

Summary
The glomerulus has been at the center of attention as the primary site of injury in diabetic nephropathy (DN). Although there is no question that there are changes seen in the glomerulus, it is also well known that tubulointerstitial changes are a prominent component of the disease, especially in patients with type 2 diabetes. The level of albuminuria and DN disease progression best correlate with tubular degeneration and interstitial fibrosis. Nephrotoxicity studies in animals reveal that albuminuria is a highly sensitive marker of early tubular toxicity even in the absence of glomerular pathology. Urinary biomarker data in human beings support the view that proximal tubule injury contributes in a primary way, rather than in a secondary manner, to the development of early DN. I present a model in which very specific injury to the proximal tubule in vivo in the mouse results in severe inflammation, loss of blood vessels, interstitial fibrosis, and glomerulosclerosis. Increased glucose levels, free glycation adducts, reactive oxygen species, and oxidized lipids result in toxicity to tubule epithelia. This results in loss of cells with a stimulus to repair the epithelium. However, because of sublethal injury there is cell-cycle arrest in epithelial cells attempting to replace damaged cells. This leads to epithelial secretion of both profibrogenic growth factors, collagens, and factors that cause pericytes to proliferate and differentiate into myofibroblasts, leading to endothelial destabilization and capillary rarefaction. Local ischemia ensues with further injury to the tubules, more profibrogenic mediators, matrix protein deposition, fibrosis, and glomerulosclerosis.
Keywords: Fibrosis, kidney, diabetic nephropathy, chronic kidney disease, KIM-1
Despite implementation within the past 20 years of treatments that were presumed to be renoprotective, diabetes mellitus continues to rank as the number one cause of end-stage renal disease (ESRD). The glomerulus has been at the center of attention as the primary site of injury that characterizes diabetic nephropathy (DN).1 Proteinuria has been linked to the degree of mesangial expansion in human beings with type 1 diabetes mellitus (DM).2 In these studies there was a striking inverse relationship between the relative surface to volume densities (S/V) of the peripheral capillary wall and the extent of mesangial expansion. Thus, mesangial expansion correlates inversely with capillary filtration surface in the glomerulus. Furthermore, the degree of mesangial expansion correlated directly with the extent of interstitial fibrosis. In their classic report, Mauer et al2 commented, “the close correlation between the mesangium and the interstitium in diabetic nephropathy suggests that studies of renal interstitial cells in diabetes could prove useful.” It generally has been accepted that the glomerular pathology is primary and the tubulointerstitial disease is secondary. However, particularly in type 2 diabetes, the progression of the disease clearly is predicted by the extent of tubular atrophy and interstitial fibrosis.3 Atubular glomeruli have been reported in both type 1 and type 2 diabetes.4,5 Atubular glomeruli are described in a number of renal diseases characterized by a severe tubulointerstitial component, with tubular atrophy and interstitial fibrosis resulting in the glomerulus losing its connection to a functional tubule.6 Many atubular glomeruli have minimal ultrastructural changes, although it is not clear how long they survive in this state. In this review we discuss the pathologic changes in kidney tubules and interstitium and explore the potential role that the primary tubulointerstitial injury might play in initiating loss of renal function with secondary sequelae that lead to chronic kidney disease, worsening of glomerular injury, and ESRD. Hopefully, this will motivate progress in therapeutics for DM that are focused on the tubulointerstitial disease.
ASSOCIATION OF INTERSTITIAL DISEASE WITH DN GLOMERULAR LESIONS, FUNCTIONAL IMPAIRMENT, AND PROTEINURIA
Although there is no question that there are changes seen in the glomerulus in patients with DN, it is also well known that tubulointerstitial changes are a prominent component of the disease, especially in patients with type 2 diabetes. The tubulointerstitium accounts for more than 90% of kidney volume. Progressive renal fibrosis is an important characteristic of DN and is believed to be an important component of the pathogenesis leading to chronic kidney disease and ultimately ESRD. In type 1 diabetes tubulointerstitial disease has been seen to occur independent of glomerular disease.1 Efferent and afferent arteriolar hyalinosis is present early in the course of diabetes in human beings.1 In 1980, Bader et al7 reported that evaluation of 103 patients with diabetic glomerulosclerosis revealed that there was a good correlation between vessel index and relative cortical interstitial volume and serum creatinine. They found that severe glomerular pathology could be found with normal creatinine levels if there was little interstitial fibrosis. On the other hand patients with mild glomerular changes, when associated with severe interstitial fibrosis, always had increased serum creatinine levels. These investigators proposed that “Alterations to postglomerular vessels by interstitial fibrotic changes result in increased resistance to renal cortical blood flow with subsequent reduction of glomerular perfusion … It is also conceivable that glomerular function is affected by the malfunctioning atrophic tubules in areas of interstitial fibrosis.”7 Mauer and colleagues8 also hypothesized that arteriolar hyalinosis could contribute to global glomerular sclerosis through compromise of glomerular capillary blood flow. Reduced glomerular surface area leads to reduced glomerular filtration rate in type 1 diabetic patients with DN.9
The level of albuminuria10 and DN disease progression11 best correlate with tubular degeneration and interstitial fibrosis. The interstitium contains multiple types of cells including pericytes, fibroblasts, dendritic cells, lymphocytes, and macrophages. When fibroblasts are activated, they become myofibroblasts and produce collagens. This activation can be related to factors that are produced by inflammatory cells or by activated tubular cells, which also can produce collagens. Local proliferation of myofibroblasts and macrophages is a prominent component of chronic kidney disease. As indicated by Humphreys in this issue of Seminars in Nephrology, it is also likely that pericytes differentiate to myofibroblasts and contribute in an important way to the fibrosis.12
Most of the therapies for DN that have been directed toward the glomerulus have been unsuccessful in stemming the course of the disease with the exception of angiotensin-converting enzyme inhibitors13,14 or angiotensin II–receptor antagonists.15,16 Angiotensin II has vasoconstrictive effects, increases intraglomerular capillary pressure,17 and alters the podocyte cytoskeleton and components of the slit diaphragm, resulting in increased proteinuria.18 In addition, however, angiotensin II can induce cell proliferation, increase the generation of reactive oxygen species, increase transforming growth factor-β1 (TGF-β) production by the proximal tubule cell, and increase matrix production.19 In collaboration with the group of Van Goor,20 we studied the effect of angiotensin-converting enzyme inhibition in Ren 2 rats, which overexpress the mouse renin gene (Ren2) and spontaneously develop kidney tubulointerstitial disease. When the biomarker for proximal tubule injury, urinary Kim-1, was measured in 8-week-old homozygous Ren2 rats with and without ramipril (1 mg/kg/d in drinking water for 4 weeks), blockade of the renin-angiotensin system resulted in a marked reduction in urinary kidney injury molecule-1 (Kim–1) levels, reduction in collagen III expression, and reduction in interstitial fibrosis. Rosolowsky et al21 recently reported data that even with careful “renoprotective therapies,” which include angiotensin II–directed therapies, the risk for ESRD in patients with type 1 diabetes has not decreased in 20 years. One could make the case that more attention should be devoted to therapies targeted to prevent the tubular injury to prevent onset and/or progression of diabetic kidney disease.
EVIDENCE THAT THE TUBULE IS A PRIMARY SITE OF INJURY IN DN
Microalbuminuria has long been associated with DN, is a predictor of long-term outcome,22 and generally has been attributed to glomerular injury. Nephrotoxicity studies in animals, however, reveal that albuminuria is a highly sensitive marker of early tubular toxicity in the absence of glomerular pathology.23 Early DN is associated with increased excretion of small-molecular-weight proteins, which may be the consequence of decreased capacity of the proximal tubule to reabsorb them.24 Hence, it is possible that the early leakage of small-molecular-weight proteins and even albumin may relate to a primary defect in the capacity of the proximal tubules to reabsorb proteins that normally pass through the glomerular filter.10 In addition, there also may be increased secretion of proteins by the proximal tubules.
Other investigators25 have argued that proteinuria derived from glomerular leakage occurs early in DN and contributes to the progression of tubulointerstitial damage. Tubulointerstitial disease has been proposed to be secondary to both enhanced protein uptake by proximal tubule cells with lysosomal rupture resulting in direct tubule toxicity, as well as cytokines and chemokines generated by the proximal tubule after albumin uptake, which enhance the inflammatory response and activate fibrotic processes in the interstitial compartment.26
Urinary biomarker data support the view that proximal tubule injury contributes in a primary way, rather than a secondary manner, to the development of early DN. Miltenyi et al27 found that urinary excretion of β2 microglobulin, γ-glutamyltransferase, leucine aminopeptidase, and N-acetyl-β-D-glucosaminidase (NAG) activities were increased during ketoacidosis in 11 patients when compared with healthy controls. All of these biomarkers are indicators of proximal tubule injury. NAG is a lysosomal enzyme that derives from the tubule and its presence in the urine indicates early tubule injury. Insulin infusion resulted in a decrease in β2 microglobulin excretion even when urinary albumin increased.28
Further evidence for proximal dysfunction was observed in 13 poorly controlled diabetic patients who had reduced tubular phosphate and sodium reabsorption.27 Watts et al29 found increased urinary NAG excretion was associated with early DN and poor long-term glycemic control in insulin-dependent diabetes mellitus. Gibb et al,30 in 1989, measured the urinary extraction of albumin, retinol binding protein, and NAG in 60 children with insulin-dependent diabetes mellitus and in 45 normal children. These investigators concluded “that tubular abnormalities are present early in the course of insulin dependent diabetes mellitus and suggest that the early increase in urinary excretion of albumin may be at least partly tubular in origin, and that glycemic control may influence this aspect of proximal tubular function.”30
By using data from the second Joslin study, we found, in a cross-sectional comparison, that urinary levels of tubular injury biomarkers, KIM-1 and NAG, were increased significantly in patients with type 1 diabetes and microalbuminuria in comparison with diabetic patients with normoalbuminuria and nondiabetic healthy controls. Low urinary KIM-1 and NAG at baseline were associated strongly with regression of microalbuminuria during a 2-year follow-up period31 (Fig. 1). Thus, less injury to the proximal tubule, as reflected by lower levels of urinary KIM-1 and NAG, is associated with regression of microalbuminuria. This effect is independent of glycemic control, or blood pressure, or treatment with angiotensin-converting enzyme inhibitors. The effects on these urinary biomarkers were independent of clinical characteristics. Hence, microalbuminuria in the absence of significant tubule injury is a more readily reversible state. The etiology of the microalbuminuria may be changes in glomerular hemodynamics or changes in functional characteristics of the proximal tubule without any structural injury. Although patients with microalbuminuria had significantly higher levels of interleukin (IL)-6, interferon-γ-induced protein-10 (IP-10), and monocyte chemoattractant protein 1 (MCP-1) levels when compared with patients with normoalbuminuria, these urinary cytokine levels were not associated with progression or regression of microalbuminuria.
Figure 1.
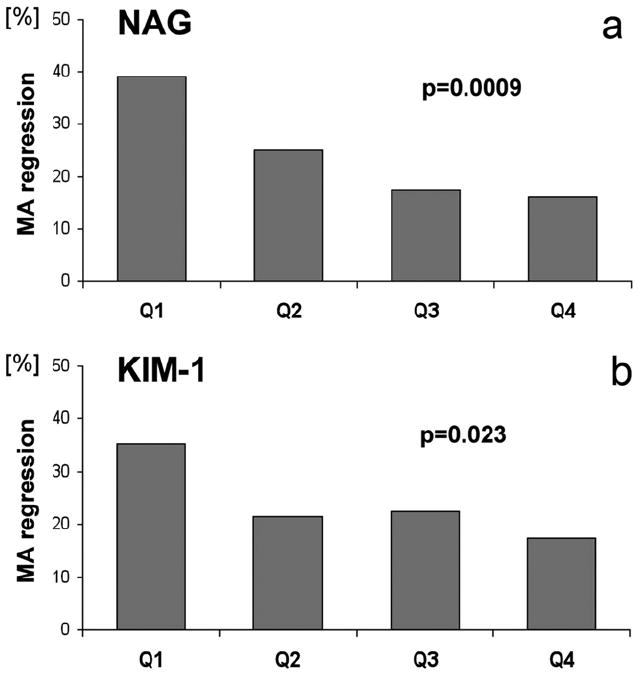
Percentage of patients who experienced a regression of microalbuminuria during the 2-year follow-up evaluation as a function of urinary concentrations of (a) NAG or (b) KIM-1, markers of tubular injury. The proportion of subjects who regressed is plotted as a function of baseline quartiles of NAG and KIM-1. MA, microalbuminuria; Q1, quartile 1; Q2, quartile 2; Q3, quartile 3; Q4, quartile 4. P values for a test of trend are presented. Reprinted with permission from Vaidya et al.31
POSSIBLE MECHANISMS FOR INJURY TO THE PROXIMAL TUBULE IN EARLY DN
High Glucose Levels
The fact that proximal32 and interstitial33 cells respond directly to high levels of glucose with production of profibrotic mediators provides a mechanism by which tubuloin-terstitial disease may occur without an increase in filtered albumin. In the streptozotocin model of diabetic nephropathy in rats, in situ hybridization studies revealed that inner cortex and outer medulla proximal tubular procollagen alpha 1(IV) messenger RNA (mRNA) levels were increased before any measurable change in glomerular levels and were reduced by insulin administration.34
Many investigators28,35 have found that the degree of tubular proteinuria is related to glycemic control. There has been some attention directed to studying the effects of hyperglycemia on the proximal tubule and how these effects might potentiate an increase in interstitial disease leading to fibrosis. Glucose is taken up into the proximal tubule primarily through two Na-glucose transporter proteins. In the early part of the proximal tubule glucose is taken up by a high-capacity, low-affinity Na+-glucose cotransporter SGLT2 (SLC5A2), whereas in the later portions of the proximal tubule, glucose uptake occurs primarily through SGLT1 (SLC5A1), a high-affinity, low-capacity Na+-glucose cotransporter. SGLT1 and 2 mRNA levels are increased in the renal cortex of streptozotocin-diabetic36 and diabetic obese Zucker rats.37
TGF-β1
When renal epithelial cells in culture are exposed to high glucose concentrations they produce increased amounts of types I and IV collagens.32 The profibrogenic cytokine, TGF-β1, has been implicated in the development of renal fibrosis in a number of pathophysiological states in the kidney. TGF-β1 has been linked closely to the development and progression of DN.38 Increased levels of glucose stimulate the production of TGF-β1 by murine mesangial cells in culture, which then acts by autocrine mechanisms to enhance collagen production.39 Increased exposure of human proximal tubule cells to high glucose concentrations in the presence of platelet-derived growth factor (PDGF) induces TGF-β1 protein synthesis. The presence of high glucose enhances TGF-β1 gene expression but the presence of PDGF is needed to facilitate TGF-β1 protein production.40 Extracellular signal-regulated kinase-p38 mitogen activated protein kinase (ERK-p38 MAPK), hydroxy-eicosatetraenoic acids (HETES), hexosamines, reactive oxygen species, and protein kinase C have been implicated in the effects of glucose to stimulate TGF-β1 production.38 In proximal tubule cells, TGF-β1 increases TGF-β1 mRNA and protein levels. TGF-β1 leads to increased ERK and p38 MAP kinases activation and increased phosphorylation of the Smads, intracellular effector molecules for TGF-β1. The transcription factor AP1 is downstream of Smad3. Smad3 is necessary but not sufficient for autoinduction of TGF-β1 because p38 and ERK activation also are required.41
The generation of TGF-β1 by the proximal tubule cell may have adverse autocrine effects on the epithelial cell in addition to its paracrine effects on interstitial cells.42,43 Tubular overexpression of TGF-β1 results in proliferation of peritubular cells and deposition of collagen. With sustained overexpression (4 days in the mouse) there is stimulation of autophagy with loss of epithelial cells and tubular atrophy. Interstitial fibroblasts proliferate with no evidence for conversion of tubular epithelial cells to myofibroblasts.
ROS
High glucose increases the generation of intracellular ROS in renal tubular epithelial cells.44 These ROS may derive from the overproduction by the mitochondria, nonenzymatic glycation, auto-oxidation of glucose, or advanced glycation end products. Han et al44 showed that the increased H2O2 generated by proximal tubule cells in response to high glucose was dependent on protein kinase C and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. ROS have been implicated also in the pathophysiology of diabetic vascular disease. ROS interact directly with nitric oxide (NO) to form peroxynitrite and reduce levels of available NO. Reduced levels of NO can lead to vasoconstriction in the peritubular vessels with secondary hypoxia to the tubules. Blood flow also can be compromised by the fact that chronic hyperglycemia can lead to increased lactate levels and decreased oxygen delivery to the tubule.
Studies have shown that the effect of hyperglycemia on ROS production persists for long periods of time even after the hyperglycemia is corrected.45 Hyperglycemia results in long-term epigenetic changes that can continue to drive ROS production and proinflammatory gene up-regulation long after the hyperglycemia has been corrected. In endothelial cells, for example, exposure to transient hyperglycemia for 16 hours resulted in a sustained increase in nuclear factor-κB p65 gene expression caused by Set7/9-mediated histone methylation. This effect was maintained for 6 days after the cells were returned to normal glucose media. During this time, two nuclear factor-κB–regulated genes involved in diabetes pathophysiology, the chemokine (C-C motif) ligand 2 gene (ccl2) (encoding MCP-1) and the vascular cell adhesion protein-1 gene (vcam-1), were activated.46 Studies using the mitochondrial antioxidant, idebenone, have shown that reduced production of ROS results in a reduction in glucose-mediated epigenetic changes.46
In a recent phase 2, placebo-controlled study of 227 patients with type 2 diabetes and chronic kidney disease, subjects treated with an oral antioxidant, bardoxolone methyl, had a higher GFR within 4 weeks and this difference was maintained for 52 weeks.47 Bardoxolone methyl activates the Keap1-Nrf2 pathway, interacting with the cysteine residues on Keap1 and thereby allowing Nrf2 to translocate to the nucleus where it up-regulates a number of cytoprotective anti-inflammatory genes.48
Advanced Glycation End Products
Chronic hyperglycemia leads to nonenzymatic glycation of proteins to form advanced glycation end products (AGEs).49 AGEs accumulate in the serum of patients with diabetes and are reabsorbed into the proximal tubules and undergo catabolism.50 There are binding sites for AGEs on proximal tubules of rodents and AGE binding is increased after development of diabetes.51 When human proximal tubules were exposed to glycated albumin there was a marked up-regulation at the mRNA and protein level of soluble intercellular adhesion molecule (ICAM-1) and IL-8.52 Methylglyoxal-BSA-AGE and BSA-AGE exposure of human proximal tubules cells resulted in up-regulation of connecting tissue growth factor (CTGF), TGF-β, vascular endothelial growth factor (VEGF), and carboxymethyl lysine-bovine serum albumin (BSA) stimulated production of IL-6, CTGF, TGF-β, VEGF, and chemokine (C-C motif) ligand 2.53 AGEs increase cytosolic phospholipase A2α activity and increase cellular phosphoinositol 4,5 bisphosphate production. Associated increased cyclooxygenase activity stimulates the production of ROS in human proximal tubule HK-2 cells in culture. Cellular Na+,K+ adenosine triphosphatase activity is reduced by the increased arachidonic acid metabolism, leading to clathrin-mediated endocytosis.54 It previously was shown55 that although total Na+,K+ adenosine triphosphatase might increase in streptozotocin-induced diabetic nephropathy in the rat, the redistribution to the cytosol suggests that the pump is less functional.
MicroRNAs
MicroRNAs (miRs) are small posttranscriptional regulators of gene expression. miRs have been isolated from paraffin-embedded biopsy specimens of patients with diabetes mellitus and nephropathy.56 Decreased levels of miR-192 were found in patients with greater amounts of kidney interstitial fibrosis with a significant inverse correlation observed between levels of miR-192 and fibrosis score. miR-192 was localized by in situ hybridization to the glomeruli and the proximal tubules. In human kidney HK-2 cells TGF-β results in a down-regulation of E-cadherin and miR-192 and induction of plasminogen activator inhibitor. If there is overexpression of miR-192 the effects of TGF-β on E cadherin are mitigated somewhat. Thus, miR-192 may be involved in changes in proximal tubule phenotype that have profibrogenic consequences in DN. Altering glucose concentration in the media of HK-2 cells grown in culture had no effect on the miR profiles of the cells.
Injury to the Proximal Tubule Can Result in Interstitial Inflammation, Fibrosis, and Glomerulosclerosis
Recent experiments from our laboratory may shed some light on the implications of primary proximal tubular injury for the subsequent development of inflammation and secondary glomerular injury, even though the model we used did not incorporate diabetes. We developed a mouse model of kidney injury using Six2-Cre-LoxP technology to selectively activate expression of the simian diphtheria toxin receptor in renal epithelia derived from the metanephric mesenchyme (DTR renal epithelial cell).57 By adjusting the timing and dose of diphtheria toxin (DT), a highly selective model of tubular injury was created to define the acute and chronic consequences of isolated epithelial injury. The DT-induced sublethal tubular epithelial injury was confined to the S1 and S2 segments of the proximal tubule rather than being widespread in the metanephric mesenchyme-derived epithelial lineage. We asked the following questions: (1) does isolated renal epithelial injury/repair affect other nonepithelial cell systems; (2) is the normal regenerative response of the renal epithelium affected after targeted disruption, and is there a difference in long-term outcome between single and multiple insults; and (3) can isolated proximal tubule injury result in tubulointerstitial fibrosis and potentially affect the glomerulus?
After a single dose of DT, acute injury promptly was followed by inflammatory cell infiltration and robust tubular cell proliferation leading to complete recovery leaving no residual injury detectable. During the acute inflammatory phase there was a focal mild to moderate increase in macrophages (F4/80+), T cells (CD3+), and neutrophils (antineutrophil antibody +). There was also a 2.5-fold (day 1) and 14-fold (day 3) increase in ICAM-1 expression in the tubulointerstitium of DT-injured kidneys. If the dosing of DT was not repeated this inflammatory response reversed and the tissue returned to normal. In striking contrast, three DT insults to renal epithelial cells at 1-week intervals resulted in maladaptive repair with tubular atrophy, interstitial capillary loss, fibrosis, proteinuria, and focal and segmental glomerulosclerosis that was highly correlated with the degree of interstitial fibrosis (Fig. 2).57 Three doses of DT over 3 weeks resulted in chronic tubulointerstitial injury including atrophic tubules and expanded tubulointerstitium (Fig. 2A). There was a clear positive correlation between the percentage of interstitial fibrosis/tubular atrophy and the number of globally or segmentally sclerosed glomeruli in these kidneys (Fig. 2B) and albuminuria in these mice Fig. 2C). There was up-regulation of TGF-β1, collagen 1α1, and fibronectin mRNA levels in the kidneys (Fig. 2D). There was increased deposition of extra-cellular matrix as shown by Masson’s trichrome staining (Fig. 2E). Staining of kidney sections with Kim-1–specific antibody revealed marked up-regulation of Kim-1 in tubules of fibrotic DT-treated kidneys (Fig. 2F) and in urine after repeated (3×) DT injection <0.15 μg/kg), but only slightly increased after one DT administration (Fig. 2G). There was no early podocyte injury even when there was significant tubular injury, whereas podocytes were clearly abnormal at later time points when fibrosis also was apparent. Thus, selective epithelial injury can drive the formation of interstitial fibrosis, capillary rarefaction, and potentially glomerulosclerosis, substantiating a direct role for damaged tubule epithelium in the pathogenesis of chronic kidney disease. Animals with advanced tubulo-interstitial damage also had increased serum creatinine levels.
Figure 2.
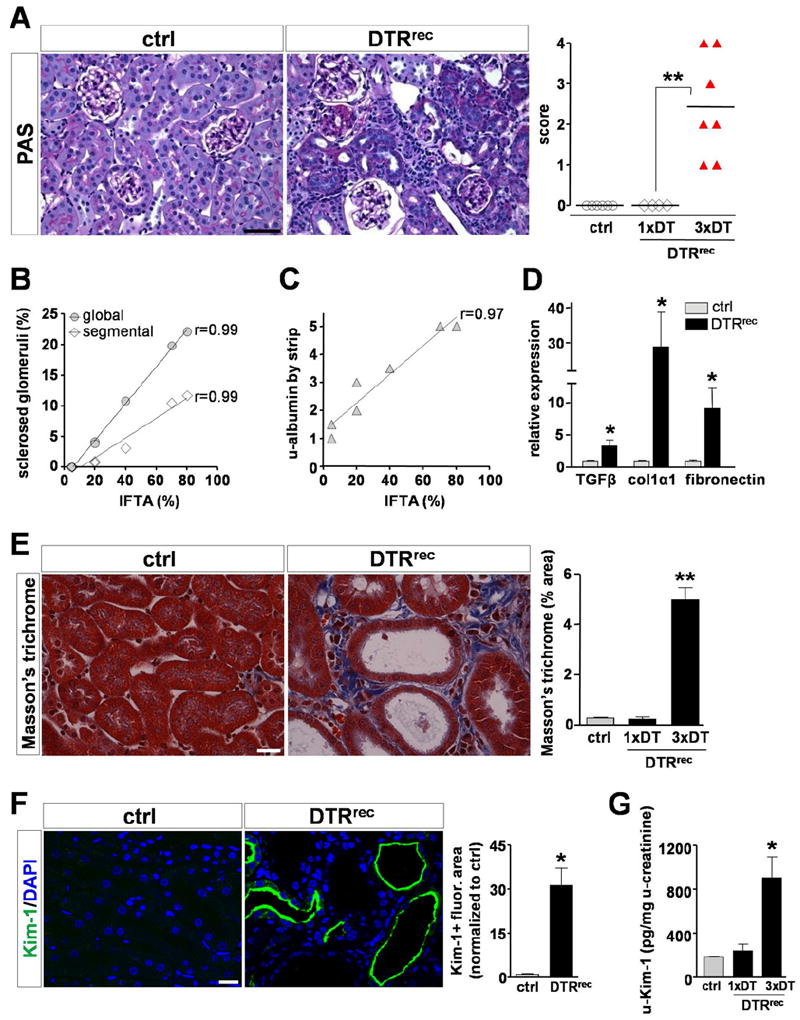
Repeated injury to proximal tubular segments S1/S2 leads to development of tubulointerstitial fibrosis with secondary glomerulosclerosis. (A) Left: Representative micrographs of periodic acid-Schiff (PAS)-stained control kidney sections and sections obtained from control animals and animals that expressed the diphtheria toxin receptor in renal epithelial cells (DTRrenal epithelial cell [DTRrec mice]). DTRrec kidneys of animals treated weekly for 3 weeks with DT developed chronic tubulointerstitial injury with atrophic tubules and expanded tubulointerstitium. Scale bar, 50 μm. Right: Scoring of chronic tubulointerstitial damage in control animals or animals treated with only one or all three doses. Data points represent individual animals. **P < .005. (B and C) The percentage of tissue interstitial fibrosis/tubular atrophy (IFTA) was correlated positively with the number of globally or segmentally sclerosed glomeruli in these kidneys and the amount of urinary (u)-albumin. Data points represent measurements of individual animals (n = 7), lines represent best fits. (D) Quantitative reverse-transcription polymerase chain reaction analysis shows up-regulation of TGF-β1, collagen 1α1, and fibronectin mRNA levels in DT-injured kidneys. *P < .05. (E) Left: Masson’s trichrome staining reveals high levels of kidney fibrosis in DT-treated animals if the DT was given three times. Scale bar, 20 μm. Right: Quantification of kidney area positive for extracellular matrix relative to total area (n = 4 to 5 for each data point). *P < .0001. (F) Left: Staining of kidney sections with Kim-1–specific antibody reveals high expression of Kim-1 in tubules of fibrotic DTRrec kidneys after repeated DT treatment. Note abnormally dilated tubules and apical localization of Kim-1. Scale bar, 20 μm. Right: Semi-automated quantitative analysis of Kim-1+ fluorescence area normalized to control. Values are shown as mean ± standard error of the mean (n = 3-5 for each data point). *P < .01. (G) Quantification of urinary Kim-1 normalized to urinary creatinine concentration after single (1×) or repeated (3×) DT injection (0.15 μg/kg). Data are given as mean ± standard error of the mean (n = 3-5 for each data point). *P < .05 versus control. DAPI, 4’6-diamino-2-phenylindole-2HCl. Reprinted with permission from Grgic et al.57
Whereas Kim-1 protein levels were increased in both kidney sections and urine of animals injected three times with DT when compared with controls, levels of urinary neutrophil gelatinase-associated lipocalin (NGAL) were only minimally increased. The increase in urinary Kim-1 in the absence of a marked change in NGAL is consistent with the morphologic findings that the injury was confined primarily to the proximal tubule and spared the distal nephron. There was a dramatic increase in the number of pericytes/perivascular fibroblasts (PDGF receptor β+), myofibroblasts (αSMA+), fibroblast specific protein-1–expressing cells (FSP-1/S100A4+), as well as macrophages (F4/80+) in DTRrenal epithelial cell kidneys after repeated DT injury. The number of endothelial cells (CD31/PECAM+) was reduced significantly in these kidneys, indicating capillary drop-out. There was increased kidney expression of the profibrotic cytokine TGF-β1 and major matrix components (collagen 1α1, fibronectin). It is possible that similar pathophysiological processes may occur in DN where, as indicated earlier, there are many components of blood and intraluminal tubular fluid that may exert toxic effects on the proximal tubule, which over time may result in cell activation, cell-cycle arrest, and, ultimately, cell loss.
HOW DOES PROXIMAL TUBULE INJURY CONTRIBUTE TO INTERSTITIAL INFLAMMATION?
High glucose and AGEs evoke the production of proinflammatory factors by the epithelial cell. Also, as indicated previously in this review, albuminuria itself has been implicated as a proinflammatory influence on the tubule.58 IL-6 and MCP-1 (also known as chemokine (C-C motif) ligand 2) are two of these proinflammatory cytokines. Macrophages accumulate in the diabetic kidney interstitium and are well documented to contribute to inflammation59 and fibrosis60 in many kidney diseases including DN. In mouse models of DN, MCP-1 production by proximal tubules is increased. MCP-1(-/-) mice are protected against the development of albuminuria in a streptozotocin model of DN. The diabetic MCP-1(-/-) mice had marked reductions in glomerular and interstitial macrophages, histologic damage, and fibrosis, and also had fewer kidney macrophages expressing markers of activation including nitric oxide synthase and sialoadhesin.61 There is also a good deal of evidence that blocking macrophage recruitment or activation can ameliorate renal inflammation and tissue fibrosis.62 Further, urine levels of MCP-1 are increased significantly in patients with DN, and its levels have been correlated significantly with albuminuria and NAG in both human and experimental DN models.61,63-65
Macrophages are not the only cells implicated in the interstitial inflammatory component of DN. There is evidence that neutrophils and T cells are involved but there are few data defining the roles of these cells in the pathobiology of DN or the roles of the proximal tubules in homing of these cells to the kidney. Abnormal activation of blood neutrophils has been reported in patients with type 1 and type 2 diabetes.66,67 AGEs stimulate proximal tubular production of IL-8 and ICAM-1, and there is colocalization of ICAM-1 and AGE in proximal tubules.68 Neutrophils are the primary target cells for the chemotactic function of IL-8. Moon et al69 recently showed that CD3+ and CD4+ T cells infiltrated the kidney in a mouse model of streptozotocin-induced DN. The recruited CD3+ cells produced interferon-γ and tumor necrosis factor-α. They also reported that CD3+ cells are found in the interstitium of patients with type 2 DN. The main site of T-cell recruitment was the interstitium rather than the glomerulus. T-cell infiltration was correlated with degree of proteinuria. It is known that in response to injury, proximal tubules can express major histocompatibility complex II, which may be involved in antigen presentation and the increased numbers of T cells in the diabetic kidney.70 It also is known that proximal tubule cells produce regulated upon activation, normal T-cell expressed, and secreted,71 which is a chemokine chemotactic for T cells, and plays a major role in recruiting these cells to sites of inflammation.
Type 2 diabetes has been proposed to be a disease of the innate immune system.72 Toll-like receptors (TLRs) are very important for the modulation of the innate immune response and inflammation. It has been reported that TLR4 is up-regulated in the renal tubules of human kidneys in patients with DN.73 Intensity of TLR4 expression correlated with the interstitial macrophage infiltration and hemoglobin A1C levels and inversely with glomerular filtration rate. The investigators also found up-regulation of the TLR4 ligand, high-mobility group box 1, in tubules of patients with DN. In vitro high glucose increased TLR4 expression by a protein kinase C–dependent process resulting in an up-regulation of IL-6 and MCP-1 in human proximal tubule cells. Silencing of TLR4 with siRNA resulted in a decrease in IL-6 and MCP-1 production.
HOW DOES PROXIMAL TUBULE INJURY LEAD TO GLOMERULOSCLEROSIS?
Glomerulosclerosis may be a consequence of the tubulointerstitium expansion, capillary loss and inflammation around the afferent and efferent glomerular vessels, as well as endovascular changes in the interlobular vessels. Horlyck et al74 noted that the glomerulosclerosis seen with type 1 diabetes was not random but clustered in a column distribution perpendicular to the surface of the kidney, a pattern that is very suggestive of a vascular etiology. In the metabolic environment of diabetes, in the setting of vascular compromise and tubular drop-out, it is possible that there is a predisposition to glomerulosclerosis.
POTENTIAL ROLE OF CELL-CYCLE ARREST IN THE DEVELOPMENT OF INTERSTITIAL FIBROSIS IN DN
A characteristic of diabetic nephropathy is enlarged kidneys with initial hyperplasia that is followed in time by hypertrophy.75 Ornithine decarboxylase, a rate-limiting enzyme in polyamine synthesis, is believed to be necessary for both the hyperplastic and the hypertrophic phases.76 Inhibiting ornithine decarboxylase decreases the GFR and the hyperreabsorption of filtered Na+,Cl- in the proximal tubule of the diabetic rat kidney. The prevalence of glomerular hyperfiltration increased with increasing stages of prediabetes and prehypertension in a subject population of 99,140 individuals in Japan.77 Accelerated proximal tubule cellular senescence has been described in diabetic nephropathy78 and it has been proposed that diabetic hypertrophy is a transitional phase to senescence.79 Senescence was described in 1965 as a process that limited the proliferation of normal human cells in culture.80 Senescence is important for controlling the proliferation of potential cancer cells but senescence is also of pathophysiological importance in aging and aging-related diseases other than cancer.
We recently proposed a hypothesis that relates cell-cycle arrest after proximal tubule injury to the generation of profibrogenic growth factors.81 We developed a method to characterize the cell-cycle profile of tubular epithelial cells in vivo at various times after an acute insult in five AKI models. The models included moderate reversible ischemia reperfusion injury (IRI), severe IRI, unilateral IRI, acute aristolochic acid toxic nephropathy, and unilateral ureteral obstruction. These models are representative of various forms of kidney disease. The models reflect some of the most common causes for human AKI: ischemia, toxic exposure, and obstruction. Each model of injury resulted in some degree of interstitial fibrosis but this was minimal in the mild bilateral IRI model. In each of the other models there was severe fibrosis that developed over time. All AKI mouse models in which prominent fibrosis developed were characterized by arrest of proximal tubular epithelial cells in the G2/M phase of the cell cycle (Fig. 3). We then showed that G2/M-arrested cultured cells had enhanced expression of the COL1A1 gene, encoding collagen 1a1, the COL4A1 gene, encoding collagen 4a1, as well as enhanced TGFB1 and CTGF gene expression in human proximal tubule cells. In each of the AKI models there were increased mRNA levels of Tgfb1, Ctgf, Col4a1, and Col1a1 in injured mouse kidney tissue. In the models in which fibrosis developed, to a significant extent the mRNA levels of these genes remained increased for at least the duration of the study (42 days in severe IRI, unilateral IRI, and acute aristolochic acid toxic nephropathy; and 14 days in unilateral ureteral obstruction).
Figure 3.
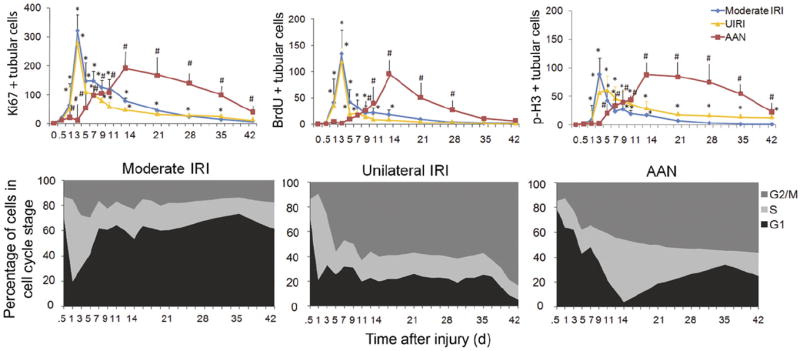
Characteristics of repair of renal tubular cells in models of AKI. (A) Number (per 400× field) of Ki-67–positive (left), bromodeoxyuridine (BrdU)-positive (middle), or p-H3–positive (right) tubular cells in moderate IRI, unilateral IRI, and aristolochic acid nephropathy (AAN) mice (n = 3 mice at each time point in each group). *P < .01 versus sham. # P < .01 versus control. Those mice in the moderate IRI and AAN groups went on to develop severe fibrosis over 42 days after the initial ischemia or dose of AA. (B) Cell-cycle distribution (G1, S, and G2/M) of tubular cells that have entered the cell cycle (Ki67+) in moderate IRI (left), unilateral IRI (middle), and AAN (right) models as a function of time after the insult (n = 3 mice at each time point in each group). Reprinted with permission from Yang et al.80
We then showed that reversal of G2/M arrest rescues the fibrogenic effects. Arrest at G2/M occurs primarily through the activation of checkpoint protein kinases Chk1 and Chk2, which are downstream of the ataxia telangiectasia, mutated (ATM) and ATM Rad3–related pathway.82,83 ATM is activated in tubular cells after injury associated with fibrosis. Blocking the ATM pathway in human HK-2 epithelial cells, with a pharmacologic inhibitor, reduced the number of cells in G2/M arrest after aristolochic acid exposure. The mRNA levels of TGFB1, CTGF, COL4A1, and COL1A1 also were down-regulated to a degree commensurate with the decrease in the number of cells blocked in G2/M. When mice undergoing unilateral IRI were pretreated with pifithrin-a, a p53 inhibitor, or when unilateral IRI mice had their untreated contralateral kidneys removed 3 days after unilateral IRI, the number of cells in G2/M arrest decreased and this was associated with reduced profibrogenic gene expression and reduced interstitial fibrosis.
Verzola et al78 found that senescence-associated β-galactosidase staining was increased in the tubules and p16INK4A was strikingly up-regulated in the nuclei of proximal tubules in kidney biopsy specimens of patients with type 2 DN. Human proximal tubule cells in primary culture that were exposed to high glucose concentrations developed accelerated senescence and reduction in mean telomere length. Satriano et al79 found that opossum kidney proximal tubule cells develop a senescent phenotype after exposure to H2O2 measured by senescence-associated β-galactosidase. On day 10 after administration of strepzotocin to rats, the kidney tissue expression of p21Cip1/Waf1 and p27Kip1 were increased markedly. p21Cip1/Waf1 is a cyclin/cyclin-dependent kinase inhibitor whereas p27Kip1 prevents the activation of cyclin E–cyclin-dependent kinase 2 and cyclin D- cyclin-dependent kinase 4, thus controlling the cell cycle at G1.
Our data associating tubular injury with cell-cycle arrest at G2/M and the generation of factors that promote pericyte conversion into myofibroblasts and interstitial fibrosis provide a potential pathophysiological pathway starting with glycation adducts and leading to vascular rarefaction, fibrosis, and secondary glomerulosclerosis. Our overall hypothesis (Fig. 4), as it relates to the association between cell-cycle arrest and diabetic interstitial fibrosis and glomerulosclerosis, is that free glycation adducts that are generated in DM, and have higher excretion rates in patients with nephropathy, are toxic to tubule epithelia. Together with hyperglycemia, the adducts induce Kim-1/Tim-1 expression in the proximal tubule and cell-cycle arrest in epithelial cells attempting to replace damaged cells. This leads to epithelial secretion of both TGF-β and CTGF, which act on interstitial pericytes to activate Wnt and FGF pathways and cause pericyte (also known as perivascular fibroblasts) proliferation and differentiation into myofibroblasts, leading to endothelial destabilization and capillary rarefaction. Local ischemia ensues with further injury to the tubules, more profibrogenic mediators, matrix protein deposition, and fibrosis. These processes may exist in DN, in which senescence is known to occur in the kidney, especially because the proximal tubule is exposed to a number of noxious influences that can lead to injury and lead to senescence. As indicated previously in this review, AGE exposure of human proximal tubules cells resulted in up-regulation of CTGF, TGF-β, and VEGF.53 CTGF is a downstream effector of TGF-β but also has TGF-β–independent effects to enhance renal fibrosis. Renal tubular expression of CTGF correlates with interstitial fibrosis in type 2 DN.84 Specific down-regulation of CTGF attenuates nephropathy in mouse models of type 1 and 2 DN.85
Figure 4.
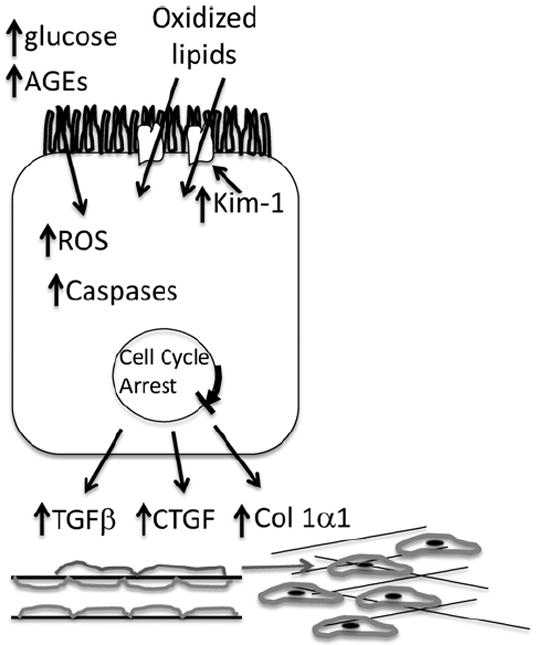
Model for tubule as a primary mediator of DM. Hyperglycemia induces the generation of ROS in the tubule. AGEs are toxic to tubule epithelia. Kim-1/Tim-1 expression is enhanced, which increases oxidized lipid uptake, which, together with the ROS and AGE, enhance caspase activity and cell death. In those cells that survive and are triggered to divide to replace adjacent cells, cell-cycle arrest occurs. This leads to epithelial secretion of both TGF-β, CTGF, and Col1α1, which act on interstitial pericytes to activate Wnt and FGF pathways and cause pericyte (also known as perivascular fibroblasts) proliferation and differentiation into myofibroblasts, leading to endothelial destabilization and capillary rarefaction. Local ischemia ensues with further injury to the tubules, more profibrogenic mediators, matrix protein deposition, and fibrosis.
CONCLUSIONS
We propose that the kidney tubule plays a critical role in the genesis of DN. Rather than the orthodox view that the glomerular injury in DN is primary with the tubular injury secondary, we take the view that in fact the tubular injury is primary. The well-accepted data that disease progression is correlated best with the degree of tubulo-interstitial disease is also consistent with a primary role for the tubule, particularly the proximal tubule. Our data on proximal tubule biomarkers of tubular injury in human beings is consistent with this view. Glomerular pathology may be contributed to by the interstitial inflammation leading to vascular rarefaction, compromise of capillary and arteriolar perfusion, and ischemia. Therapies targeted to the mitigation of injury to the proximal tubule and the subsequent inflammation and fibrosis may be more successful in slowing down the progression of diabetic nephropathy, which is the single most important contributor to the epidemic of end stage kidney disease worldwide.
Acknowledgments
Financial support: Supported by National Institutes of Health grants DK39773, DK72381, and DK054741.
Footnotes
Conflict of interest statement: The author is a co-inventor on kidney injury molecule-1 patents that are assigned to Partners Healthcare and licensed by Partners Healthcare to Johnson and Johnson, Sekisui, BiogenIdec, and a number of research reagent companies. The author also is a consultant for Sekisui.
References
- 1.Fioretto P, Mauer M. Histopathology of diabetic nephropathy. Semin Nephrol. 2007;27:195–207. doi: 10.1016/j.semnephrol.2007.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mauer SM, Steffes MW, Ellis EN, et al. Structural-functional relationships in diabetic nephropathy. J Clin Invest. 1984;74:1143–55. doi: 10.1172/JCI111523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White KE, Bilous RW. Type 2 diabetic patients with nephropathy show structural-functional relationships that are similar to type 1 disease. J Am Soc Nephrol. 2000;11:1667–73. doi: 10.1681/ASN.V1191667. [DOI] [PubMed] [Google Scholar]
- 4.Najafian B, Kim Y, Crosson JT, Mauer M. Atubular glomeruli and glomerulotubular junction abnormalities in diabetic nephropathy. J Am Soc Nephrol. 2003;14:908–17. doi: 10.1097/01.asn.0000057854.32413.81. [DOI] [PubMed] [Google Scholar]
- 5.White KE, Marshall SM, Bilous RW. Prevalence of atubular glomeruli in type 2 diabetic patients with nephropathy. Nephrol Dial Transplant. 2008;23:3539–45. doi: 10.1093/ndt/gfn351. [DOI] [PubMed] [Google Scholar]
- 6.Marcussen N. Atubular glomeruli and the structural basis for chronic renal failure. Lab Invest. 1992;66:265–84. [PubMed] [Google Scholar]
- 7.Bader R, Bader H, Grund KE, et al. Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathol Res Pract. 1980;167:204–16. doi: 10.1016/S0344-0338(80)80051-3. [DOI] [PubMed] [Google Scholar]
- 8.Harris RD, Steffes MW, Bilous RW, Sutherland DE, Mauer SM. Global glomerular sclerosis and glomerular arteriolar hyalinosis in insulin dependent diabetes. Kidney Int. 1991;40:107–14. doi: 10.1038/ki.1991.187. [DOI] [PubMed] [Google Scholar]
- 9.Osterby R, Parving HH, Nyberg G, et al. A strong correlation between glomerular filtration rate and filtration surface in diabetic nephropathy. Diabetologia. 1988;31:265–70. doi: 10.1007/BF00277406. [DOI] [PubMed] [Google Scholar]
- 10.Ziyadeh FN, Goldfarb S. The renal tubulointerstitium in diabetes mellitus. Kidney Int. 1991;39:464–75. doi: 10.1038/ki.1991.57. [DOI] [PubMed] [Google Scholar]
- 11.Rodriguez-Iturbe B, Johnson RJ, Herrera-Acosta J. Tubulointerstitial damage and progression of renal failure. Kidney Int Suppl. 2005;99:S82–6. doi: 10.1111/j.1523-1755.2005.09915.x. [DOI] [PubMed] [Google Scholar]
- 12.Humphreys BD. Targeting pericyte differentation as a strategy to modulate kidney fibrosis in diabetic nephropathy. Semin Nephrol. 2012;32:463–70. doi: 10.1016/j.semnephrol.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456–62. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 14.Gurley SB, Coffman TM. The renin-angiotensin system and diabetic nephropathy. Semin Nephrol. 2007;27:144–52. doi: 10.1016/j.semnephrol.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 15.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–9. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 16.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–60. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 17.Zatz R, Dunn BR, Meyer TW, et al. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J Clin Invest. 1986;77:1925–30. doi: 10.1172/JCI112521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perico N, Benigni A, Remuzzi G. Present and future drug treatments for chronic kidney diseases: evolving targets in renoprotection. Nat Rev Drug Discov. 2008;7:936–53. doi: 10.1038/nrd2685. [DOI] [PubMed] [Google Scholar]
- 19.Decleves AE, Sharma K. New pharmacological treatments for improving renal outcomes in diabetes. Nat Rev. 2010;6:371–80. doi: 10.1038/nrneph.2010.57. [DOI] [PubMed] [Google Scholar]
- 20.de Borst MH, van Timmeren MM, Vaidya VS, et al. Induction of kidney injury molecule-1 in homozygous Ren2 rats is attenuated by blockade of the renin-angiotensin system or p38 MAP kinase. Am J Physiol. 2007;292:F313–20. doi: 10.1152/ajprenal.00180.2006. [DOI] [PubMed] [Google Scholar]
- 21.Rosolowsky ET, Skupien J, Smiles AM, et al. Risk for ESRD in type 1 diabetes remains high despite renoprotection. J Am Soc Nephrol. 2011;22:545–53. doi: 10.1681/ASN.2010040354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Zeeuw D. Albuminuria: a target for treatment of type 2 diabetic nephropathy. Semin Nephrol. 2007;27:172–81. doi: 10.1016/j.semnephrol.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Yu Y, Jin H, Holder D, et al. Urinary biomarkers trefoil factor 3 and albumin enable early detection of kidney tubular injury. Nat Biotechnol. 2010;28:470–7. doi: 10.1038/nbt.1624. [DOI] [PubMed] [Google Scholar]
- 24.Abrass CK. Diabetic proteinuria. Glomerular or tubular in origin? Am J Nephrol. 1984;4:337–46. doi: 10.1159/000166849. [DOI] [PubMed] [Google Scholar]
- 25.Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol. 2006;17:2974–84. doi: 10.1681/ASN.2006040377. [DOI] [PubMed] [Google Scholar]
- 26.Strutz FM. EMT and proteinuria as progression factors. Kidney Int. 2009;75:475–81. doi: 10.1038/ki.2008.425. [DOI] [PubMed] [Google Scholar]
- 27.Miltenyi M, Korner A, Tulassay T, Szabo A. Tubular dysfunction in type I diabetes mellitus. Arch Dis Child. 1985;60:929–31. doi: 10.1136/adc.60.10.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mogensen CE, Christensen NJ, Gundersen HJ. The acute effect of insulin on renal haemodynamics and protein excretion in diabetics. Diabetologia. 1978;15:153–7. doi: 10.1007/BF00421231. [DOI] [PubMed] [Google Scholar]
- 29.Watts GF, Vlitos MA, Morris RW, Price RG. Urinary N-acetyl-beta-D-glucosaminidase excretion in insulin-dependent diabetes mellitus: relation to microalbuminuria, retinopathy and glycaemic control. Diabete Metab. 1988;14:653–8. [PubMed] [Google Scholar]
- 30.Gibb DM, Tomlinson PA, Dalton NR, et al. Renal tubular proteinuria and microalbuminuria in diabetic patients. Arch Dis Child. 1989;64:129–34. doi: 10.1136/adc.64.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vaidya VS, Niewczas MA, Ficociello LH, et al. Regression of microalbuminuria in type 1 diabetes is associated with lower levels of urinary tubular injury biomarkers, kidney injury molecule-1, and N-acetyl-beta-D-glucosaminidase. Kidney Int. 2011;79:464–70. doi: 10.1038/ki.2010.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ziyadeh FN, Snipes ER, Watanabe M, et al. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am J Physiol. 1990;259:F704–14. doi: 10.1152/ajprenal.1990.259.4.F704. [DOI] [PubMed] [Google Scholar]
- 33.Polhill TS, Saad S, Poronnik P, Fulcher GR, Pollock CA. Short-term peaks in glucose promote renal fibrogenesis independently of total glucose exposure. Am J Physiol. 2004;287:F268–73. doi: 10.1152/ajprenal.00084.2004. [DOI] [PubMed] [Google Scholar]
- 34.Ihm CG, Lee GS, Nast CC, et al. Early increased renal procollagen alpha 1(IV) mRNA levels in streptozotocin induced diabetes. Kidney Int. 1992;41:768–77. doi: 10.1038/ki.1992.120. [DOI] [PubMed] [Google Scholar]
- 35.Christiansen JS, Frandsen M, Parving HH. Effect of intravenous glucose infusion on renal function in normal man and in insulin-dependent diabetics. Diabetologia. 1981;21:368–73. doi: 10.1007/BF00252683. [DOI] [PubMed] [Google Scholar]
- 36.Vestri S, Okamoto MM, de Freitas HS, et al. Changes in sodium or glucose filtration rate modulate expression of glucose transporters in renal proximal tubular cells of rat. J Membr Biol. 2001;182:105–12. doi: 10.1007/s00232-001-0036-y. [DOI] [PubMed] [Google Scholar]
- 37.Tabatabai NM, Sharma M, Blumenthal SS, Petering DH. Enhanced expressions of sodium-glucose cotransporters in the kidneys of diabetic Zucker rats. Diabetes Res Clin Pract. 2009;83:e27–30. doi: 10.1016/j.diabres.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Y, Usui HK, Sharma K. Regulation of transforming growth factor beta in diabetic nephropathy: implications for treatment. Semin Nephrol. 2007;27:153–60. doi: 10.1016/j.semnephrol.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ziyadeh FN, Sharma K, Ericksen M, Wolf G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-beta. J Clin Invest. 1994;93:536–42. doi: 10.1172/JCI117004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser D, Brunskill N, Ito T, Phillips A. Long-term exposure of proximal tubular epithelial cells to glucose induces transforming growth factor-beta 1 synthesis via an autocrine PDGF loop. Am J Pathol. 2003;163:2565–74. doi: 10.1016/s0002-9440(10)63611-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M, Fraser D, Phillips A. ERK, p38, and Smad signaling pathways differentially regulate transforming growth factor-beta1 autoinduction in proximal tubular epithelial cells. Am J Pathol. 2006;169:1282–93. doi: 10.2353/ajpath.2006.050921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koesters R, Kaissling B, Lehir M, et al. Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol. 2010;177:632–43. doi: 10.2353/ajpath.2010.091012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips AO, Steadman R. Diabetic nephropathy: the central role of renal proximal tubular cells in tubulointerstitial injury. Histol Histopathol. 2002;17:247–52. doi: 10.14670/HH-17.247. [DOI] [PubMed] [Google Scholar]
- 44.Han HJ, Lee YJ, Park SH, Lee JH, Taub M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am J Physiol. 2005;288:F988–96. doi: 10.1152/ajprenal.00327.2004. [DOI] [PubMed] [Google Scholar]
- 45.Zhong Q, Kowluru RA. Epigenetic changes in mitochondrial superoxide dismutase in the retina and the development of diabetic retinopathy. Diabetes. 2011;60:1304–13. doi: 10.2337/db10-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tonna S, El-Osta A, Cooper ME, Tikellis C. Metabolic memory and diabetic nephropathy: potential role for epigenetic mechanisms. Nat Rev. 2010;6:332–41. doi: 10.1038/nrneph.2010.55. [DOI] [PubMed] [Google Scholar]
- 47.Pergola PE, Raskin P, Toto RD, et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med. 2011;365:327–36. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 48.Dinkova-Kostova AT, Liby KT, Stephenson KK, et al. Extremely potent triterpenoid inducers of the phase 2 response: correlations of protection against oxidant and inflammatory stress. Proc Natl Acad Sci U S A. 2005;102:4584–9. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tan AL, Forbes JM, Cooper ME. AGE, RAGE, and ROS in diabetic nephropathy. Semin Nephrol. 2007;27:130–43. doi: 10.1016/j.semnephrol.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 50.Gugliucci A, Bendayan M. Renal fate of circulating advanced glycated end products (AGE): evidence for reabsorption and catabolism of AGE-peptides by renal proximal tubular cells. Diabetologia. 1996;39:149–60. doi: 10.1007/BF00403957. [DOI] [PubMed] [Google Scholar]
- 51.Youssef S, Nguyen DT, Soulis T, et al. Effect of diabetes and aminoguanidine therapy on renal advanced glycation end-product binding. Kidney Int. 1999;55:907–16. doi: 10.1046/j.1523-1755.1999.055003907.x. [DOI] [PubMed] [Google Scholar]
- 52.Tang SC, Leung JC, Chan LY, Tsang AW, Lai KN. Activation of tubular epithelial cells in diabetic nephropathy and the role of the peroxisome proliferator-activated receptor-gamma agonist. J Am Soc Nephrol. 2006;17:1633–43. doi: 10.1681/ASN.2005101113. [DOI] [PubMed] [Google Scholar]
- 53.Tang SC, Chan LY, Leung JC, et al. Differential effects of advanced glycation end-products on renal tubular cell inflammation. Nephrology (Carlton) 2011;16:417–25. doi: 10.1111/j.1440-1797.2010.01437.x. [DOI] [PubMed] [Google Scholar]
- 54.Gallicchio MA, Bach LA. Advanced glycation end products inhibit Na+ K+ ATPase in proximal tubule epithelial cells: role of cytosolic phospholipase A2alpha and phosphatidylinositol 4-phosphate 5-kinase gamma. Biochim Biophys Acta. 2010;1803:19–930. doi: 10.1016/j.bbamcr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 55.Fekete A, Rosta K, Wagner L, et al. Na+,K+-ATPase is modulated by angiotensin II in diabetic rat kidney—another reason for diabetic nephropathy? J Physiol. 2008;586:5337–48. doi: 10.1113/jphysiol.2008.156703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanchez Mejia RO, Lam BK, Arm JP. Matrix-associated transforming growth factor-beta1 primes mouse bone marrow-derived mast cells for increased high-affinity Fc receptor for immunoglobulin E-dependent eicosanoid biosynthesis. Am J Respir Cell Mol Biol. 2000;22:557–65. doi: 10.1165/ajrcmb.22.5.3902. [DOI] [PubMed] [Google Scholar]
- 57.Grgic I, Campanholle G, Bijol V, et al. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012;82:172–83. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tang S, Leung JC, Abe K, et al. Albumin stimulates interleukin-8 expression in proximal tubular epithelial cells in vitro and in vivo. J Clin Invest. 2003;111:515–27. doi: 10.1172/JCI16079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30:234–54. doi: 10.1016/j.semnephrol.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vernon MA, Mylonas KJ, Hughes J. Macrophages and renal fibrosis. Semin Nephrol. 2010;30:302–17. doi: 10.1016/j.semnephrol.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 61.Chow FY, Nikolic-Paterson DJ, Ozols E, et al. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int. 2006;69:73–80. doi: 10.1038/sj.ki.5000014. [DOI] [PubMed] [Google Scholar]
- 62.Vielhauer V, Kulkarni O, Reichel CA, Anders HJ. Targeting the recruitment of monocytes and macrophages in renal disease. Semin Nephrol. 2010;30:318–33. doi: 10.1016/j.semnephrol.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 63.Banba N, Nakamura T, Matsumura M, et al. Possible relationship of monocyte chemoattractant protein-1 with diabetic nephropathy. Kidney Int. 2000;58:684–90. doi: 10.1046/j.1523-1755.2000.00214.x. [DOI] [PubMed] [Google Scholar]
- 64.Morii T, Fujita H, Narita T, et al. Association of monocyte chemoattractant protein-1 with renal tubular damage in diabetic nephropathy. J Diabetes Complications. 2003;17:11–5. doi: 10.1016/s1056-8727(02)00176-9. [DOI] [PubMed] [Google Scholar]
- 65.Takebayashi K, Matsumoto S, Aso Y, Inukai T. Aldosterone blockade attenuates urinary monocyte chemoattractant protein-1 and oxidative stress in patients with type 2 diabetes complicated by diabetic nephropathy. J Clin Endocrinol Metab. 2006;91:2214–7. doi: 10.1210/jc.2005-1718. [DOI] [PubMed] [Google Scholar]
- 66.Fardon NJ, Wilkinson R, Thomas TH. Abnormalities in primary granule exocytosis in neutrophils from type I diabetic patients with nephropathy. Clin Sci (Lond) 2002;102:69–75. [PubMed] [Google Scholar]
- 67.Takahashi T, Hato F, Yamane T, et al. Increased spontaneous adherence of neutrophils from type 2 diabetic patients with overt proteinuria: possible role of the progression of diabetic nephropathy. Diabetes Care. 2000;23:417–8. doi: 10.2337/diacare.23.3.417. [DOI] [PubMed] [Google Scholar]
- 68.Tang SC, Leung JC, Lai KN. Diabetic tubulopathy: an emerging entity. Contrib Nephrol. 2011;170:124–34. doi: 10.1159/000325647. [DOI] [PubMed] [Google Scholar]
- 69.Moon JY, Jeong KH, Lee TW, et al. Aberrant recruitment and activation of T cells in diabetic nephropathy. Am J Nephrol. 2012;35:164–94. doi: 10.1159/000334928. [DOI] [PubMed] [Google Scholar]
- 70.Hagerty DT, Allen PM. Processing and presentation of self and foreign antigens by the renal proximal tubule. J Immunol. 1992;148:2324–30. [PubMed] [Google Scholar]
- 71.Lai KN, Leung JC, Chan LY, Guo H, Tang SC. Interaction between proximal tubular epithelial cells and infiltrating monocytes/T cells in the proteinuric state. Kidney Int. 2007;71:526–38. doi: 10.1038/sj.ki.5002091. [DOI] [PubMed] [Google Scholar]
- 72.Pickup JC, Mattock MB, Chusney GD, Burt D. NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia. 1997;40:1286–92. doi: 10.1007/s001250050822. [DOI] [PubMed] [Google Scholar]
- 73.Lin M, Yiu WH, Wu HJ, et al. Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. J Am Soc Nephrol. 2012;23:86–102. doi: 10.1681/ASN.2010111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Horlyck A, Gundersen HJ, Osterby R. The cortical distribution pattern of diabetic glomerulopathy. Diabetologia. 1986;29:146–50. doi: 10.1007/BF02427084. [DOI] [PubMed] [Google Scholar]
- 75.Huang HC, Preisig PA. G1 kinases and transforming growth factor-beta signaling are associated with a growth pattern switch in diabetes-induced renal growth. Kidney Int. 2000;58:162–72. doi: 10.1046/j.1523-1755.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- 76.Thomson SC, Deng A, Bao D, et al. Ornithine decarboxylase, kidney size, and the tubular hypothesis of glomerular hyperfiltration in experimental diabetes. J Clin Invest. 2001;107:217–24. doi: 10.1172/JCI10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Okada R, Yasuda Y, Tsushita K, et al. Glomerular hyperfiltration in prediabetes and prehypertension. Nephrol Dial Transplant. 2012;27:1821–5. doi: 10.1093/ndt/gfr651. [DOI] [PubMed] [Google Scholar]
- 78.Verzola D, Gandolfo MT, Gaetani G, et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol. 2008;295:F1563–73. doi: 10.1152/ajprenal.90302.2008. [DOI] [PubMed] [Google Scholar]
- 79.Satriano J, Mansoury H, Deng A, et al. Transition of kidney tubule cells to a senescent phenotype in early experimental diabetes. Am J Physiol Cell Physiol. 2010;299:C374–80. doi: 10.1152/ajpcell.00096.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 81.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–43. doi: 10.1038/nm.2144. 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 83.Goodarzi AA, Block WD, Lees-Miller SP. The role of ATM and ATR in DNA damage-induced cell cycle control. Prog Cell Cycle Res. 2003;5:393–411. [PubMed] [Google Scholar]
- 84.Kobayashi T, Okada H, Inoue T, Kanno Y, Suzuki H. Tubular expression of connective tissue growth factor correlates with interstitial fibrosis in type 2 diabetic nephropathy. Nephrol Dial Transplant. 2006;21:548–9. doi: 10.1093/ndt/gfi194. [DOI] [PubMed] [Google Scholar]
- 85.Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21:3355–68. doi: 10.1096/fj.06-6713com. [DOI] [PubMed] [Google Scholar]