

Abstract
The precise link between hyperglycemia and its deleterious effects on retinal and kidney microvasculature, and more specifically loss of retinal perivascular supporting cells including smooth muscle cell/pericytes (SMC/PC), in diabetes are not completely understood. We hypothesized that differential cellular proteasome activity contributes to sensitivity of PC to high glucose-mediated oxidative stress and vascular rarefaction. Here we show that retinal endothelial cells (EC) have significantly higher proteasome peptidase activity compared to PC. High glucose treatment (HGT) increased the level of total ubiquitin-conjugated proteins in cultured retinal PC and EC, but not photoreceptor cells. In addition, in vitro proteasome activity assays showed significant impairment of proteasome chymotrypsin-like peptidase activity in PC, but not EC. The PA28-α/-β and PA28-β/-γ protein levels were also higher in the retina and kidney glomeruli of diabetic mice, respectively. Our results demonstrate, for the first time, that high glucose has direct biological effects on cellular proteasome function, and this modulation might be protective against cellular stress or damage induced by high glucose.
Keywords: Hyperglycemia, Diabetic retinopathy, Diabetic nephropathy, Pericytes, PA28 proteasome regulator
1. Introduction
Diabetic retinopathy (DR) and diabetic nephropathy (DN) are the leading causes of blindness and end-stage renal failure in diabetic patients, respectively [1–3]. Diminished number of retinal PC [4–5] and hypertrophy of the renal mesangial cells, as perivascular cells of the glomerulus with similar functions as PC, are hallmarks of established DR and DN [1–3, 6–7]. The HGT of both retinal PC and EC promote oxidative stress, endoplasmic reticulum (ER) stress, and apoptosis in PC, but not in EC [8–9]. However, the molecular basis for selective vulnerability of PC in diabetic retinal and kidney complications, and higher sensitivity of retinal PC to high glucose compared to EC are not completely understood. We hypothesized that differential cellular proteasome activity contributes to selective sensitivity of PC to hyperglycemia.
Ubiquitin Proteasome System (UPS) is responsible for protein quality control and degradation. The conjugation of ubiquitin to target proteins primes them for UPS-mediated degradation [10]. The proteasomes are multiprotein barrel shaped assemblies with an approximate molecular weight of 2500 kDa and are considered the proteolytic machinery, which regulates the turnover of eukaryotic proteins. Proteasomes are composed of 20S core subunit and 19S or 11S regulatory subunits capable of binding to both ends of the barrel shaped core subunit and stimulating the proteolytic activity of the proteasomes. UPS also comprises many other enzymes involved in the ubiquitin activation (E1s), conjugation (E2s), ligation (E3s) and removal from target proteins by deubiquitylating enzymes (DUBs) [10].
The 11S proteasome regulatory subunit comprises the PA28-α, PA28-β and the PA28-γ proteins encoded by three different genes. All these proteins form a heptameric ring-shaped complex and, while the PA28-α and PA28-β proteins form a heptameric complex together (α3β4), the PA28-γ only exists in homoheptameric form. Although the important role of PA28 proteins in intracellular antigen processing and presentation to immune cells has been demonstrated, the PA28-α and PA28-β proteins can also mediate cellular response to oxidative stress [11–12].
Recent studies have demonstrated that high glucose, the glucose-mediated post-translational protein modifications, and diabetes are modulators of proteasome activity [13–15]. These reports link diabetes or hyperglycemia to UPS and raise many questions on how diabetes or hyperglycemia can modulate proteasome targeting and activity, and whether this modulation occurs in a cell-specific manner.
2. Material and methods
2.1. Cell culture
The isolation and culture of mouse retinal PC and EC were previously described [16–17]. Choroid EC (ChEC) were isolated from the mouse choroid using magnetic beads coated with anti-PECAM-1 antibody and grown similar to retinal EC. NIH3T3 cells and 661W mouse photoreceptor cells were obtained from ATCC (Manassas, VA). Low glucose level was 5 mM and for HGT the cells were treated for 5 days with 30 mM D-glucose (Sigma, St. Louis, MO). For osmolarity control the cells were treated with 5 mM D-glucose and 25 mM L-glucose (Sigma). For cycloheximide (CHX; Sigma) treatment, cells were treated at final concentration of 100 μg/mL for indicated times.
2.2. Western blotting
For immunoblots, the cultured cells and the retina tissue were lysed in RIPA lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 5mM NaF, 1 mM Na3VO4) supplemented with protease inhibitor cocktail (Roche, Nutley, NJ). The lysates were collected in 1.5 ml tubes, sonicated, and protein concentration was measured using BCA Protein Assay Kit (Pierce; Rockford, IL). Total protein (50 μg) was separated by SDS–PAGE (4–20% Tris-Glycine gels; Invitrogen, Carlsbad CA) and transferred to nitrocellulose membrane. Protein expression was analyzed by incubating with specific primary antibodies (supplementary Table 1) at 4°C overnight. Blots were washed, incubated with appropriate HRP-conjugated secondary antibody and developed using ECL. Mouse β-actin was used for loading control.
2.3. In vitro proteasome peptidase assay
Proteasome peptidase assays were performed as described by Bech-Otschir et al. [18]. The cells or tissues were lysed in the lysis buffer containing 50 mM Tris/HCl pH7.4, 1 mM ATP, 10% glycerol, 0.1% NP40, 2 mM MgCl2, 1.5 mM DTT and 0.03% SDS. In total, 50 μg (1 μg/μl) of protein lysates were loaded into a dark 96-well microplate and probed with 100 μM of fluorogenic peptide substrates: Suc-Leu-Leu-Val-Tyr-AMC for chymotrypsin-like, Ac-Arg-Leu-Arg-AMC for trypsin-like, and Z-Leu-Leu-Glu-AMC for caspase-like (Enzo Life Sciences, Farmingdale, NY). The plates were then incubated at 37°C for 30 minutes, and the reaction was stopped by the addition of 200 μl of 100% ethanol. The fluorescence signal intensity of the samples was measured using a luminescence plate reader (PerkinElmer, Wellesley, MA). The excitation and emission wavelengths were 355 and 460 nm, respectively. Boiled protein lysates were used as blank samples and test values were normalized to the respective blanks.
2.4. Mice
The wild type and heterozygous Akita/+ (Ins2Akita/+) male mice on C57BL/6j background were purchased from Jackson Laboratories (Bar Harbor, ME) and maintained in our facility. Genotyping was performed by PCR using specific primers (supplementary Table 2). All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin School of Medicine and Public Health. Only male Akita/+ mice develop diabetes by 4-weeks of age and have an average life expectancy of 10 moths. They develop some of the early non-proliferative retinopathies by six-months of age [19].
2.5. Immunofluorescence studies
The cryosections, prepared from eyes and kidneys harvested from age-matched male wild type and Akita/+ mice, were fixed in ice-cold acetone for 30 min, blocked (3% BSA, 0.1% Tween-20) for 1h and incubated with the desired primary (Supplementary Table 1) and secondary antibodies (Alexa Fluor -488 or -594 conjugated; Invitrogen). Control samples were incubated with relevant IgG and nuclei were counterstained with DAPI (Invitrogen). Microscopic slides were photographed using a Zeiss fluorescence microscope in digital format (Carl Zeiss, Thornwood, NY).
2.6. Statistical analysis
For Western blot densitometry analysis the wand tool of Image-J, and for statistics the GraphPad Prism software were used, respectively. Data are expressed as mean ± sem. Statistical significance was assessed by unpaired t test. Values were considered statistically significant at P < 0.05.
3. Results
3.1. Retinal EC exhibited enhanced proteasome peptidase activity
To test the effects of HGT on proteasomal degradation capacity of retinal PC, EC and retinas, synthetic fluorogenic proteasome substrate peptides were assayed in the presence of ATP [18]. Regardless of any change in the peptidase activity by HGT, we found that retinal EC had substantially higher peptidase activity for all the substrates compared to other cells (Fig. 1A–C). Retinal EC, compared to PC, showed approximately 3.4-, 8.1- and 6.5- fold higher chymotrypsin-like, trypsin-like and caspase-like peptidase activities, respectively (Fig. 1A–C). HGT of cultured cells resulted in a modest reduction of peptidase activity for most of the tested substrates, except for EC, which showed a significant rise in peptidase activity after HGT. Retinal PC after HGT showed a significant reduction in chymotrypsin-like peptide degradation. Similar to EC, but to a lower extent, the Ins2Akita/+ retinas also showed a significant increase in chymotrypsin-like and trypsin-like peptidase activity compared to age-matched control retinas (Fig. 1A–C). Surprisingly, the ChEC responsible for the nourishment of outer retina did not show as high of peptidase activity as retinal EC (Fig. 1A–C). This may stem from the differences in the physiology of these EC, and suggest that ChEC are more susceptible to cytotoxic effects of hyperglycemia.
Fig. 1.
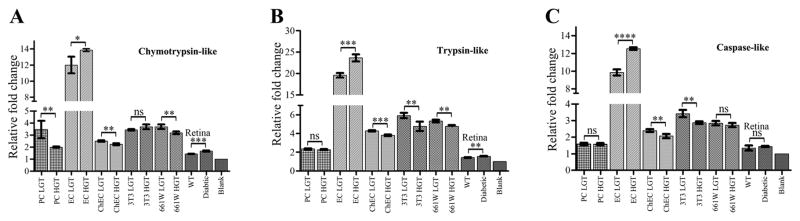
The in vitro proteasome peptidase activity of cultured retinal cells and retinas under low and high glucose conditions. (A) Chymotrypsin-like activity was significantly reduced by HGT in RPC (p<0.0066), ChEC (p<0.0026) and 661W (p<0.0045) whereas HGT increased the degradation activity in REC (p<0.0120) and retina (p<0.0003). REC in LGT showed 3.4 fold and 4.8 fold higher chymotrypsin-like activity compared to RPC and ChEC cells, respectively. (B) Trypsin-like degradation was increased in REC (p<0.0002) and retina (p<0.0012), and reduced by HGT in ChEC (p<0.0002), NIH3T3 cells (p<0.0071) and 661W cells (p<0.0015). The REC showed 8.1 and 4.6-fold higher trypsin-like activity than RPC and ChEC under LGT conditions, respectively. (C) Significant increases in caspase-like activity by HGT in EC (p<0.0001), and reduction in ChEC (p<0.0056) and NIH3T3 (p<0.0031) cells. The REC showed approximately 6.5- and 4-fold higher caspase-like activity compared to RPC and ChEC, respectively. Sample number for all assays (n=3). Error bars, Mean ± SEM; *Statistically significant; ns, not-significant.
3.2. HGT increased the total levels of ubiquitinated proteins and PA28-α/-β levels in retinal vascular cells
Using Western blot to detect universal ubiquitination in cultured cells, we found that HGT resulted in elevation of total protein ubiquitination in retinal PC, EC, and NIH3T3 cells, but not in 661W photoreceptor cells (Fig. 2A). The observed rise in ubiquitination following HGT was similar to the increased ubiquitination reported for cultured myocytes incubated with oxidant reagent (H2O2), which was due to attenuation of proteasome activity [11].
Fig. 2.


Effect of HGT on ubiquitination and PA28-α/-β protein levels in cultured retinal cells. (A) Western blot for ubiquitinated and PA28-α/-β proteins in retinal PC and EC, NIH3T3, and 661W cells. (B) The quantification of Western blots for PA28-β protein under LGT and HGT showing the percent change in RPC and NIH3T3.
We next determined whether HGT-mediated impairment of proteasome activity was responsible for increased levels of ubiquitinated proteins by analyzing the clearance of ubiquitinated proteins after CHX treatment and inhibition of new protein synthesis. Western blot for ubiquitin after 12 h of CHX treatment showed reduction in total amount of ubiquitinated proteins in HGT similar to normal glucose conditions in all cell types examined (Fig. 2A). Thus, HGT enhanced protein ubiquitination with minimal effect on the clearance of the ubiquitinated proteins by proteasomes.
We then asked whether HGT could also influence the composition of the proteasome regulatory subunits to handle the rise in substrate levels. Thus, we investigated the levels of 11S proteasome regulatory subunits (PA28-α/-β) in the retinal vascular cells after HGT. Indeed, HGT elevated the levels of PA28-α/-β proteins in retinal PC and NIH3T3 cells by 36% and 310%, respectively (Fig. 2B), but apparently not in retinal EC and 661W cells (Fig. 2A and not shown). These results suggested differential requirement for cells of 11S regulatory proteasome subunits under HGT conditions to accommodate the rise in ubiquitination. In addition, the parallel increase in both PA28-α and PA28-β levels by HGT in retinal PC and NIH3T3 cells was also in agreement with the ability of PA28-α and PA28-β to form a heptameric (α3β4) complex allowing their concerted stability and activity [20–21].
3.3. Diabetic mice had higher levels of PA28-α/-β proteins in their retinas
We next asked whether the retinas from Ins2Akita/+ mice with established diabetes demonstrate any changes in expression of PA28-α/-β proteins. Western blot analysis of the total retinal lysates from adult Ins2Akita/+ mice with established diabetes showed significant increase in the PA28-α/-β protein levels compared with non-diabetic mice (Fig. 3A and B). However, contrary to HGT-induced rise in ubiquitination in retinal PC and EC, the retina lysates from adult Ins2Akita/+ mice did not show any difference in total ubiquitination levels compared to control animals (not shown). This suggested that hyperglycemia-mediated rise in the PA28-α/-β protein was independent of universal ubiquitination in the diabetic retina.
Fig. 3.


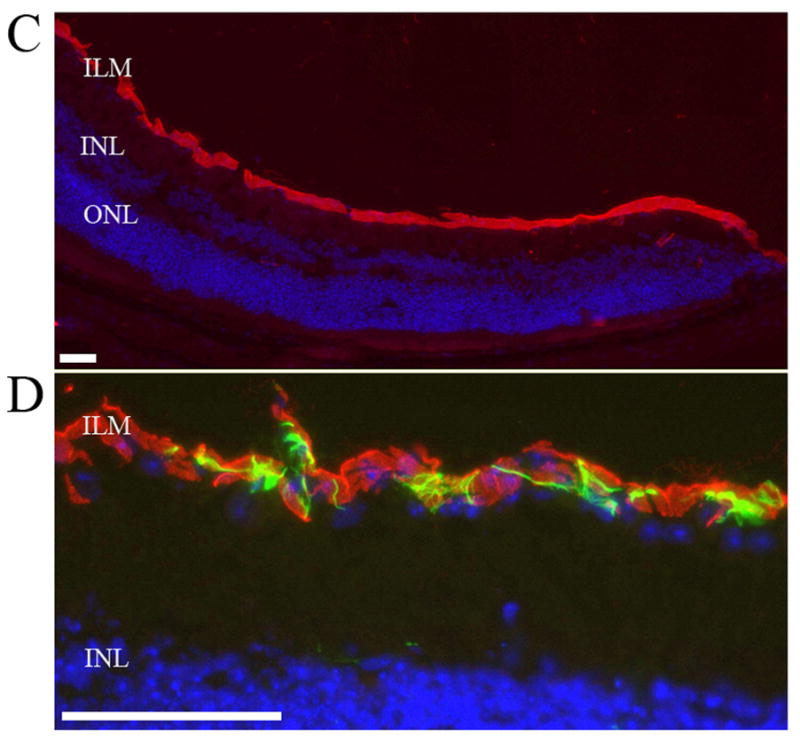
Increased levels of PA28-α/-β proteins in the retinas of diabetic mice. (A) Western blot analysis showed increased retinal PA28-α/-β levels in 7-month old Ins2Akita/+ mice. (B) Quantification of the combined mean value of PA28-β levels after normalizing to β–actin band intensity showing fold change between wild type and Ins2Akita/+ mice. (C) PA28-β immunoreactivity (red) in the inner limiting membrane (ILM) layer of the wild type mouse retina. Cells in the outer nuclear layer (ONL) or inner nuclear layer (INL) of retina did not show any immunoreactivity for PA28-β. (D) High magnification of the retinal ILM layer showing GFAP-positive astrocytes (green) surrounding the PA28-β positive cells. Nuclei are counterstained with DAPI. Scale bar 25 μm.
Following the observed rise in PA28-α/-β levels in diabetic retina, we used immunofluorescence staining to detect PA28-β in the retina of adult wild type and Ins2Akita/+ mice. Similar to adult 8-month old wild type mice, the retinal PA28-β expression in Ins2Akita/+ mice was only restricted to the superficial nucleated layer of the retina in the inner limiting membrane (ILM), which appeared to be only one layer thick and were covered by GFAP positive processes of the astrocytes (Fig. 3C and D) [22]. We could also detect focal signal for PA28-β on retinal blood vessels in retinal flatmounts (Supplement Fig. 1). The immunoreactivity site for the PA28-β in retina suggested that the PA28-β positive cells might be retinal ganglion cell, therefore we performed co-staining with βIII-tubulin [23] but there was no overlapped staining (Supplementary Fig. 2A). Co-staining with PA28-β and PC marker PDGF-Rβ (20) (Supplementary Fig. 2B), and microglial marker Iba1 [24](not shown), also did not show overlapped staining. Altogether, the retinal PA28-β immunofluorescence signal labeled a one-layer thick nucleated compartment restricted to the ILM layer, did not vary between wild type and diabetic mice, and did not co-localize with RGC, PC, microglia, and astrocytes. There was no detectable fluorescence signal for the PA28-γ subunit of the 11S proteasome in the mouse retina sections (not shown).
3.4. Diabetic mice had higher levels of PA28-β/-γ, Cul1 and Cul3 proteins in their intraglomerular capillaries
Kidney sections from Ins2Akita/+ mice were also analyzed by immunofluorescence for variation in 11S regulatory subunits of the UPS. The immunostaining pattern for 11S proteasome regulatory subunits in the kidney sections varied between the wild type and Ins2Akita/+ mice, and among the Ins2Akita/+ mice with longer duration of diabetes and severity of nephropathy. The severity of the nephropathy was judged both visually based on the degree of kidney atrophy upon dissection and microscopically based on the glomerular hypertrophy revealed by immunofluorescence. The Kidneys from 3-month old wild type and Ins2Akita/+ mice did not show any detectable staining for PA28-β or PA28-γ (not shown). The adult 8-month or older non-diabetic mice did not reveal any detectable immunoreactivity in their kidney sections for 11S regulatory subunits (Fig. 4A and C). In contrast, 8-month old Ins2Akita/+ mice demonstrated PA28-β/-γ staining in the glomeruli, which co-stained with PDGF-Rβ (a mesangial cell marker; Fig. 4B and D) [25–26]. We also observed a similar co-staining pattern with VE-cadherin as EC marker and PA28-β/-γ in adult 8-month old InsAkita/+ mice (Supplementary Fig. 3A and B). Collectively the 11S regulatory proteins were increased in the vascular units of the glomerulus. Thus, severe nephropathy was associated with more robust 11S proteasome regulatory subunit accumulation in the glomerular capillaries. The increased 11S proteasome regulatory subunits in the diabetic kidney glomeruli were suggestive of enhanced proteasome activation.
Fig. 4.
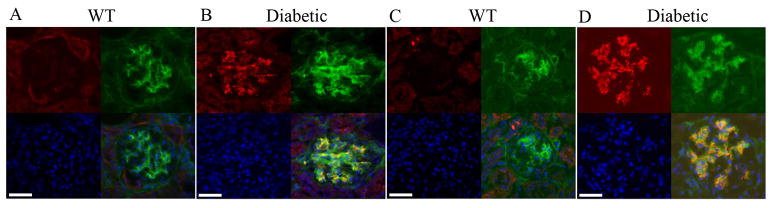
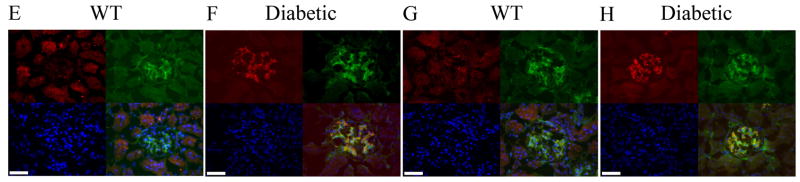
Increased immunoreactivity for PA28 and Cul1/Cul3 proteins in the glomeruli of Ins2Akita/+ mice. No immunoreactivity for PA28-β (red, A) and PA28-γ (red, C) in the 7-month old wild type glomeruli. Ins2Akita/+ mice showed high immunoreactivity for both PA28-β (red, B) and PA28-γ (red, D) which overlapped with PDGF-Rβ labeling of the mesangial cells (green). Immunoreactivity for Cul1 (red, E) and Cul3 (red, G) were determined in 7-month-old wild type (non-diabetic) glomeruli. Ins2Akita/+ mice showed high immunostaining for both Cul1 (red, F) and Cul3 (red, H) which overlaped with PDGF-Rβ labeling of the mesangial cells (green). Nuclei are counterstained with DAPI. Scale bar 25 μm.
We then examined other components of the UPS in diabetic kidneys, namely the Cul1 and Cul3, which are E3 ubiquitin ligase members of Cullin family [27–28]. A member of cullin family is shown in complex with O-GlcNAcase, an enzyme which regulates protein O-glycosylation events [29]. Thus, it is possible that cullins may be involved in specific targeting of glycosylated proteins for proteasome degradation. The antibodies against Cul1 and Cul3 in kidney sections showed similar pattern of immunoreactivity analogous to 11S proteins (Fig. 4E–H). The Cul1 and Cul3 expression was almost undetectable in wild type mice (Fig. 4E and G) whereas Ins2Akita/+ mice showed strong immunoreactivity and co-localization for these proteins in the glomeruli with PDGF-Rβ (Fig. 4F and H), which correlated with the severity of nephropathy. Similar to 11S proteins, we did not find any obvious changes in immunostaining pattern of tubular components of the kidneys for the Cul1 and Cul3 (not shown), indicating that glomerular capillaries of the kidney are targets of altered proteasome activity in diabetes.
4. Discussion
The exceptionally higher peptidase activity together with the lack of increase in PA28-α/-β levels following HGT in EC compared with PC indicated that retinal EC may possess higher proteasomal efficiency for clearance of damaged proteins compared with PC, even without a requirement for rise in 11S proteasome subunits. These observations are consistent with recognition of retinal PC, but not EC, as the early target of hyperglycemia damage and endoplasmic reticulum stress in retinal vasculature during diabetes [9, 30]. Elevated ubiquitination in retinal vascular cells induced by HGT provides higher amounts of proteasome substrate for degradation. Efficient clearance of the ubiquitin-conjugated proteins after 12 h of CHX treatment indicated that HGT did not impair proteasome activity in PC and EC. However, the modest but statistically significant decrease in in vitro proteasome activity in PC, ChEC, NIH3T3, and 661W cells pointed to the impairment of the catalytic activities of the proteasome by HGT. The interpretation of the differential peptidase activity among these cells requires more functional assessment of the proteasome activity and characterization of the proteasome quantity, subunit composition, and adjunct proteasome-associated proteins.
Photoreceptor cells did not show elevated HGT-mediated ubiquitination compared to PC and EC, which was suggestive of differences in the metabolic responsiveness to HGT among retinal neuronal and non-neuronal cells. These results also suggest that retinal neuronal cells may not be the primary target of hyperglycemia and pathogenesis of DR. The link between HGT and ubiquitination is unclear but protein modifications such as protein oxidation [31] and O-glycosylation [15, 32] target proteins for ubiquitination. The rise in PA28-α/-β levels in diabetic retinas provided an in vivo link between diabetes and modulation of proteasome activity.
The observed increase in immunoreactivity for PA28 and Cullin proteins in the intraglomerular capillaries of Ins2Akita/+ mice and its correlation with the severity of the DN is the first report correlating the proteasome activation to DN. The primary site of DN damage in patients is within the glomeruli with the net outcome of reduced levels of glomerular filtration rate and proteinuria [33]. Thus, overlapping rise in immunofluorescence signal for UPS components in glomerular capillaries implies a significant role for proteasomes in pathogenesis of DN, and changes in expression/levels of these proteins may provide a suitable marker for detection of nephropathy. However, it is not clear whether increased proteasome activity is protecting the glomeruli from diabetes or contributes to the exacerbation of nephropathy. A recent study showed that MG132 mediated blockage of proteasome in diabetic rats was protective against renal oxidative stress and DN [34]. Based on the role of 11S subunits in protection against oxidative stress, and the reported improvement of DN symptoms with proteasome inhibition [34], it is possible that primary activation of the proteasome under diabetic conditions in vulnerable organs provides temporary protection against oxidative stress. However, its chronic activation may ultimately result in more devastating tissue damage in vulnerable organs including kidney.
In summary, the observed HGT-mediated modulation of UPS components or UPS-related substrate levels in vitro, and also in tissues vulnerable to diabetes damage including retina and kidney, identify hyperglycemia as key modulator of proteasome activity. Thus, differential activation of the 11S proteasome activator in certain subset of vulnerable cells may provide protection against damage caused by glucose-mediated protein modifications including O-glycosylation [15] and protein oxidation [31]. However, the direct contribution of these protein modifications and their clearance by proteasome to pathogenesis of DR and DN awaits future investigation.
Supplementary Material
Highlights.
Retinal endothelial cells exhibit higher proteasome peptidase activity compared to pericytes.
High glucose treatment increased the levels of ubiquitinated proteins in retinal vascular cells.
High glucose treatment enhanced the PA28 protein levels in pericytes but not endothelial cells.
Diabetic retina and kidney have higher levels of PA28 proteasome regulatory proteins.
The UPS components are increased within the glomerular capillaries of diabetic kidney.
Acknowledgments
This work was supported by grants EY016995, EY021357, and P30-EY016665, from the National Institutes of Health and an unrestricted departmental award from Research to Prevent Blindness. NS is a recipient of a Research Award from American Diabetes Association, 1-10-BS-160 and Retina Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 2.Aronson D. Hyperglycemia and the pathobiology of diabetic complications. Adv Cardiol. 2008;45:1–16. doi: 10.1159/000115118. [DOI] [PubMed] [Google Scholar]
- 3.Curtis TM, Gardiner TA, Stitt AW. Microvascular lesions of diabetic retinopathy: clues towards understanding pathogenesis? Eye (Lond) 2009;23:1496–1508. doi: 10.1038/eye.2009.108. [DOI] [PubMed] [Google Scholar]
- 4.Denis U, Lecomte M, Paget C, Ruggiero D, Wiernsperger N, Lagarde M. Advanced glycation end-products induce apoptosis of bovine retinal pericytes in culture: involvement of diacylglycerol/ceramide production and oxidative stress induction. Free Radical Biology and Medicine. 2002;33:236–247. doi: 10.1016/s0891-5849(02)00879-1. [DOI] [PubMed] [Google Scholar]
- 5.Liu B, Bhat M, Padival AK, Smith DG, Nagaraj RH. Effect of dicarbonyl modification of fibronectin on retinal capillary pericytes. Invest Ophthalmol Vis Sci. 2004;45:1983–1995. doi: 10.1167/iovs.03-0995. [DOI] [PubMed] [Google Scholar]
- 6.Frank RN. Diabetic Retinopathy. N Engl J Med. 2004;350:48–58. doi: 10.1056/NEJMra021678. [DOI] [PubMed] [Google Scholar]
- 7.Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395–423. doi: 10.1146/annurev.pathol.4.110807.092150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong Y, Wang JJ, Zhang SX. Intermittent but not constant high glucose induces ER stress and inflammation in human retinal pericytes. Adv Exp Med Biol. 2012;723:285–292. doi: 10.1007/978-1-4614-0631-0_37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Q, Sheibani N. High glucose promotes retinal endothelial cell migration through activation of Src, PI3K/Akt1/eNOS, and ERKs. Am J Physiol Cell Physiol. 2008;295:C1647–1657. doi: 10.1152/ajpcell.00322.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metzger MB, Hristova VA, Weissman AM. HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci. 2012;125:531–537. doi: 10.1242/jcs.091777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong X, Liu J, Zheng H, Glasford JW, Huang W, Chen QH, Harden NR, Li F, Gerdes AM, Wang X. In situ dynamically monitoring the proteolytic function of the ubiquitin-proteasome system in cultured cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1417–1425. doi: 10.1152/ajpheart.01233.2003. [DOI] [PubMed] [Google Scholar]
- 12.Pickering AM, Linder RA, Zhang H, Forman HJ, Davies KJ. Nrf2-dependent induction of proteasome and Pa28alphabeta regulator are required for adaptation to oxidative stress. J Biol Chem. 2012;287:10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami T, Frey T, Lin C, Antonetti DA. Protein kinase cbeta phosphorylates occludin regulating tight junction trafficking in vascular endothelial growth factor-induced permeability in vivo. Diabetes. 2012;61:1573–1583. doi: 10.2337/db11-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H, Yu S, Xu W, Xu J. Enhancement of 26S Proteasome Functionality Connects Oxidative Stress and Vascular Endothelial Inflammatory Response in Diabetes Mellitus. Arterioscler Thromb Vasc Biol. 2012 doi: 10.1161/ATVBAHA.112.253385. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K, Kanno J, Ohtake F, Kitagawa H, Roeder RG, Brown M, Kato S. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature. 2011;480:557–560. doi: 10.1038/nature10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheef EA, Sorenson CM, Sheibani N. Attenuation of proliferation and migration of retinal pericytes in the absence of thrombospondin-1. Am J Physiol Cell Physiol. 2009;296:C724–734. doi: 10.1152/ajpcell.00409.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su X, Sorenson CM, Sheibani N. Isolation and characterization of murine retinal endothelial cells. Mol Vis. 2003;9:171–178. [PubMed] [Google Scholar]
- 18.Bech-Otschir D, Helfrich A, Enenkel C, Consiglieri G, Seeger M, Holzhutter HG, Dahlmann B, Kloetzel PM. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat Struct Mol Biol. 2009;16:219–225. doi: 10.1038/nsmb.1547. [DOI] [PubMed] [Google Scholar]
- 19.Barber AJ, Antonetti DA, Kern TS, Reiter CEN, Soans RS, Krady JK, Levison SW, Gardner TW, Bronson SK. The Ins2Akita Mouse as a Model of Early Retinal Complications in Diabetes. Invest Ophthalmol Vis Sci. 2005;46:2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 20.Knowlton JR, Johnston SC, Whitby FG, Realini C, Zhang Z, Rechsteiner M, Hill CP. Structure of the proteasome activator REGalpha (PA28alpha) Nature. 1997;390:639–643. doi: 10.1038/37670. [DOI] [PubMed] [Google Scholar]
- 21.Cascio P, Goldberg AL. Preparation of hybrid (19S-20S-PA28) proteasome complexes and analysis of peptides generated during protein degradation. Methods Enzymol. 2005;398:336–352. doi: 10.1016/S0076-6879(05)98028-2. [DOI] [PubMed] [Google Scholar]
- 22.Kinouchi R, Takeda M, Yang L, Wilhelmsson U, Lundkvist A, Pekny M, Chen DF. Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat Neurosci. 2003;6:863–868. doi: 10.1038/nn1088. [DOI] [PubMed] [Google Scholar]
- 23.Hellstrom M, Ruitenberg MJ, Pollett MA, Ehlert EM, Twisk J, Verhaagen J, Harvey AR. Cellular tropism and transduction properties of seven adeno-associated viral vector serotypes in adult retina after intravitreal injection. Gene Ther. 2009;16:521–532. doi: 10.1038/gt.2008.178. [DOI] [PubMed] [Google Scholar]
- 24.Ibrahim AS, El-Remessy AB, Matragoon S, Zhang W, Patel Y, Khan S, Al-Gayyar MM, El-Shishtawy MM, Liou GI. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes. 2011;60:1122–1133. doi: 10.2337/db10-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alpers CE, Seifert RA, Hudkins KL, Johnson RJ, Bowen-Pope DF. PDGF-receptor localizes to mesangial, parietal epithelial, and interstitial cells in human and primate kidneys. Kidney Int. 1993;43:286–294. doi: 10.1038/ki.1993.45. [DOI] [PubMed] [Google Scholar]
- 26.Lindahl P, Hellstrom M, Kalen M, Karlsson L, Pekny M, Pekna M, Soriano P, Betsholtz C. Paracrine PDGF-B/PDGF-Rbeta signaling controls mesangial cell development in kidney glomeruli. Development. 1998;125:3313–3322. doi: 10.1242/dev.125.17.3313. [DOI] [PubMed] [Google Scholar]
- 27.Emanuele MJ, Elia AE, Xu Q, Thoma CR, Izhar L, Leng Y, Guo A, Chen YN, Rush J, Hsu PW, Yen HC, Elledge SJ. Global identification of modular cullin-RING ligase substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 29.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-Glycosylation of Nuclear and Cytosolic Proteins: CLONING AND CHARACTERIZATION OF A NEUTRAL, CYTOSOLIC β-N-ACETYLGLUCOSAMINIDASE FROM HUMAN BRAIN. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- 30.Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL. Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med. 2009;15:1298–1306. doi: 10.1038/nm.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grune T, Merker K, Sandig G, Davies KJ. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochemical and Biophysical Research Communications. 2003;305:709–718. doi: 10.1016/s0006-291x(03)00809-x. [DOI] [PubMed] [Google Scholar]
- 32.Shrikhande GV, Scali ST, da Silva CG, Damrauer SM, Csizmadia E, Putheti P, Matthey M, Arjoon R, Patel R, Siracuse JJ, Maccariello ER, Andersen ND, Monahan T, Peterson C, Essayagh S, Studer P, Guedes RP, Kocher O, Usheva A, Veves A, Kaczmarek E, Ferran C. O-glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PLoS ONE. 2010;5:e14240. doi: 10.1371/journal.pone.0014240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caramori ML, Fioretto P, Mauer M. Low glomerular filtration rate in normoalbuminuric type 1 diabetic patients: an indicator of more advanced glomerular lesions. Diabetes. 2003;52:1036–1040. doi: 10.2337/diabetes.52.4.1036. [DOI] [PubMed] [Google Scholar]
- 34.Luo ZF, Qi W, Feng B, Mu J, Zeng W, Guo YH, Pang Q, Ye ZL, Liu L, Yuan FH. Prevention of diabetic nephropathy in rats through enhanced renal antioxidative capacity by inhibition of the proteasome. Life Sci. 2011;88:512–520. doi: 10.1016/j.lfs.2010.12.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.