

Abstract
p21 is a member of the cyclin kinase inhibitor family of proteins and plays pivotal roles in cellular proliferation as well as in the regulation of apoptosis, and thus has diverse functions in diseases as varied as cancer and atherosclerosis. In light of its pleiotropic effects and potential clinical relevance, new methods of attenuation of p21 protein levels by selective inhibitors are therefore powerful tools to probe malignant, infectious and other diseases. Here we introduce a novel p21 attenuator, UC2288, which possesses consistent and relatively selective activity for p21. UC2288 was synthesized based on the chemical model of sorafenib, a multikinase inhibitor that also attenuates p21, but unlike sorafenib, UC2288 did not inhibit Raf kinases or alter p-ERK protein levels. UC2288 decreased p21 mRNA expression independently of p53, and attenuated p21 protein levels with minimal effect on p21 protein stability. In addition, UC2288 inhibits cell growth in the kidney cancer cell lines (GI50 = approximately 10 µM) as well as multiple other cancer cell lines. Thus, this novel p21 inhibitor will be indispensable for exploring the function of p21, and upon further study may be translatable to the clinic.
Keywords: p21 inhibitor, p21, sorafenib, proliferation, apoptosis
Introduction
p21 is a member of the cip/kip family of cyclin dependent kinase inhibitors and has shown pleiotropic effects on cell growth and apoptosis in both malignant and non-malignant cells and tissues.1-5 Due to its pivotal roles in cellular proliferation and in the regulation of apoptosis, p21 possesses diverse physiological functions in various diseases including vascular smooth muscle cell proliferation (an important step for cancer angiogenesis as well as atherosclerosis),6 cancer progression,7 and viral infection.4,6,7 The initial descriptions of p21 have been focused on its location in the tumor suppressor protein signaling pathways downstream of p53, and thus on its function as a conveyor of the signaling properties of this cancer-relevant protein.8 However, recent studies have demonstrated an anti-apoptotic function of p21 when localized to the cytosol,9 and for these reasons p21 has been studied as a marker of cancer prognosis10 and has been suggested to be a novel target for cancer therapy.1
Overexpression of p21 is associated with poor prognosis in many cancers such as renal cell carcinoma (RCC),11 acute myeloid leukemia,12 ovarian cancer,13 prostate cancer,14 and breast cancer.1 Consistent with these findings and with the anti-apoptotic function of p21, attenuation of p21 levels in RCC cell lines in vitro has been shown to sensitize the cells to apoptosis in response to conventional DNA damaging chemotherapy.15 In addition, attenuation of p21 in CD4+ T-cells with an earlier inhibitor resulted in a marked increase of viral reverse transcripts and mRNA production in these cells.4 Furthermore, it has been recently reported that p21 not only inhibits cellular apoptosis but also induces stress-induced premature senescence (SIPS) after DNA damage (reviewed in16); SIPS is a sustained growth-arrested state in which cells are alive and secrete factors which may promote cancer progression.16 For these reasons, selective p21 inhibitors would be useful for analyzing its role in physiological function, progression of disease, drug resistance and cancer therapy. While antisense oligodeoxynucleotides and siRNA to p21 are commercially available for in vitro use and have been investigated in our laboratory, these tools have proven difficult to translate to the clinic due to transient expression, insufficient efficacy and resistance.17
Our laboratory has previously discovered small molecule p21 inhibitors using a combinatorial approach. Utilizing an on-bead enzyme-linked colorimetric binding assay, compounds which bind p21 were identified through screening of a diverse one-bead-one-compound combinatorial small molecule library.18 Of these 12 candidate compounds, three had high p21 binding affinity and yielded similar chemical structure. While useful in some early studies, these inhibitors proved to be less stable than the new p21 inhibitor described in this study and, more importantly, activity of the earlier inhibitors is inadequate for ultimate clinical utility, since the concentration required to attenuate p21 was an order of magnitude higher for the earlier inhibitors.
In an attempt to improve upon our previous p21 inhibitors, we have capitalized on our previous work with sorafenib in which we found that sorafenib possesses p21 inhibitory function at 10 µM;19 thus, the structure of sorafenib was utilized to synthesize a selective p21 inhibitor with higher inhibitory activity toward p21. We now show that this newer generation p21 inhibitor, UC2288, attenuates p21 protein abundance at 10 µM, independently of p53 activity and at the level of transcription or post-transcription. In addition, UC2288 decreases cytosolic p21 protein level in an RCC cell line, which is consistent with the inhibitor’s cytotoxic effect,9 and inhibits survival in many of the NCI60 panel cell lines. Thus, UC2288 is a valuable addition to the available armamentarium for further exploration of p21 function in the physiology of cancer and other diseases, and may ultimately lead to clinical utility of p21 attenuators.
Results
UC2288 decreases p21 protein level independently of Raf kinase inhibition
We have previously shown that the multi-kinase inhibitor sorafenib (Nexavar®) consistently attenuates p21 protein levels in various kidney and liver cancer cell lines.19 Using the structure of sorafenib as a template, we synthesized multiple compounds which are structurally related to sorafenib. These compounds were screened for attenuation of p21, p-Akt and p-ERK protein levels by immunoblotting (Table S1) and UC2288 (Fig. 1) was chosen as a potential p21 inhibitor for further analysis due to its p21 attenuation with no p-Akt or p-ERK attenuation among all the compounds. p21 protein levels after incubation of several cancer cell lines and a “normal” kidney proximal tubule epithelial cell line with UC2288 were evaluated. All cell lines tested in this manner showed markedly decreased p21 levels (Fig. 2a).
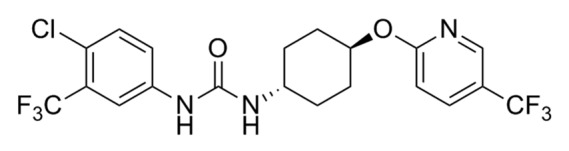
Figure 1. Structure of UC2288.

Figure 2. UC2288 consistently decreases p21 protein level, but not levels of other proteins. Cancer cells and those derived from a normal kidney [HK2 (normal kidney), 786-O (RCC), Caki-1 (RCC), ACHN (RCC) and HEY (ovarian cancer)] were grown to confluence in 10% serum-containing media and treated with either UC2288, sorafenib, or vehicle (DMSO) at the concentrations indicated for 24 h. The cells were harvested and immunoblotted with the antibodies indicated. (A) p21 and (B) p-Akt, p-ERK, t-Akt, t-ERK and p53 proteins are shown. β-actin is a loading control for each membrane. The experiment shown is representative of at least 3 separate experiments.
Due to its global inhibitory properties, sorafenib affects a variety of signaling proteins (e.g., Raf/MEK kinases20), and in light of the structural similarity between UC2288 and sorafenib, we asked if UC2288 could inhibit mitogenic- and apoptotic-relevant kinases. While sorafenib showed the expected vascular endothelial growth factor receptor 2 (VEGFR2) and Raf kinase inhibitory activity, UC2288 had minimal effects on these kinases (Table 1). To confirm this result, we determined phosphorylation levels of downstream p-ERK in UC2288 treated cells. Changes of p-ERK levels were inconsistent among the cell lines examined supporting the likelihood that UC2288 has minimal Raf kinase inhibitory function (Fig. 2b).
Table 1. UC2288 has no inhibition of VEGFR2 and Raf kinases.
| IC50 (nM) | VEGFR2 inhibition at 10 µM (%) | ||
|---|---|---|---|
| |
C-Raf |
B-RafV600E |
|
| Sorafenib |
45 ± 5 |
13 ± 2 |
100 |
| UC2288 | > 10000 | > 10000 | 0 |
Kinase assays were performed with UC2288 as described in the Materials and Methods. UC2288 showed no inhibition against C-Raf, B-Rafv600E and VEGFR2.
UC2288 decreases p21 mRNA independent of p53 expression
Our first-described p21 inhibitor, which was discovered by high-throughput screening of one-bead one-compound combinatorial library (see Introduction),18 attenuated p21 protein level by accelerating its degradation. Because UC2288 has no structural similarity to the earlier inhibitors, we asked whether it has a mechanism of action distinct from those compounds. In addition to p21 protein levels earlier discussed (see Figure 2a), mRNA expression for p21 was decreased by UC2288 in the p53-mutant21 RCC cell line 786-O (Fig. 3a), demonstrating that UC2288 repressed p21 mRNA transcriptionally or post-transcriptionally but independently of p53. Consistent with these data, p53 protein, the major transcription factor of p21,8 was not changed after UC2288 treatment in p53-wt cell lines (see Fig. 2b), suggesting that the mechanism of p21 attenuation is independent of p53.

Figure 3. UC2288 decreases p21 mRNA expression with no effect on p21 protein stability. 786-O cells were grown to confluence in 10% serum-containing media and: (A) treated with UC2288 at 10 µM concentration or vehicle (DMSO) for the indicated time. mRNA was isolated and RT-PCR was performed as described in Materials and Methods. GAPDH is a loading control. (B) exposed to CHX (35 µM), UC2288 (10 µM), CHX (10 µg/ml) and UC2288 (10 µM), or vehicle (DMSO) for the indicated times and immunoblotting was performed with the antibodies indicated. β-actin is a loading control. The experiments shown are representative of at least 3 separate experiments.
To evaluate p21 stability in the presence of UC2288, we utilized cycloheximide (CHX), an inhibitor of protein translation. p21 levels were decreased after four hours of UC2288 incubation (Fig. 3b), yet incubation with both UC2288 and CHX did not change the stability of p21 as compared with CHX incubation alone, suggesting that, unlike earlier p21 inhibitory compounds, UC2288 does not affect p21 degradation (Fig. 3b). In support of this finding, we evaluated changes in the levels of p-Akt, a protein which is known to stabilize p21.22 p-Akt levels were inconsistently altered among the cell lines, showing that the p21 decrease by UC2288 is not due to degradation and is independent of p-Akt (Fig. 2b).
UC2288 inhibits cell growth of various cancer cell lines
Since we have previously shown that attenuation of p21 protein level by antisense ODN inhibits RCC cell growth,15 we asked whether UC2288 behaves similarly which would suggest potential clinical utility. NCI60 data showed that in most cell lines tested, UC2288 incubation resulted in approximately 50% cell growth inhibition at 10 µM (Fig. 4), a concentration at which p21 was attenuated (see Fig. 2a).

Figure 4. UC2288 inhibits cancer cell growth. TCA assay was conducted in the NCI60 cell lines treated with UC2288 as described in the Materials and Methods. UC2288 inhibited various cancer cell growth.
It is known that cytosolic p21 inhibits apoptosis while nuclear p21 causes cell cycle arrest8,9 and can contribute to the regulation of growth of cancer23 and other24 cells. To ascertain whether UC2288 differentially affects subcellular localization in at least one situation, 786-O cells were incubated with UC2288 and subjected to immunofluorescence for p21 (Fig. 5). Surprisingly, cytosolic but not nuclear p21 levels were markedly attenuated (note decreased green cytosolic staining in “Merge” panels in Fig. 5).

Figure 5. UC2288 decreases cytosolic p21. 786-O cells were grown to confluence on 8 well chamber slides and treated with either UC2288 at 10 µM concentration or vehicle (DMSO) for 24 h. Immunofluorescence staining of p21 was visualized by confocal microscopy as described in Materials and Methods. p21 is green and the nuclear dye (DAPI) is blue. The experiment shown is representative of at least 3 separate experiments.
Discussion
Since p21 is a key regulator of both cell proliferation and apoptosis, this protein plays an integral role in the regulation of many physiologic processes as well as in the pathophysiology of many and varied diseases such as cancer, atherosclerosis and human immunodeficiency virus (HIV).4,5,7-9 Thus, means of attenuation of p21 level using a selective inhibitor has been and continues to be useful for probing cell cycle regulation and apoptosis. In this study, we introduce and validate a novel selective inhibitor of p21.
While we have previously developed and reported other potential p21 inhibitors,18 activities against p21 of those compounds were insufficient for clinical translation and, furthermore, those compounds proved to be somewhat unstable over long storage times. Based on our recent finding that sorafenib inhibits p21,19 we designed several new compounds based on the structure of sorafenib which retain p21 activity but which have variable and inconsistent kinase inhibitory properties. Of these compounds, UC2288 (t-CTTU) was shown to be the most promising as a p21 inhibitor and was further studied in this work.
While the mechanism by which UC2288 attenuates p21 protein level has not been exhaustively determined in this study, it is clear from our data that attenuation occurs by means of transcriptional or post-transcriptional regulation and not via protein degradation. Surprisingly, in light of the known transcriptional activation of p21 by p53, UC2288 downregulated p21 levels without altering p53 levels, and UC2288 furthermore attenuated p21 in p53 mutated cell lines (786-O); thus UC2288 functions independently of p53, a property that could be clinically useful as p53 is truly the “guardian of the genome” and thus its levels and function should be preserved in any novel therapy including those that may include p21 attenuation.25,26 Further studies to validate the effects of UC2288 on p21 transcription and post-transcription are necessary.
Independently of its p21 inhibitory activity, sorafenib is known to possess multi-kinase and soluble epoxide hydrolase (sEH) inhibitory functions.27,28 Our goal in this project was to separate out the p21 inhibitory effect of sorafenib from its other properties which lead to the synthesis of UC2288. There were inconsistent decreases in the p-ERK pathways by UC2288, and Raf kinases inhibition was minimal while the attenuation of p21 was consistent among the cell lines. These findings suggest that p21 attenuation by UC2288 is not dependent on kinase inhibition and that the changes in p-ERK are likely indirect effects of UC2288 rather than through its direct attenuation of p21. Furthermore, while sorafenib has been shown to efficiently inhibit sEH, it is clear that such inhibition does not affect p21 levels;19 these findings show that p21 attenuation by UC2288 is independent from multi kinase as well as sEH inhibition.
In light of the known important role of p21 in RCC,11,15 including the finding that overexpression of p21 correlates with poor prognosis in this and other cancers (reviewed in7), we utilized 786-O cells, the p53-mutant RCC cell line, to explore such therapeutic potential. UC2288 in fact caused significant growth inhibition in these cells, a finding consistent with our previous work using antisense p21 oligodeoxynucleotides.29 UC2288 decreased cell viability approximately 20% even at the low concentration which does not inhibit p21; thus it is possible that this cytotoxicity at this concentration is due to off-targets effects of UC2288 and this finding suggests that further studies are necessary before the clinical potential of UC2288 for cancer therapy can be fully realized.
In summary, we introduce and validate a novel p21 inhibitor, UC2288, which functions independently of the classical p21 regulator proteins p53 and p-Akt. UC2288 is important research tool in the available armamentarium useful for investigating p21 related pathophysiology and may ultimately lead to useful treatments for those cancers in which overexpressed p21 plays a role in pathogenicity.
Materials and Methods
Cell lines
A normal human kidney primary proximal tubule epithelial cell line (HK2) and three human proximal tubule epithelial cancer cell lines (786-O, Caki-1 and ACHN) were obtained from the American Type Culture Collection (Rockville, MD), and the human ovarian carcinoma cell line (Hey) was generously provided by Dr. Erin Dickerson, University of Minnesota. HK2 and ACHN cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 100 units/mL streptomycin and 100 mg/mL penicillin. 786-O, Caki-1 and Hey cells were maintained in RPMI supplemented with 10% FBS, 100 units/mL streptomycin and 100 mg/mL penicillin. Cells were maintained at 5% CO2 at 37°C.
Materials
CHX and dimethyl sulfoxide (DMSO) was obtained from Sigma (St. Louis, MO, USA). UC2288 and CHX were dissolved in DMSO. Mouse monoclonal anti-p21WAF1/Cip antibody was obtained from Millipore (Billerica, MA, USA). Mouse monoclonal anti-phosphor Akt antibody, mouse monoclonal anti-p53 antibody, rabbit monoclonal anti-phospho ERK, rabbit monoclonal anti- p21WAF1/Cip antibody and anti-rabbit IgG (H+L), F(ab')2 Fragment (Alexa Fluor® 488 Conjugate) antibody were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). Goat anti-mouse and goat anti-rabbit HRP conjugated IgG were obtained from Bio-Rad (Hercules, CA, USA). ECL Plus Western Blotting Detection Reagents was obtained from GE Healthcare (Piscataway, NJ, USA). VECTASHIELD HardSet Mounting Medium with DAPI was obtained from Vector Laboratories (Burlingame, CA, USA).
The synthesis of UC2288
trans-1-(4-chloro-3-trifluoromethyl-phenyl)-3-(4-hydroxy-cyclohexyl)-urea (1).
To a solution of 4-chloro-3-(trifluoromethyl)phenyl isocyanate (5 g, 22.6 mmol) in DMF (35 mL) were added trans-4-aminocyclohexanol hydrochloride (3.8 g, 24.8 mmol) and Et3N (3.5 mL, 24.8 mmol) at 0°C. The reaction mixture was warmed up to room temperature and stirred overnight. After adding 1N HCl (40 mL) and water, the resulting white precipitates were collected by suction filtration. The collected solid was thoroughly washed with water. The combined white powers collected were dissolved in EtOH (200 mL) then aqueous 30% NaOH (25 mL) was added. The reaction mixture was refluxed overnight. EtOH was removed in vacuo. The precipitates were filtered and washed with water. Recrystallization from methanol afforded 6.3 g (82%) of the title compound as a white solid. mp: 239.5 241.1°C. 1HNMR (DMSO-d6): δ 8.79 (s, 1H), 8.06 (d, J = 2 Hz, 1H), 7.55–7.47 (m, 2H), 6.20 (d, J = 8 Hz, 1H), 4.54 (d, J = 4 Hz, 1H), 3.45–3.34 (m, 2H), 1.86–1.75 (m, 4H), 1.28–1.11 (m, 4H).
trans-1-(4-chloro-3-trifluoromethyl-phenyl)-3-[4-(5-trifluoromethyl-pyridin-2-yloxy)-cyclohexyl]-urea (UC2288)
To a solution of 1 (0.17 g, 0.5 mmol) in DMF (5 mL) was added 60% sodium hydride in oil (0.03 g, 0.75 mmol) portionwise at 0°C. After 10 min, 2-chloro-5-(trifluoromethyl)pyridine (0.14 g, 0.75 mmol) was added. The reaction mixture was allowed to slowly warm to room temperature overnight. The reaction was quenched by adding water and the resulting white precipitates were collected and washed with water. The collected solid was recrystallized from methanol to give the title compound, 0.2 g (83%) as a white solid. mp 176.5–177.9°C. 1H NMR (DMSO-d6): δ 8.82 (s, 1H), 8.58–8.54 (m, 1H), 8.08–8.06 (m, 1H), 8.04 (dd, J = 9 and 3 Hz, 1H), 7.54–7.52 (m, 2H), 6.96 (d, J = 9 Hz, 1H), 6.30 (d, J = 8 Hz, 1H), 5.13–4.99 (m, 1H), 3.64–3.48 (m, 1H), 2.15–1.89 (m, 4H), 1.63–1.30 (m, 4H). MS (ESI) m/z: 480.1 (M-H)-
All reagents and solvents used for synthesis of UC2288 were obtained from commercial suppliers and were used without further purification. All reactions, unless otherwise described, were performed under an inert atmosphere of dry nitrogen. Melting points were determined on an OptiMelt melting point apparatus and are uncorrected. 1H NMR spectra were recorded on a Varian QE-300 spectrometer at 300 MHz. Mass spectra were measured by LC-MS equipped with a Waters 2790 and a Waters PDA 996 using electrospray (-) ionization.
Assay for recombinant Raf kinase inhibition
IC50 values were calculated by quantifying the end-point ADP production from each kinase reaction using the ADP-GloTM Kinase Assay kit (Promega, Madison, WI) according to the manufacturer’s instructions. Reactions were performed in Tris buffer (50 mM pH 7.5, rt) containing 20 mM MgCl2 and 0.1% bovine serum albumin. Full length recombinant Raf-1 and b-Raf (V600E) kinases, and their respective substrate, recombinant MEK-1, were purchased from US Biological (Swampscott, Mass.). All assays were performed using 10 nM Raf kinase, 1 µM MEK-1, 10 µM ATP, at 22°C for one hour. Inhibitors were dissolved in DMSO and IC50 values were obtained by measuring the change in the ADP production (luminescent signal intensity) at various inhibitor concentrations as compared with the control. Individual data sets were performed in duplicate and each IC50 value was determined from three separate experiments. The data were fit to a saturation curve using KaleidaGraph graphing program (Synergy Software) to determine the IC50 values.
Inhibitory assay of recombinant VEGFR-2 activity
Inhibition of kinases was screened by the KinaseSeekerTM assay with Luceome Biotechnologies, LLC (Tucson, AZ) as previously described.30 Briefly, 10 mM stocks of inhibitors were diluted in DMSO to a concentration of 250 µM. Prior to initiating a profiling campaign, the compounds were evaluated for false positive against split-luciferase. The inhibitors were then each screened in duplicate against VEGFR-2. For kinase assays, each Cfluc-Kinase was translated along with Fos-Nfluc using a cell-free system (rabbit reticulocyte lysate) at 30°C for 90 min. Twenty-four µL aliquot of this lysate containing a kinase specific probe was added to 1 µL of either DMSO (for no-inhibitor control) or a 250 µM inhibitor solution in DMSO (final concentration of 10 µM) and incubated for 1 h at room temperature. 80 µL of luciferin assay reagent was added to each solution and luminescence immediately measured on a luminometer. The % Inhibition was calculated using the following equation:
| % Inhibition = | ALUcontrol - ALUsample |
x 100 |
| ALUcontrol |
Immunoblotting
Immunoblotting was performed as previously described.19 Briefly, after appropriate treatments, the cells were washed with phosphate buffered saline (PBS) and lysed in lysis buffer. Cell lysates were immunoblotted. Membranes were blocked in 5% nonfat dry milk for one hour at room temperature and probed with appropriate antibodies. Membranes were then probed with horseradish peroxidase tagged anti-mouse or anti-rabbit IgG antibodies. Signal was detected using ECL solutions.
Reverse transcriptase-PCR
Total mRNA was collected and cDNA synthesized using a Qiagen RNeasykit (Valencia, CA) following manufacture’s protocol. The PCR primers used are 5′-ACCATGTGGACCTGTCACTGTCT-3′ (p21 sense), 5′-AGAAGATGTAGAGCGGGCCTTTGA-3′ (p21 antisense), 5′-ACGCATTTGGTCGTATTGGG-3′ (GAPDH sense) and 5′-TGATTTTGGAGGGATCTCGC-3′ (GAPDH antisense). Reverse-transcribed cDNA was subjected to 40 cycles. Thermal cycling conditions are as follows: denaturation for 30 sec at 94°C annealing for 30 sec at 56.5°C and elongation for one min at 72°C DNA was analyzed by 2% ethidium bromide agarose gel electrophoresis.
Cell viability assay
TCA assay was conducted by NCI Developmental Therapeutics Program. Briefly, cells were plated in 96-well plates, and after appropriate treatments, the plates are incubated for an additional 48 h at 37°C, 5% CO2, 95% air and 100% relative humidity. The assay was terminated by the addition of cold TCA. Cells were fixed by the addition of 50 µl of cold 50% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 min at 4°C. The plates were washed water and air-dried. Sulforhodamine B (SRB) solution (100 µl) at 0.4% (w/v) in 1% acetic acid was added to each well, and plates were incubated for 10 min at room temperature. After staining, the plates were washed with 1% acetic acid and air-dried. Bound stain was subsequently solubilized with 10 mM trizma base, and the absorbance was read on an automated plate reader at a wavelength of 515 nm.
Immunofluorescence
After appropriate treatments in 8 well chamber slides, immunofluorescence was conducted as described previously.19 Briefly, the cells were fixed in 2% paraformaldehyde and blocked in the blocking buffer. After blocking, the cells were incubated with appropriate antibody, incubated with anti-mouse or anti-rabbit IgG (H+L), F(ab')2 Fragment (Alexa Fluor® 488 Conjugate), and coverslipped with vectashield with DAPI. The specimens were examined by confocal microscopy.
Supplementary Material
Acknowledgments
This work was supported by NIH grants 5UO1CA86402 (Early Detection Research Network), 1R01CA135401–01A1 and 1R01DK082690–01A1 (to R.H.W.) and the Medical Service of the US Department of Veterans’ Affairs (to R.H.W.); and by NIEHS grant ES02710, NIEHS Superfund grant P42 ES04699 and NIHLB grant HL059699 (to B.D.H.). Aaron T. Wecksler was supported by Award Number T32CA108459 from the National Institutes of Health. Bruce D. Hammock is a George and Judy Senior Fellow of the American Asthma Society. The NCI60 Cell Screen was performed by the NCI Developmental Therapeutics program (http://dtp.cancer.gov/branches/btb/ivclsp.html).
Glossary
Abbreviations:
- RCC
renal cell carcinoma
- HIV
human immunodeficiency virus
- CHX
cycloheximide
- sEH
soluble epoxide hydrolase
- FBS
fetal bovine serum
- DMSO
dimethyl sulfoxide
- PBS
phosphate buffered saline
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/23374
References
- 1.Weiss RH. p21Waf1/Cip1 as a therapeutic target in breast and other cancers. Cancer Cell. 2003;4:425–9. doi: 10.1016/S1535-6108(03)00308-8. [DOI] [PubMed] [Google Scholar]
- 2.Yeh JR, Mohan R, Crews CM. The antiangiogenic agent TNP-470 requires p53 and p21CIP/WAF for endothelial cell growth arrest. Proc Natl Acad Sci U S A. 2000;97:12782–7. doi: 10.1073/pnas.97.23.12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin SV, Shankland SJ. Not just an inhibitor: a role for p21 beyond the cell cycle-“The truth is rarely pure and never simple”. J Am Soc Nephrol. 2004;15:825–6. doi: 10.1097/01.ASN.0000117896.79378.43. [DOI] [PubMed] [Google Scholar]
- 4.Chen H, Li C, Huang J, Cung T, Seiss K, Beamon J, et al. CD4+ T cells from elite controllers resist HIV-1 infection by selective upregulation of p21. J Clin Invest. 2011;121:1549–60. doi: 10.1172/JCI44539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bedelbaeva K, Snyder A, Gourevitch D, Clark L, Zhang XM, Leferovich J, et al. Lack of p21 expression links cell cycle control and appendage regeneration in mice. Proc Natl Acad Sci U S A. 2010;107:5845–50. doi: 10.1073/pnas.1000830107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss RH, Joo A, Randour C. p21(Waf1/Cip1) is an assembly factor required for platelet-derived growth factor-induced vascular smooth muscle cell proliferation. J Biol Chem. 2000;275:10285–90. doi: 10.1074/jbc.275.14.10285. [DOI] [PubMed] [Google Scholar]
- 7.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-P. [DOI] [PubMed] [Google Scholar]
- 9.Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–34. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Winters ZE, Leek RD, Bradburn MJ, Norbury CJ, Harris AL. Cytoplasmic p21WAF1/CIP1 expression is correlated with HER-2/ neu in breast cancer and is an independent predictor of prognosis. Breast Cancer Res. 2003;5:R242–9. doi: 10.1186/bcr654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss RH, Borowsky AD, Seligson D, Lin PY, Dillard-Telm L, Belldegrun AS, et al. p21 is a prognostic marker for renal cell carcinoma: implications for novel therapeutic approaches. J Urol. 2007;177:63–8, discussion 68-9. doi: 10.1016/j.juro.2006.08.073. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W., et al. High levels of constitutive WAF1/Cip1 protein are associated with chemoresistance in acute myelogenous leukemia. Clin Cancer Res. 1995;1:1051–7. [PubMed] [Google Scholar]
- 13.Ferrandina G, Stoler A, Fagotti A, Fanfani F, Sacco R, De Pasqua A, et al. p21WAF1/CIP1 protein expression in primary ovarian cancer. Int J Oncol. 2000;17:1231–5. doi: 10.3892/ijo.17.6.1231. [DOI] [PubMed] [Google Scholar]
- 14.Baretton GB, Klenk U, Diebold J, Schmeller N, Löhrs U. Proliferation- and apoptosis-associated factors in advanced prostatic carcinomas before and after androgen deprivation therapy: prognostic significance of p21/WAF1/CIP1 expression. Br J Cancer. 1999;80:546–55. doi: 10.1038/sj.bjc.6690390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park SH, Park JY, Weiss RH. Antisense attenuation of p21 sensitizes kidney cancer to apoptosis in response to conventional DNA damaging chemotherapy associated with enhancement of phospho-p53. J Urol. 2008;180:352–60. doi: 10.1016/j.juro.2008.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J Biomed Biotechnol. 2012;2012:170325. doi: 10.1155/2012/170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burnett JC, Rossi JJ, Tiemann K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol J. 2011;6:1130–46. doi: 10.1002/biot.201100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park SH, Wang X, Liu R, Lam KS, Weiss RH. High throughput screening of a small molecule one-bead-one-compound combinatorial library to identify attenuators of p21 as chemotherapy sensitizers. Cancer Biol Ther. 2008;7:2015–22. doi: 10.4161/cbt.7.12.7069. [DOI] [PubMed] [Google Scholar]
- 19.Inoue H, Hwang SH, Wecksler AT, Hammock BD, Weiss RH. Sorafenib attenuates p21 in kidney cancer cells and augments cell death in combination with DNA-damaging chemotherapy. Cancer Biol Ther. 2011;12:827–36. doi: 10.4161/cbt.12.9.17680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–44. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- 21.O’Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997;57:4285–300. [PubMed] [Google Scholar]
- 22.Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002;277:11352–61. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 23.Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–52. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]
- 24.Dong Y, Chi SL, Borowsky AD, Fan Y, Weiss RH. Cytosolic p21Waf1/Cip1 increases cell cycle transit in vascular smooth muscle cells. Cell Signal. 2004;16:263–9. doi: 10.1016/S0898-6568(03)00136-0. [DOI] [PubMed] [Google Scholar]
- 25.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–6. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 26.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–78. [PubMed] [Google Scholar]
- 27.Motzer RJ, Bukowski RM. Targeted therapy for metastatic renal cell carcinoma. J Clin Onco. 2006;24:5601–8. doi: 10.1200/JCO.2006.08.5415. [DOI] [PubMed] [Google Scholar]
- 28.Liu JY, Park SH, Morisseau C, Hwang SH, Hammock BD, Weiss RH. Sorafenib has soluble epoxide hydrolase inhibitory activity, which contributes to its effect profile in vivo. Mol Cancer Ther. 2009;8:2193–203. doi: 10.1158/1535-7163.MCT-09-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan Y, Borowsky AD, Weiss RH. An antisense oligodeoxynucleotide to p21(Waf1/Cip1) causes apoptosis in human breast cancer cells. Mol Cancer Ther. 2003;2:773–82. [PubMed] [Google Scholar]
- 30.Jester BW, Cox KJ, Gaj A, Shomin CD, Porter JR, Ghosh I. A coiled-coil enabled split-luciferase three-hybrid system: applied toward profiling inhibitors of protein kinases. J Am Chem Soc. 2010;132:11727–35. doi: 10.1021/ja104491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.