

Abstract
Cadherin-17 (CDH17), as a structurally unique member of the cadherin superfamily, has been identified to predict a poor prognosis for gastric cancer (GC). Our previous study demonstrated the positive correlation between CDH17 and lymph node micrometastasis in GC. We sought to further identify the role of CDH17 in the tumorigenesis and lymphatic metastasis of GC. Hence, we inhibited the CDH17 expression in MKN-45 gastric cancer cells by using RNA interference. Consequently, the malignant potency of cancer cells was evaluated, and the change in NFκB signaling pathway was also probed. Tumor growth and lymphatic metastasis model were conducted in nude mice to confirm the hypothesis. Downregulation of CDH17 not only suppressed the proliferation, adherence and invasion potency of MKN-45 cells, but also induced cell cycle arrest. Meanwhile, the NFκB signaling pathway was inactivated as well, with the reductions of downstream proteins including VEGF-C and MMP-9. Moreover, silencing CDH17 inhibited tumor growth in vivo significantly, and there was no lymph node metastasis detected in the mice without CDH17 expression, as opposed to the positive nodes found in controls. CDH17 is a novel oncogene in gastric cancer cells, which is associated with lymphatic metastasis and proliferation strongly. The inactivation of NFκB signaling pathway might be involved in targeting CDH17 in GC. On the whole, CDH17 is proposed to serve as a biomarker and attractive therapeutic target in GC.
Keywords: NFκB signaling pathway, RNA interference, cadherin-17, gastric cancer, lymphatic metastasis
Introduction
Gastric cancer is one of the most common alimentary malignancies, the motality due to which is only inferior to that of lung cancer in the world.1 Although early gastric cancer offers an excellent chance of cure based on surgical resection,2 the prognosis for advanced cancer remains poor, with a five year survival rate less than 40%,3 even in the patients receiving redical gastrectomy and chemotherapy.4 When it comes to those metastatic cases, that rate is ranging 5–15%.5 Lymph node metastasis (LNM) is generally responsible for the high mortality rate of gastric cancer.6 Especially in recent years, the importance of lymph node micrometastasis (LNMM) has been increasingly recognized,7 which is regarded as the supplement of current staging systems for predicting prognosis or planning adjuvant therapy.8 However, it remains difficult to diagnose lymphatic metastasis preoperatively or to supervise its recurrence postoperatively by imaging tests,9 let alone to diagnose the micrometastasis. Therefore, to identify the metastasis related biomarkers as auxiliary examination or therapeutic target trends to be essential.
Since the gastric cancer SAGE library has been created, more and more aberrant expressions and mutations of molecules associated with GC are being uncovered.10 By comparing gene expression profiles of gastric cancer at early and advanced stage, CDH17 is found to be the most upregulated gene in advanced gastric cancer.11 CDH17, as a unique member of cadherin family mediating cell-cell adhesion, expresses exclusively in human intestinal and pancreatic ductal epithelial cells,12 which is not supposed to be found in healthy stomach.13 A serial studies have identified the overexpression of CDH17 in gastric cancer predicted a poorer prognosis, which is also associated with deeper invasion and lymph node metastasis.14-17 However, there is inconsistency in recent research that CDH17 is a prognostic marker for early stage or node-negative gastric cancer.18
Respecting its controversial role in LNM, we investigated the relationship between CDH17 and LNMM in our earlier study. As results, we found CDH17 was the independent risk factor of LNMM, and associated with lymph vessel invasion closely, which suggested it initiating the process of LNM.19 Nevertheless, the biochemical mechanism of CDH17 promoting invasion and metastasis in GC remains incompletely defined. Hence, we knocked down CDH17 in gastric cancer cells by RNA interference and conducted multiple assays including proliferation, adhesion, invasion and cell cycle. We also analyzed the changes in NFκB signaling pathway, which is constitutively activated in GC and specifically regulates key mediators of lymphangiogenesis.20-23 Besides, in vivo assays of tumor growth and lymphatic metastasis were implemented as well.
Results
Stable knockdown of CDH17 in MKN-45 cell line
The sequences of four plasmids including pRI-CMV/eGFP-CDH17-miR-1, -2, -3 and -neg confirmed by DNA sequencing were consistent with the designed miRNA insert fragments (Fig. 1A). Compared with the MKN-45 cells receiving no treatment, the lentiviral vector expression cassette allowed for the permanent expression of green fluorescent protein (GFP) in transfection cells (Fig. 1B). The percentage of GFP positive cells was 94–98% (Fig. 1C). Moreover, the pRI-CMV/eGFP-CDH17-miR-2 group yielded the highest interference efficacy (80% in suppression) of CDH17 in both real time RT-PCR and western blot analyses (Fig. 1D and E), which was defined as knockdown group (miR-CDH17).

Figure 1. RNAi-mediated stable knockdown of CDH17 in MKN-45 cells. (A) The plasmids of pRI-CMV/eGFP-CDH17-miR-1, -2, -3 and -neg detected by DNA sequencing were consistent with the designed miRNA insert fragments. (B) High expression of GFP was observed in lentiviral vector transfected cells, as opposed to the negative expression of it in MKN-45 cells receiving no treatment (100μm). (C) 94–98% of transfected cells are GFP positive, determined by flow cytometry analysis. (D) Real time RT-PCR indicated that the miR-CDH17(2) was the optimal sequence, with 80% interference efficacy of CDH17, which was confirmed by (E) western blot analysis.
Targeting CDH17 repressed the malignant potency of MKN-45 cells
CDH17 was significantly downregulated in miR-CDH17 cells, as opposed to the high expression in both MKN-45 cells and miR-neg cells, confirmed by immunofluorescence staining (Fig. 2A). Furthermore, miR-CDH17 cells exhibited a considerable inhibition of cancer cell proliferation at the time points of 48h, 72h, 96h and 120h in MTT assay (p < 0.001) (Fig. 2B), and there were less miR-CDH17 cells sticking on the plastic plates coated with matrigel at the time points of 20min, 40min and 60min (p < 0.05) (Fig. 2C). Cell cycle analysis showed that more miR-CDH17 cells were arrested in G0/G1 phase (59.7 ± 1.23%, p < 0.001), with fewer cells in S phase (34.4 ± 1.77%, p = 0.004), compared with the other two control groups (Fig. 2D). In transwell assay, the migrated cells in miR-CDH17 group was less than those in miR-neg group and MKN-45 group (12.50 ± 3.55/HP vs. 50.33 ± 4.04/HP and 52.33 ± 3.51/HP, p < 0.001), which suggested that the invasion potency of MKN-45 cells was suppressed considerably by knocking down CDH17 (Fig. 2E). No significant difference was found between MKN-45 group and the miR-neg group.

Figure 2. Inhibition of CDH17 repressed the malignant potency of MKN-45 cells and alleviated the activation of NFκB signaling pathway. (A) Immunofluorescence staining demonstrated that CDH17 was significantly downregulated in miR-CDH17 cells, compared with the high expression in both MKN-45 and miR-neg cells (100μm). (B) Proliferation assay. (C) Adhesion assay. (D) Cell cycle analysis. (E) Invasion assay (100μm). (F) In western blot analysis, there was less expression of CDH17, nuclear p65, MMP-9 and VEGF-C, but more expression of IκB-α in miR-CDH17 group. There was no difference in total p65 among each group.
Inactivation of the NFκB signaling pathway aroused by targeting CDH17
The p65, as the active subunit of NF-κB, decreased significantly in the nucleus of miR-CDH17 cells, compared with those in controls (MKN-45 and miR-neg cells). Whereas, there was less degradation of IκB-α in miR-CDH17 cells, which kept NFκB as stable triple complex (IκB-α/p50/p65) in cytoplasm. Additionally, the down stream proteins regulated by NFκB containing MMP-9 and VEGF-C were also suppressed in miR-CDH17 cells (Fig. 2F). All the differences in the protein expressions indicated that the anti-tumor effects of targeting CDH17 on MKN-45 cells were probably via inactivating the NFκB signaling pathway.
Knockdown of CDH17 alleviated tumor growth and lymph node metastasis in vivo
On the post-inoculation day seven–nine, tumors of the two control groups were palpable in succession, which were also detected by optical in vivo imaging. Whereas the tumors of the miR-CDH17 group could be observed until two weeks after the inoculation, and grew more slowly than the other two controls, as shown in Figure 3A, B. Upon the termination of in vivo assay, tumor volumes of mock and miR-neg groups were larger than that of miR-CDH17 group (1551.67 ± 80.28 mm3 and 1079.83 ± 12.73 mm3 vs.148.50 ± 9.07 mm3, p < 0.001), as shown in Figure 3C and D.

Figure 3. Knockdown of CDH17 inhibited tumor growth and the expressions of NF-κB, MMP-9 and VEGF in tumor tissue. (A) Upon the termination, masses with fluorescence were larger in mock and miR-neg groups than those in miR-CDH17 group, which was coincident with the gross specimen (B, C). (D) The line chart illustrated that the tumor volume of miR-CDH17 was smaller than the other two groups consecutively, p < 0.05. (E) Expressions of p65 (NFκB), VEGF-C and MMP-9 were all suppressed in tumor tissue of miR-CDH17 nude mice (100μm). There was no significant difference between those in mock group and miR-neg group.
Furthermore, expressions of p65, VEGF-C and MMP-9 were all suppressed in tumor tissues of miR-CDH17 group (Fig. 3E), which was consistent with the results in vitro. It is worth of mention that VEGF-C was just proved to be one of the most eminent lymphatic endothelial growth factors.9,23
Figure 4A showed the morphological characteristics of claw pads in each group at the beginning and the end of lymphogenesis assay. Massive tumor nodules grew in the claws of the mock and miR-neg mice, which were larger than those of the miR-CDH17 mice. Of great interest, four mice developed metastasis in the inguinal region at fifth–sixth week after injection (two in mock group and two in miR-neg group), as shown by the presence of GFP-positive tumor cells, and there was no more new metastatic lesion detected until the termination. By contrast, mice of miR-CDH17 group had no metastasis consistently (Fig. 4B). Meanwhile, the anatomical gross specimens were consistent with the results from the in vivo imaging study, as shown in Figure 4C.

Figure 4. Knockdown of CDH17 expression inhibited lymph node metastasis in vivo. (A) The morphological characteristics of claw pads in each group were similar at the beginning, and then tumor nodules of the controls grew much larger than those of the miR-CDH17 group. (B) Two mice in both mock group and miR-neg group developed metastasis in the inguinal region, as shown by the presence of GFP-positive tumor cells. However, there was no fluorescent lesion detected in the mice of miR-CDH17 group, which was confirmed by (C) the anatomical gross specimens. (D) HE and immunohistochemisty of AE1/AE3 staining showed that the enlarged lymph nodes in mock and miR-neg group were occupied by tumor cells, as opposed to the normal lymph node in miR-CDH17 group. Additionally, CDH17 in lymphatic lesions expressed as high as it in primary ones (100 μm).
HE and immunohistochemical staining of AE1/AE3 confirmed that the enlarged lymph nodes of mock and miR-neg groups were occupied by tumor cells. Additionally, high expression of CDH17 in positive nodes demonstrated that the metastatic lesions had the similarities with the primary ones (Fig. 4D).
Discussion
Although CDH17 belongs to the cadherin superfamily accounting for the intercellular conjunction, unlike the classical cadherins, CDH17 is seemingly distinct that it can still retain its adhesive function without interacting with other cytoplasmic components.25 It has recently become more evident that such adhesion molecule plays an important role during the process of invasion or metastasis in colorectal, liver and gastric cancers,13,26-28 not only considered as a static intercellular glue. During the last couple of years, accumulating evidence suggested that high expression of CDH17 is associated with high metastatic potential, positive lymph node metastasis and short overall survival in gastric cancer patients.14,15 However, several researchers hold the opposite opinion that CDH17 predicts a better prognosis in GC with N0 status.18
In the context of controversial role of CDH17 in LNM of GC, we explored the relationship between CDH17 and LNMM to figure out whether it participated in the initiating process of LNM. As a result, we found that CDH17 was the independent risk factor of LNMM, predicting a poor prognosis for GC.19 Accordingly, our present study demonstrates the knockdown of CDH17 in a poorly differentiated GC cell line (MKN-45) by using RNAi, rendered the cancer cells less proliferative and invasive both in vitro and in vivo, which implies that CDH17 could be an attractive therapeutic target for GC.
Up to now, the Met and Wnt signaling pathways have been reported to interact with CDH17 in hepatic cell carcinoma.29,30 Despite that, the mechanism of CDH17 activation in GC has not been well elucidated yet.31,32 NFκB signaling pathway, as an eminent key factor regulating inflammation-induced carcinogenesis and lymphangiogenesis, is consecutively activated in GC.20,22,33 The primary NFκB family consists of RelA (p65), NF-κB1 (p50), RelB, c-Rel and NFκB2 (p52), among which the p50/p65 heterodimers are the main complexes regulating the transcription of responsive genes.34 Comparison with adjacent normal epithelial cells, increased nuclear translocation of p65 in GC is correlated with the depth of invasion, tumor size and metastases.35 Especially in recent study, Flister et al. suggests that the induction of p50/p65 enhances the responsiveness of preexisting lymphatic endothelium to VEGF-C and VEGF-D, ultimately resulting in robust lymphangiogenesis.23 Taken together, we presumed the implicit relationship between CDH17 and the NFκB signaling pathway.
Based on the alterations of proteins in vitro and in vivo, we inference that the NFκB signaling pathway might be the key mediator in the process of CDH17 affecting gastric cancer, on the other hand, knocking down CDH17 expression by miRNA is responsible for the retention of NFκB and a concomitant reduction in downstream proteins (e.g., VEGF-C), as presented in Figure 5. It is of note that VEGF-C has been identified as major regulator of the development of lymphatic vessels (lymphangiogenesis),36 expressing positively in LNM of human cancers including thyroid, prostate, gastric, colorectal and lung cancers.37 Although there has been considerable effort to decipher the specific activation mechanisms and contribution of NFκB to oncogenesis and metastasis, the crosstalk between NFκB and other important oncogenic signaling pathways pathways including Ras, p53, Notch,38 TLR4,39 and LTβR signalings,40 remains poorly defined. Additionally, there is also considerable conservation of signaling intermediates upstream of the IKK complex, such as the receptor-interacting proteins (RIPs) and TNFR-associated factors (TRAFs) which are critical to IKK activation,38 hence it is still difficult to assert that CDH17 regulates NFκB directly.
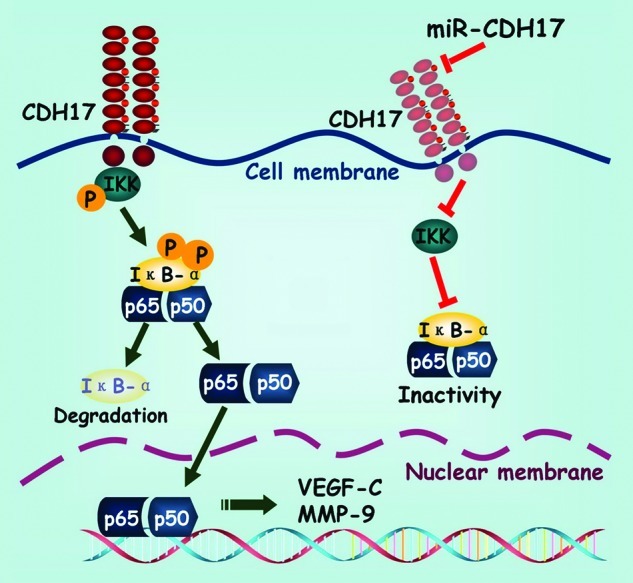
Figure 5. Schematic exhibition of the proposed signaling pathway modulated by CDH17 in gastric cancer cells. Coupling CDH17 activates the IKK complex, which subsequently phosphorylates the IκB-α. Then the phosphorylated IκB-α undergoes proteasome-dependent decomposition, which releases the heterodimers of p65/p50 into cytoplasm and transferred into the nucleus. Finally, the p65 binds to its responsive gene and promotes the transcription of downstream proteins including VEGF-C and MMP-9. On the other hand, RNAi mediated inhibition of CDH17 attenuates the activation of NFκB in gastric cancer cells, leading to a concomitant reduction in downstream proteins.
The dorsal subcutaneous xenografted tumor model was used to evaluate the effect of CDH17 on proliferative potency of gastric cancer. On the other hand, the claw pad lymph node metastasis model was selected specifically to probe the association between CDH17 and LNM, since we identified that CDH17 was the independent risk factor of LNMM previously.19 That is why we employed two different animal models to investigate the impact of CDH17 on tumorigenesis and metastasis respectively. Although orthotopic model is established to simulate metastasis more distinctively, the heterotopic subcutaneous lymphatic metastasis model including implantation in back, claw pad, tail, penis or femoribus internus, remains less invasive and more convenient to be monitored. Among them, subcutaneous implantation in claw pad is a mouse model with moderate frequency of lymph node metastasis, of which the tumor cells are supposed to shift from the lymphoreticular tissue in claw to the popliteal LN, inguinal LN, axillary LN and lung tissue.41,42 With abundant lymphatic capillary networks and explicit drainage, the claw pad lymphatic metastasis model is superior to the other models in construction and inspection. From our analysis, node-positive mice in control groups accounted for 33%. By contrast, there was no metastasis in miR-CDH17 group. Owing to the relatively small sample capacity, statistical difference was not found between the control groups and the knockdown group (p = 0.276). Nevertheless, the trend that antagonizing CDH17 inhibits LNM could still be observed, which shed light on the further investigations.
Conclusion
Although many molecular factors have been identified from microarray expression profiling, much remains to be confirmed to identify their mechanisms and clinical significance. Our data showed that RNAi-mediated knockdown of CDH17 inhibited proliferation and lymph node metastasis of GC both in vitro and in vivo. We also demonstrate for the first time that CDH17 modulates the p65 dynamics and, subsequently, the transactivation of the NFκB target genes. Hopefully, CDH17 could serve as a candidate biomarker and an attractive therapeutic target for progression and metastasis in GC.
Materials and Methods
Cell lines and animals
Human gastric cancer cell line MKN-45 was chosen as our investigating object, which was established from the lymph node metastatic foci of poorly differentiated gastric adenocarcinoma, and provided by the Cell Centre of Chinese Academy of Medical Sciences. The MKN-45 cells were cultured in RPMI1640 medium with 10% fetal bovine serum (Hyclone) and incubated in a humidified incubator (37°C, containing 5% CO2). The 293FT cells (Inovogen) were cultured in complete medium containing geneticin (Gibco).
A total of 36 four-week-old male BALB/c-nu/nu nude mice were purchased from the Experimental Animal Centre of the National Institute for the Control of Pharmaceutical and Biological Products (NICPBP) and maintained under specific pathogen-free conditions. All experiments were performed with the approval of the Animal Experimental Ethics Committee of Peking Union Medical College Hospital and complied with the Guide for the Care and Use of Laboratory Animals.
Selection of the optimal miRNA sequence targeting CDH17 and stable transfection of MKN-45 cells
Three precursor miRNA sequences (miR-1,-2,-3) targeting CDH17 (GenBank accession number NM_004063) and its mismatch mutants (miR-neg) as negative control were designed using the RNAi search software (Inovogen). These double-stranded oligonucleotides were synthesized and inserted into pRI-CMV/eGFP-miRNA expression vector (Inovogen) to construct lentiviral recombinant plasmids pRI-CMV/eGFP-CDH17-miR-1,-2,-3 and -neg respectively, which were then transformed into DH5a-competent E. coli cells. The positive recombinants were identified and extracted for sequence detection. We packaged the recombinants using the four plasmid system (Inovogen) and then co-transfected these admixtures into 293FT cells with Lipofectamine 2000 (Invitrogen). 48 h later, lentivirus supernatant was harvested and transfected into MKN-45 cells, which were consequently cultured in medium containing geneticin (Gibco) for two weeks to select the stable transfected cells. Flow cytometry was performed to detect transfection efficiency.
CDH17 expressions of MKN-45 cells transfected with different plasmids were examined by real-time RT-PCR using SYBR Green master mix kit and calculated based on the method of 2-△△Ct. The forward primer of CDH17 was 5′-GGACAGAGAAGCCGGAAGTC-3′ and the reverse one was 5′-GAACAAGCCCGTGTAGTCCTT-3′. Western blot was also performed. Thereupon, the sequence yielding the highest interference efficacy of CDH17 in both analyses was defined as knockdown group (miR-CDH17). The MKN-45 cell receiving either no treatment or mismatch mutants was defined as control group (MKN-45) or negative control group (miR-neg).
Cell immunofluorescence, proliferation, adhesion, invasion and cell cycle assays
For each group, cells were incubated by primary monoclonal antibody against CDH17 (dilutions 1:100, R&D) and Dylight™594-labeled secondary antibody (dilutions 1:200, ZSGB-BIO), counterstained with DAPI (ZSGB-BIOa). Fluorescence was detected by EVOS fl microscopy (AMG).
The cellular proliferation was measured via MTT [3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay performing standard method in 96-well microtiter plates. After incubation for 12, 24, 48, 72, 96 and 120 h, the optical absorbance of each group at 570 nm was read by a microplate reader (Bio-rad 680).
To evaluate cellular adhesion, 96-well microtiter plates was precoated by incubating with 70μl Matrigel (10 μg/ml) at 4°C overnight. Cells were suspended and cultured for 20min, 40min and 60min respectively. After that, nonadherent cells were removed by washing three times with sterile PBS. The relative quantity of adherent cells was calculated by the MTT method as described above.
The invasive potency was assessed in transwell cell-culture chambers containing 6.5 mm-diameter polycarbonate membrane filters with 8-μm-pore (Costar). Five × 105 cells were cultured in the upper chamber with 0.5% RPMI-1640 medium, and the lower chambers were filled with 20% FBS RPMI-1640 medium. After 48h of incubation at 37°C, non-invading cells remaining on the upper surface of the filter were removed using cotton swab. The migrated cells on the underside of the membrane were fixed with 4% paraformaldehyde and stained by Crystal Violet Staining Solution (Beyotime). Cells in 10 random fields of view at × 200 magnifications per well were counted.
Cell cycle was estimated by using propidium iodide staining. Chiefly, cells were fixed in 70% ethanol at 4°C overnight, and then resuspended with PBS containing 50 μg/ml propidium iodide and 500μg/ml RNase A. After incubation for 60 min, cell population in each phase was analyzed by flow cytometry using Coulter EPICS XL (Beckman Coulter). All these assays were repeated thrice independently.
Detection of NFκB signaling pathway
The canonical NFκB activation pathway consists of IKK (IκB kinase), IκB-α and NFκB (typically a p65/p50 heterodimer). Upon activation, the catalytic IKK phosphorylates the serine residues in IκB-α, triggering ubiquitin-dependent degradation of IκB-α, so as to expose the NLS signal on p50, ensuring nuclear translocation of p65 to promote gene transcription.24 To test our hypothesis, CDH17 (R&D Inc., 1:200) and components of NFκB signaling pathway in each group were detected by western blot. The nuclear p65 expression (Abcam, 1:200) is the key point to evaluate the activation of NFκB signaling pathway. On the other hand, MMP-9 (Cell Signaling Technology, 1:1000) and VEGF-C (Abcam, 1:100) are identified as the downstream targets in this pathway. Histone (Abcam, 1:1000) and β-actin (Abgent, 1:500) were used as endogenous proteins for normalization.
Tumorigenesis assay in vivo
Eighteen nude mice were randomly assigned to three groups, which were inoculated with MKN-45 cells trasfected either by pRI-CMV/eGFP vector (mock group), non-targeted RNAi vector (miR-neg group) or the CDH17-targeted RNAi vector (miR-CDH17 group) respectively. 200 μl suspension of tumor cells (1 × 107 cells/ml) was injected in the right dorsal area subcutaneously. To supervise tumor growth in vivo dynamically and non-invasively, optical in vivo imaging (NightOWL II LB 983, Berthod) was applicated. The animals were monitored every three days, and tumor volume was calculated as follows: volume (mm3) = (shortest diameter)2 × (longest diameter)/2. Eight weeks after inoculation, primary tumors were excised and evaluated pathologically. To confirm the hypothetic signaling pathway, the expressions of p65, MMP-9 and VEGF-C in tumor tissues were detected by immunohistochemical staining as well.
Lymphatic metastasis assay in vivo
Another 18 nude mice were randomized into three groups according to the principle in tumorigenesis assay. To induce LNM, 50μl suspension of tumor cells (1 × 108 cells/ml) was injected in the left claw pad subcutaneously.41 The development of LNM was monitored externally and noninvasively with the in vivo imaging system every three days. Eight weeks later, all the mice were dissected to obtain the lymph nodes or the lesions with fluoresce, which consequently received HE and immunohistochemistry staining to confirm the metastasis. The CDH17 expression in metastatic lesion was examined as well.
Statistical analysis
All statistical analyses and graphics were performed with the SPSS 16.0 statistical package (SPSS Inc.). Continuous variables were expressed as mean ± standard deviation, and difference in each assay was determined by ANOVA, followed by LSD’s t-test. Enumeration data was analyzed by Chi-square test. The significance of difference was accepted at p < 0.05.
Acknowledgments
We sincerely appreciate Professor Jin Gao for his contribution to the claw pad lymphatic metastasis mouse model, who went in for the gastric cancer research throughout his life and died of gastric cancer several years ago. We also thank Wen-Jin Zhao and Professor Pei Gu for providing the technical support generously.
Disclosure of Potential Conflicts of Interest
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/23299
References
- 1.Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009;374:477–90. doi: 10.1016/S0140-6736(09)60617-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reshamwala PA, Darwin PE. Endoscopic management of early gastric cancer. Curr Opin Gastroenterol. 2006;22:541–5. doi: 10.1097/01.mog.0000239870.04457.80. [DOI] [PubMed] [Google Scholar]
- 3.Catalano V, Labianca R, Beretta GD, Gatta G, de Braud F, Van Cutsem E. Gastric cancer. Crit Rev Oncol Hematol. 2009;71:127–64. doi: 10.1016/j.critrevonc.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Yokota T, Ishiyama S, Saito T, Teshima S, Narushima Y, Murata K, et al. Lymph node metastasis as a significant prognostic factor in gastric cancer: a multiple logistic regression analysis. Scand J Gastroenterol. 2004;39:380–4. doi: 10.1080/00365520310008629. [DOI] [PubMed] [Google Scholar]
- 5.Oñate-Ocaña LF, Méndez-Cruz G, Hernández-Ramos R, Becker M, Carrillo JF, Herrera-Goepfert R, et al. Experience of surgical morbidity after palliative surgery in patients with gastric carcinoma. Gastric Cancer. 2007;10:215–20. doi: 10.1007/s10120-007-0437-4. [DOI] [PubMed] [Google Scholar]
- 6.Namikawa T, Kitagawa H, Iwabu J, Okabayashi T, Sugimoto T, Kobayashi M, et al. Clinicopathological properties of the superficial spreading type early gastric cancer. J Gastrointest Surg. 2010;14:52–7. doi: 10.1007/s11605-009-1059-4. [DOI] [PubMed] [Google Scholar]
- 7.Kim JH, Park JM, Jung CW, Park SS, Kim SJ, Mok YJ, et al. The significances of lymph node micrometastasis and its correlation with E-cadherin expression in pT1-T3N0 gastric adenocarcinoma. J Surg Oncol. 2008;97:125–30. doi: 10.1002/jso.20937. [DOI] [PubMed] [Google Scholar]
- 8.Kim JH, Park SS, Park SH, Kim SJ, Mok YJ, Kim CS, et al. Clinical significance of immunohistochemically-identified lymphatic and/or blood vessel tumor invasion in gastric cancer. J Surg Res. 2010;162:177–83. doi: 10.1016/j.jss.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Arigami T, Natsugoe S, Uenosono Y, Yanagita S, Ehi K, Arima H, et al. Vascular endothelial growth factor-C and -D expression correlates with lymph node micrometastasis in pN0 early gastric cancer. J Surg Oncol. 2009;99:148–53. doi: 10.1002/jso.21228. [DOI] [PubMed] [Google Scholar]
- 10.Yasui W, Oue N, Sentani K, Sakamoto N, Motoshita J. Transcriptome dissection of gastric cancer: identification of novel diagnostic and therapeutic targets from pathology specimens. Pathol Int. 2009;59:121–36. doi: 10.1111/j.1440-1827.2009.02329.x. [DOI] [PubMed] [Google Scholar]
- 11.Yasui W, Oue N, Ito R, Kuraoka K, Nakayama H. Search for new biomarkers of gastric cancer through serial analysis of gene expression and its clinical implications. Cancer Sci. 2004;95:385–92. doi: 10.1111/j.1349-7006.2004.tb03220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang XQ, Luk JM, Leung PP, Wong BW, Stanbridge EJ, Fan ST. Alternative mRNA splicing of liver intestine-cadherin in hepatocellular carcinoma. Clin Cancer Res. 2005;11:483–9. [PubMed] [Google Scholar]
- 13.Hinoi T, Lucas PC, Kuick R, Hanash S, Cho KR, Fearon ER. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology. 2002;123:1565–77. doi: 10.1053/gast.2002.36598. [DOI] [PubMed] [Google Scholar]
- 14.Ito R, Oue N, Yoshida K, Kunimitsu K, Nakayama H, Nakachi K, et al. Clinicopathological significant and prognostic influence of cadherin-17 expression in gastric cancer. Virchows Arch. 2005;447:717–22. doi: 10.1007/s00428-005-0015-2. [DOI] [PubMed] [Google Scholar]
- 15.Ko S, Chu KM, Luk JM, Wong BW, Yuen ST, Leung SY, et al. Overexpression of LI-cadherin in gastric cancer is associated with lymph node metastasis. Biochem Biophys Res Commun. 2004;319:562–8. doi: 10.1016/j.bbrc.2004.04.197. [DOI] [PubMed] [Google Scholar]
- 16.Okayama H, Kumamoto K, Saitou K, Hayase S, Kofunato Y, Sato Y, et al. CD44v6, MMP-7 and nuclear Cdx2 are significant biomarkers for prediction of lymph node metastasis in primary gastric cancer. Oncol Rep. 2009;22:745–55. doi: 10.3892/or_00000496. [DOI] [PubMed] [Google Scholar]
- 17.Suh YS, Lee HJ, Jung EJ, Kim MA, Nam KT, Goldenring JR, et al. The combined expression of metaplasia biomarkers predicts the prognosis of gastric cancer. Ann Surg Oncol. 2012;19:1240–9. doi: 10.1245/s10434-011-2125-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee HJ, Nam KT, Park HS, Kim MA, Lafleur BJ, Aburatani H, et al. Gene expression profiling of metaplastic lineages identifies CDH17 as a prognostic marker in early stage gastric cancer. Gastroenterology. 2010;139:213–25, e3. doi: 10.1053/j.gastro.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Yu JC, Kang WM, Wang WZ, Liu YQ, Gu P. The predictive effect of cadherin-17 on lymph node micrometastasis in pN0 gastric cancer. Ann Surg Oncol. 2012;19:1529–34. doi: 10.1245/s10434-011-2115-3. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y, Bai L, Chen W, Xu S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010;14:45–55. doi: 10.1517/14728220903431069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 22.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–6. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 23.Flister MJ, Wilber A, Hall KL, Iwata C, Miyazono K, Nisato RE, et al. Inflammation induces lymphangiogenesis through up-regulation of VEGFR-3 mediated by NF-kappaB and Prox1. Blood. 2010;115:418–29. doi: 10.1182/blood-2008-12-196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 25.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ko S, Chu KM, Luk JM, Wong BW, Yuen ST, Leung SY, et al. CDX2 co-localizes with liver-intestine cadherin in intestinal metaplasia and adenocarcinoma of the stomach. J Pathol. 2005;205:615–22. doi: 10.1002/path.1741. [DOI] [PubMed] [Google Scholar]
- 27.Lee NP, Poon RT, Shek FH, Ng IO, Luk JM. Role of cadherin-17 in oncogenesis and potential therapeutic implications in hepatocellular carcinoma. Biochim Biophys Acta. 2010;1806:138–45. doi: 10.1016/j.bbcan.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Wang XQ, Luk JM, Garcia-Barcelo M, Miao X, Leung PP, Ho DW, et al. Liver intestine-cadherin (CDH17) haplotype is associated with increased risk of hepatocellular carcinoma. Clin Cancer Res. 2006;12:5248–52. doi: 10.1158/1078-0432.CCR-06-0558. [DOI] [PubMed] [Google Scholar]
- 29.Kaposi-Novak P, Lee JS, Gòmez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest. 2006;116:1582–95. doi: 10.1172/JCI27236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu LX, Lee NP, Chan VW, Xue W, Zender L, Zhang C, et al. Targeting cadherin-17 inactivates Wnt signaling and inhibits tumor growth in liver carcinoma. Hepatology. 2009;50:1453–63. doi: 10.1002/hep.23143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu QS, Zhang J, Liu M, Dong WG. Lentiviral-mediated miRNA against liver-intestine cadherin suppresses tumor growth and invasiveness of human gastric cancer. Cancer Sci. 2010;101:1807–12. doi: 10.1111/j.1349-7006.2010.01600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang J, Liu QS, Dong WG. Blockade of proliferation and migration of gastric cancer via targeting CDH17 with an artificial microRNA. Med Oncol. 2011;28:494–501. doi: 10.1007/s12032-010-9489-0. [DOI] [PubMed] [Google Scholar]
- 33.Yu YY, Li Q, Zhu ZG. NF-kappaB as a molecular target in adjuvant therapy of gastrointestinal carcinomas. Eur J Surg Oncol. 2005;31:386–92. doi: 10.1016/j.ejso.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Beinke S, Ley SC. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem J. 2004;382:393–409. doi: 10.1042/BJ20040544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolcet X, Llobet D, Pallares J, Matias-Guiu XNF. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–82. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 36.Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, et al. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290–8. [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu K, Kubo H, Yamaguchi K, Kawashima K, Ueda Y, Matsuo K, et al. Suppression of VEGFR-3 signaling inhibits lymph node metastasis in gastric cancer. Cancer Sci. 2004;95:328–33. doi: 10.1111/j.1349-7006.2004.tb03211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 39.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–7. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 40.Basak S, Hoffmann A. Crosstalk via the NF-kappaB signaling system. Cytokine Growth Factor Rev. 2008;19:187–97. doi: 10.1016/j.cytogfr.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao J. Construction of lymphatic metastasis model. In: Gao J, Zhang JB, eds. Basic and clinical research on invasion and metastasis of carcinoma. Beijing: Science press, 2003:67-74. [Google Scholar]
- 42.Kozaki K, Miyaishi O, Tsukamoto T, Tatematsu Y, Hida T, Takahashi T, et al. Establishment and characterization of a human lung cancer cell line NCI-H460-LNM35 with consistent lymphogenous metastasis via both subcutaneous and orthotopic propagation. Cancer Res. 2000;60:2535–40. [PubMed] [Google Scholar]