 EWSR1-FLI1 and EWSR1-ERG transgenic MSC mimic essential steps of the histogenesis of EFT quite well. The EWSR1-ETS target gene EZH2 has been described as highly specific for EFT.10 EZH2 is known to be essential for maintaining the malignant phenotype of EFT14,26 and might, therefore, be an ideal target for immunotherapy.16
Another interesting target is LIPI. LIPI might be involved in oncogenesis by initiating metastasis and invasivity through its enzymatic production of lysophosphatidic acid (LPA).27 LPA receptors 1–6 were detected in EFT.12 These receptors are critical in activating important signaling pathways for proliferation, tumor cell migration and invasion. Moreover the activation of LPA receptor 1 advances the process of neoangiogenesis and enhances permeability of endothelial cells.27 In the present paper we demonstrate that the peptides LDY and NLL from LIPI were able to induce cellular immune responses in vitro. Both epitopes seem to be presented on MHC molecules by A673 cells. Lysis of A673 cells was achieved by NLL- and LDY-primed CTL. Both peptides were selected on their binding to HLA-A2. Consequently only the subpopulation of HLA-A2-positive patients would be amenable for an immunotherapy with these LIPI peptides. Identification of other immunogenic epitopes is needed for other HLA alleles. CTL which were able to induce specific immune responses were derived from healthy donors. CTL that are derived from patients suffering from EFT could be even primed against diverse CTA in vivo. Boosting a specific immune response by autologous T-cells bearing an adequate T cell receptor for a CTA could be easier achievable in EFT-patients for that reason.28 The investigated peptides of CCND1 that also induced cellular immune responses are not specific for tumor cells and could induce autoimmune phenomena. Tumor cell lysate represents the complete undefined repertoire of tumor associated antigens including the unique mutations of the individual neoplasm. But substantial disadvantages in using tumor cell lysate are the need of autologous tumor cells, the often inappropriate concentration of tumor associated antigens leading to deletion or apoptosis of specific T cells and the risk of cross reactions with ubiquitously expressed antigens. This risk is decreased by using CTA with a highly specific expression in malignancies but absent expression in most tissues of the NBA. Immunization against distinct CTA could induce selection of tumor cells that does not present corresponding epitopes. But CTA often participate in malignant transformation and proliferation so that a loss of them is likely to conduct to loss of the malignant phenotype either. The risk of potential loss of antigens by using such targets can be bypassed also by immunization against diverse epitopes derived from different CTA.29 A polyvalent vaccine should provide diverse tumor-specific epitopes in a natural protein context, e.g., by using recombinant proteins. Those peptides are processed and presented exclusively by professional APC, leading to more effective, sustained T-cell responses.30,31 Restriction to distinct HLA-haplotypes could be avoided as well by using polyvalent vaccines. There exist several clinical trials supporting the high efficiency of adoptive T-cell-transfer.32 Adoptive cell transfers of in vitro expanded autologous tumor-infiltrating lymphocytes or lymphocytes engineered to recognize e.g., CTA can be employed as effective, safe vaccines.32 Some CTA are expressed in distinct normal cell populations, e.g., mesenchymal stem cells.4 The expression of tumor associated antigens in normal cells can be a limitation for immunotherapy by initiating severe autoimmune effects. Indeed, autoimmune phenomena have been observed as side effects from immunotherapy.33 Especially in the case of CTA, immunotherapy might negatively influence fertility. However, clinical observations indicate only low toxicities of CTA-specific T cells.8,34,35 This provides promising forecasts using CTA-specific T-cells for immunotherapy. Thus identification of highly tumor-specific CTA is a precious contribution to immunotherapeutic strategies.
Material and Methods
Cells and cell culture
PBMC, DC, EFT cell lines A67336 and SK-N-MC37 were maintained in RPMI medium (Biochrom) supplemented with 10% fetal calf serum (Biochrom), penicillin (10,000 U/mL) and streptomycin (10,000 μg/mL; Biochrom). HLA-haplotypes of EFT cells lines were determined as described.21 The HLA-A2-positive Epstein Barr virus-immortalized LCL A2+2310 was established in our laboratory38 and maintained in X-VIVO 15 Medium (Lonza). LCL were used as APC in IFNγ-ELISPOT assay (Becton-Dickinson) or for re-stimulation of PBMC. LCL were inactivated by treatment with mitomycin C (50 mg/mL, 30 min, 37°C; Roche).
PBMC from healthy HLA-A2-positive donors were isolated as described39 with informed consent and approval by the ethics committee of the Medical Faculty of the Martin-Luther-University Halle-Wittenberg.
Generation of EFT-specific T cells
PBMC were enriched for CD8 positive cells by magnetic cell sorting (Miltenyi). CD8 positive cells were afterwards mixed with fresh autologous PBMC, so that the ratio between CD4-positive and CD8-positive cells was about 1:2. Cells were analyzed using flow cytometry (FACScan, CellQuestPro-Software, Becton-Dickinson).38
DC were obtained from the adherent fraction of PBMC and were used for priming of PBMC. On day one and three, 1000 U/mL granulocyte-macrophage colony stimulating factor (Leukine, Berlex Laboratories) and 300 U/mL interleukin 4 (IL, R&D Systems) were added to DC. 50,000 DC/mL were pulsed with 0.5 μg/mL of peptides. The following peptides were used: NLLKHGASL (derived from LIPI, amino acids 163–171), LDYTDAKFV (derived from LIPI, 219–227), SLSVHIKNC (derived from LIPI, 156–164), RLTRFLSRV (derived from CCND1, 228–236), LLGATCMFV (derived from CCND1, 101–109). Peptide-synthesis was conducted by WITA GmbH, Telto, Germany. In silico prediction of the probability to bind HLA-A2 was achieved by using the SYFPEITHI-database.40
Cell lysates from A673 cells and SK-N-MC cells were generated by three cycles of freezing and thawing. A673 lysates und SK-N-MC lysates were used to pulse DC. A ratio of 50,000 DC/mL to 8,000 lysed tumor cells/mL was applied. Moreover, 3 μg/mL β2‑microglobulin (Roche Diagnostics) were added. Antigen exposition was performed for a total of 6 to 7 h. In order to promote maturation of DC, after two hours of antigen exposition 1 μg/mL lipopolysaccharide (Sigma-Aldrich) and 20 ng/mL IFNγ (R&D Systems) were added. Afterwards 1.3 × 106 PBMC enriched for CD8-positive cells and 10 ng/mL IL-7 (R&D Systems) were added to 50,000 DC. Twenty-four and 48 h after stimulation, 10 U/mL IL-2 (R&D Systems) were added. Seven days after priming of naive PBMC, re-stimulation was accomplished with the appropriate antigen. Further re-stimulations were done weekly. For re-stimulation, antigen-pulsed and mitomycin C-inactivated LCL were applied. 200,000 LCL in 500 μL X-VIVO 15 medium were pulsed with 4 μg/mL peptide or with cell lysates (according to 8,000 lysed cells/mL) and 3 μg/mL β2-microglobulin. LCL were added to PBMC after two hours of pulsing. The first and the second day after re-stimulation, 10 U/mL IL2 were added.
PBMC were phenotypically characterized after the first successful re-stimulation by flow cytometry.
Measurement of cell-mediated cytotoxicity and EFT-specific IFNγ-release
PBMC of 34 HLA-A2 positive donors were used. In 13 cases proliferation was strong enough to carry out further characterization in IFN γ ELISPOT (Becton-Dickinson) or LDH assay (Roche Diagnostics). PBMC of 5 donors were investigated in IFNγ ELISPOT and PBMC derived from 8 donors were investigated in LDH assay. IFNγ ELISPOT was performed following the instructions of the manufacturer. CD8-enriched PBMC intended to be analyzed by IFNγ ELISPOT were primed with SK-N-MC lysate-pulsed DC. IFNγ release was measured one week after first re-stimulation during exposition to peptides derived from LIPI or CCND1 presented on allogeneic HLA-A2 positive LCL or to HLA-A2 positive A673 or HLA-A2 negative21 SK-N-MC tumor cells. Negative controls include CD8-enriched PBMC without re-stimulation during IFNγ ELISPOT. Experiments were accomplished in triplicates.
Autologous DC intended to apply in LDH assay were pulsed with A673 lysate, SK-N-MC lysate, peptides derived from CCND1 or LIPI, respectively. Un-pulsed DC served as negative controls. Seven days after second re-stimulation with the appropriate antigen presented on allogeneic LCL, measurement of cell-mediated cytotoxicity by CTL during exposure to living A673 cells was accomplished using LDH assay. This assay was performed following the manufacturers’ instructions.
Identification of CTA-specific expression profiles
For identification of CTA-specific expression profiles, microarray data sets from Affymetrix HG_U133Plus2.0 microarrays including expression data from EFT and normal tissues NBA were analyzed. Expression data were obtained from the Gene Expression Omnibus (GEO) database.42 The used data sets19,43-55 are summarized in Table 1. Primary data analysis was performed with Expression Console (Affymetrix) applying the Microarray Suite 5 algorithm. All microarray data sets were scaled to the same target intensity of 500. Large data tables were processed for subsequent analysis by using TableButler.56 Data visualization and cluster analysis was performed with Genesis.57
Two approaches were pursued in order to filter the microarray-data sets for CTA-like expression patterns. First, genes showing high signal intensities in testes and EFT and at the same time low signal intensities in the NBA were identified in microarray-data sets. Genes were proposed to show overexpression in EFT or testes, when the median of signal intensities in EFT and the median of signal intensities in testes were 2.5 times higher than the 90th percentile of the reference tissues of the NBA. Cell lines and sperm were not included in the database that was used for identification of putative new CTA but were included in subsequent cluster analysis. Calculations were performed with the MAfilter software.21 In a second approach, already known CTA that are listed in the CTA-database of the Ludwig Institute for Cancer Research7 were checked for expression in EFT in microarray-data sets. Instead of using a fixed threshold with supposed expression of a gene when this threshold is exceeded, expression of a known CTA was assessed employing a relative threshold. This relative threshold implied the relation between expression values of the NBA tissue and EFT. When the expression value of a known CTA in one EFT exceeded 2.5 times the 90.percentile of all NBA tissues, overexpression of this CTA was presumed. The percentage of EFT-samples with overexpression of a distinct known CTA was assessed. Pathways of newly identified CTA-candidates and known CTA (but mentioned the first time in EFT) were determined according to the SOURCE database.58
Expression analysis of EWSR1-ETS transgenic MSC
In order to investigate the impact of EFT-specific fusion proteins on the expression of CTA, GEO data sets from EWS-ETS transgenic MSC were analyzed.19,42 Expression data sets from EWSR1-FLI1 transgenic MSC and expression data sets from EWSR1-ERG transgenic MSC were analyzed. Probe sets with signal intensities below 100 in the experimental situations were excluded from analysis. Genes were proposed to show overexpression in TET-ETS transgenic cells when the mean of signal intensities in transgenic cells was 2 times higher than the mean of the control transfected cells from the same experiment.
Supplementary Material
Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflict of interest.
Acknowledgments
We thank Ines Volkmer for grateful technical assistance.
Glossary
Abbreviations:
- APC
antigen presenting cells
- ARX
aristaless related homeobox
- CCND1
cyclin D1
- CTA
cancer/testis antigens
- CTL
cytotoxic T-lymphocytes
- DC
dendritic cells
- ETS
E-twenty-six
- EFT
Ewing family tumor
- ERG
v-ets related gene
- EWSR1
Ewing sarcoma breakpoint region 1
- EZH2
enhancer of zeste homologue 2
- FLI1
Friend leukemia virus integration 1
- GEO
Gene Expression Omnibus
- IFNγ ELISPOT
interferon γ enzyme-linked immunospot assay
- IL
interleukin
- LCL
lymphoblastoid B cell line
- LDH assay
lactate dehydrogenase release assay
- LDY
LDYTDAKFV
- LIPI
lipase I (membrane-associated phospholipase A1-beta)
- MSC
mesenchymal stem cells
- NBA
normal body atlas tissue
- NLL
NLLKHGASL
- PBMC
peripheral blood mononuclear cells
- SLS
SLSVHIKNC
- TET
translocated in liposarcoma, Ewing sarcoma breakpoint region 1, TATA box binding protein-associated factor 15
- TEX15
testis expressed 15
- XAGE1
X antigen family, member 1
Supplemental Materials
Supplemental materials may be found here:
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/23298
References
- 1.Staege M, Max D. Genetics and epigenetics of the TET-ETS translocation network. Genetics and Epigenetics. 2009;2:1–15. [Google Scholar]
- 2.Laurence V, Pierga JY, Barthier S, Babinet A, Alapetite C, Palangié T, et al. Long-term follow up of high-dose chemotherapy with autologous stem cell rescue in adults with Ewing tumor. Am J Clin Oncol. 2005;28:301–9. doi: 10.1097/01.coc.0000156921.28880.e1. [DOI] [PubMed] [Google Scholar]
- 3.Hawkins DS. Sarcomas gone bad: what to do about recurrent Ewing sarcoma. Pediatr Blood Cancer. 2011;57:535–6. doi: 10.1002/pbc.23126. [DOI] [PubMed] [Google Scholar]
- 4.Costa FF, Le Blanc K, Brodin B. Concise review: cancer/testis antigens, stem cells, and cancer. Stem Cells. 2007;25:707–11. doi: 10.1634/stemcells.2006-0469. [DOI] [PubMed] [Google Scholar]
- 5.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–25. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 6.Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014–21. doi: 10.1111/j.1349-7006.2009.01303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Almeida LG, Sakabe NJ, deOliveira AR, Silva MC, Mundstein AS, Cohen T, et al. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009;37(Database issue):D816–9. doi: 10.1093/nar/gkn673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kikuchi E, Yamazaki K, Nakayama E, Sato S, Uenaka A, Yamada N, et al. Prolonged survival of patients with lung adenocarcinoma expressing XAGE-1b and HLA class I antigens. Cancer Immun. 2008;8:13. [PMC free article] [PubMed] [Google Scholar]
- 9.Pfeifle C, Reinhardt K, Heins S, Burdach S, Staege MS. Development and characterization of HAT-sensitive Ewing tumour cells for immunotherapy. Anticancer Res. 2009;29:4489–96. [PubMed] [Google Scholar]
- 10.Staege MS, Hutter C, Neumann I, Foja S, Hattenhorst UE, Hansen G, et al. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res. 2004;64:8213–21. doi: 10.1158/0008-5472.CAN-03-4059. [DOI] [PubMed] [Google Scholar]
- 11.Foell JL, Hesse M, Volkmer I, Schmiedel BJ, Neumann I, Staege MS. Membrane-associated phospholipase A1 beta (LIPI) Is an Ewing tumour-associated cancer/testis antigen. Pediatr Blood Cancer. 2008;51:228–34. doi: 10.1002/pbc.21602. [DOI] [PubMed] [Google Scholar]
- 12.Schmiedel BJ, Hutter C, Hesse M, Staege MS. Expression of multiple membrane-associated phospholipase A1 beta transcript variants and lysophosphatidic acid receptors in Ewing tumor cells. Mol Biol Rep. 2011;38:4619–28. doi: 10.1007/s11033-010-0595-z. [DOI] [PubMed] [Google Scholar]
- 13.Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–81. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- 14.Burdach S, Plehm S, Unland R, Dirksen U, Borkhardt A, Staege MS, et al. Epigenetic maintenance of stemness and malignancy in peripheral neuroectodermal tumors by EZH2. Cell Cycle. 2009;8:1991–6. doi: 10.4161/cc.8.13.8929. [DOI] [PubMed] [Google Scholar]
- 15.Richter GH, Plehm S, Fasan A, Rössler S, Unland R, Bennani-Baiti IM, et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. 2009;106:5324–9. doi: 10.1073/pnas.0810759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiel U, Pirson S, Müller-Spahn C, Conrad H, Busch DH, Bernhard H, et al. Specific recognition and inhibition of Ewing tumour growth by antigen-specific allo-restricted cytotoxic T cells. Br J Cancer. 2011;104:948–56. doi: 10.1038/bjc.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu XF, Helman LJ, Yeung C, Bera TK, Lee B, Pastan I. XAGE-1, a new gene that is frequently expressed in Ewing’s sarcoma. Cancer Res. 2000;60:4752–5. [PubMed] [Google Scholar]
- 18.Brinkmann U, Vasmatzis G, Lee B, Pastan I. Novel genes in the PAGE and GAGE family of tumor antigens found by homology walking in the dbEST database. Cancer Res. 1999;59:1445–8. [PubMed] [Google Scholar]
- 19.Riggi N, Suvà ML, De Vito C, Provero P, Stehle JC, Baumer K, et al. EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate mesenchymal stem cell reprogramming toward Ewing sarcoma cancer stem cells. Genes Dev. 2010;24:916–32. doi: 10.1101/gad.1899710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Bergwelt-Baildon MS, Shimabukuro-Vornhagen A, Wendtner CM, Kondo E. Identification of native, immunogenic peptides from Cyclin D1. Leukemia. 2010;24:209–11. doi: 10.1038/leu.2009.184. [DOI] [PubMed] [Google Scholar]
- 21.Winkler C, Steingrube DS, Altermann W, Schlaf G, Max D, Kewitz S, et al. Hodgkin’s lymphoma RNA-transfected dendritic cells induce cancer/testis antigen-specific immune responses. Cancer. Immunol Immunother. 2012;March 15:1–11. doi: 10.1007/s00262-012-1239-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Staege MS, Hansen G, Baersch G, Burdach S. Functional and molecular characterization of interleukin-2 transgenic Ewing tumor cells for in vivo immunotherapy. Pediatr Blood Cancer. 2004;43:23–34. doi: 10.1002/pbc.20013. [DOI] [PubMed] [Google Scholar]
- 23.Staege MS, Gorelov V, Bulankin A, Fischer U, Dumon K, Hohndorf L, et al. Stable transgenic expression of IL-2 and HSV1-tk by single and fusion tumor cell lines bearing EWS/FLI-1 chimeric genes. Pediatr Hematol Oncol. 2003;20:119–40. doi: 10.1080/0880010390158612. [DOI] [PubMed] [Google Scholar]
- 24.Schaefer KL, Eisenacher M, Braun Y, Brachwitz K, Wai DH, Dirksen U, et al. Microarray analysis of Ewing’s sarcoma family of tumours reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur J Cancer. 2008;44:699–709. doi: 10.1016/j.ejca.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Hofmann O, Caballero OL, Stevenson BJ, Chen YT, Cohen T, Chua R, et al. Genome-wide analysis of cancer/testis gene expression. Proc Natl Acad Sci U S A. 2008;105:20422–7. doi: 10.1073/pnas.0810777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sankar S, Lessnick SL. Promiscuous partnerships in Ewing’s sarcoma. Cancer Genet. 2011;204:351–65. doi: 10.1016/j.cancergen.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Meeteren LA, Moolenaar WH. Regulation and biological activities of the autotaxin-LPA axis. Prog Lipid Res. 2007;46:145–60. doi: 10.1016/j.plipres.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Quintarelli C, Dotti G, De Angelis B, Hoyos V, Mims M, Luciano L, et al. Cytotoxic T lymphocytes directed to the preferentially expressed antigen of melanoma (PRAME) target chronic myeloid leukemia. Blood. 2008;112:1876–85. doi: 10.1182/blood-2008-04-150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kessler JH, Melief CJ. Identification of T-cell epitopes for cancer immunotherapy. Leukemia. 2007;21:1859–74. doi: 10.1038/sj.leu.2404787. [DOI] [PubMed] [Google Scholar]
- 30.Zwaveling S, Vierboom MP, Ferreira Mota SC, Hendriks JA, Ooms ME, Sutmuller RP, et al. Antitumor efficacy of wild-type p53-specific CD4(+) T-helper cells. Cancer Res. 2002;62:6187–93. [PubMed] [Google Scholar]
- 31.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turcotte S, Rosenberg SA. Immunotherapy for metastatic solid cancers. Adv Surg. 2011;45:341–60. doi: 10.1016/j.yasu.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amos SM, Duong CP, Westwood JA, Ritchie DS, Junghans RP, Darcy PK, et al. Autoimmunity associated with immunotherapy of cancer. Blood. 2011;118:499–509. doi: 10.1182/blood-2011-01-325266. [DOI] [PubMed] [Google Scholar]
- 34.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kono K, Iinuma H, Akutsu Y, Tanaka H, Hayashi N, Uchikado Y, et al. Multicenter, phase II clinical trial of cancer vaccination for advanced esophageal cancer with three peptides derived from novel cancer-testis antigens. J Transl Med. 2012;10:141. doi: 10.1186/1479-5876-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, et al. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. J Natl Cancer Inst. 1973;51:1417–23. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 37.Biedler JL, Helson L, Spengler BA. Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res. 1973;33:2643–52. [PubMed] [Google Scholar]
- 38.Hoennscheidt C, Max D, Richter N, Staege MS. Expression of CD4 on Epstein-Barr virus-immortalized B cells. Scand J Immunol. 2009;70:216–25. doi: 10.1111/j.1365-3083.2009.02286.x. [DOI] [PubMed] [Google Scholar]
- 39.Foell JL, Volkmer I, Giersberg C, Kornhuber M, Horneff G, Staege MS. Loss of detectability of Charcot-Leyden crystal protein transcripts in blood cells after treatment with dimethyl sulfoxide. J Immunol Methods. 2008;339:99–103. doi: 10.1016/j.jim.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 40.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–9. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 41.Barrett T, Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 2006;411:352–69. doi: 10.1016/S0076-6879(06)11019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyagawa Y, Okita H, Nakaijima H, Horiuchi Y, Sato B, Taguchi T, et al. Inducible expression of chimeric EWS/ETS proteins confers Ewing’s family tumor-like phenotypes to human mesenchymal progenitor cells. Mol Cell Biol. 2008;28:2125–37. doi: 10.1128/MCB.00740-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scotlandi K, Remondini D, Castellani G, Manara MC, Nardi F, Cantiani L, et al. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol. 2009;27:2209–16. doi: 10.1200/JCO.2008.19.2542. [DOI] [PubMed] [Google Scholar]
- 44.Savola S, Klami A, Myllykangas S, Manara C, Scotlandi K, Picci P, et al. High expression of complement component 5 (C5) at tumor site associates with superior survival in Ewing's sarcoma family of tumour patients. ISRN Oncol 2011; 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Postel-Vinay S, Véron AS, Tirode F, Pierron G, Reynaud S, Kovar H, et al. Common variants near TARDBP and EGR2 are associated with susceptibility to Ewing sarcoma. Nat Genet. 2012;44:323–7. doi: 10.1038/ng.1085. [DOI] [PubMed] [Google Scholar]
- 46.Pierron G, Tirode F, Lucchesi C, Reynaud S, Ballet S, Cohen-Gogo S, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet. 2012;44:461–6. doi: 10.1038/ng.1107. [DOI] [PubMed] [Google Scholar]
- 47.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC, et al. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7:67–80. doi: 10.1007/s10048-006-0032-6. [DOI] [PubMed] [Google Scholar]
- 49.Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, et al. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol. 2008;10:513–26. doi: 10.1038/ncb1714. [DOI] [PubMed] [Google Scholar]
- 50.Johnson LA, Clasper S, Holt AP, Lalor PF, Baban D, Jackson DG. An inflammation-induced mechanism for leukocyte transmigration across lymphatic vessel endothelium. J Exp Med. 2006;203:2763–77. doi: 10.1084/jem.20051759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Denis JA, Rochon-Beaucourt C, Champon B, Pietu G. Global transcriptional profiling of neural and mesenchymal progenitors derived from human embryonic stem cells reveals alternative developmental signaling pathways. Stem Cells Dev. 2011;20:1395–409. doi: 10.1089/scd.2010.0331. [DOI] [PubMed] [Google Scholar]
- 52.Chin MH, Pellegrini M, Plath K, Lowry WE. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell. 2010;7:263–9. doi: 10.1016/j.stem.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang P, Rodriguez RT, Wang J, Ghodasara A, Kim SK. Targeting SOX17 in human embryonic stem cells creates unique strategies for isolating and analyzing developing endoderm. Cell Stem Cell. 2011;8:335–46. doi: 10.1016/j.stem.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Platts AE, Dix DJ, Chemes HE, Thompson KE, Goodrich R, Rockett JC, et al. Success and failure in human spermatogenesis as revealed by teratozoospermic RNAs. Hum Mol Genet. 2007;16:763–73. doi: 10.1093/hmg/ddm012. [DOI] [PubMed] [Google Scholar]
- 55.Larson BL, Ylöstalo J, Prockop DJ. Human multipotent stromal cells undergo sharp transition from division to development in culture. Stem Cells. 2008;26:193–201. doi: 10.1634/stemcells.2007-0524. [DOI] [PubMed] [Google Scholar]
- 56.Schwager C, Wirkner U, Abdollahi A, Huber PE. TableButler - a Windows based tool for processing large data tables generated with high-throughput methods. BMC Bioinformatics. 2009;10:235. doi: 10.1186/1471-2105-10-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sturn A, Quackenbush J, Trajanoski Z. Genesis: cluster analysis of microarray data. Bioinformatics. 2002;18:207–8. doi: 10.1093/bioinformatics/18.1.207. [DOI] [PubMed] [Google Scholar]
- 58.Diehn M, Sherlock G, Binkley G, Jin H, Matese JC, Hernandez-Boussard T, et al. SOURCE: a unified genomic resource of functional annotations, ontologies, and gene expression data. Nucleic Acids Res. 2003;31:219–23. doi: 10.1093/nar/gkg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.