

Abstract
Previous studies have demonstrated that a small subset of cancer cells is capable of tumor initiation. The existence of tumor initiating cancer stem cells (CSCs) has several implications in terms of future cancer treatment and therapies. However, recently, several researchers proposed that differentiated cancer cells (non-CSCs) can convert to stem-like cells to maintain equilibrium. These results imply that removing CSCs may prompt non-CSCs in the tumor to convert into stem cells to maintain the equilibrium. Interleukin-6 (IL-6) has been found to play an important role in the inducible formation of CSCs and their dynamic equilibrium with non-stem cells. In this study, we used CSC-like human breast cancer cells and their alternate subset non-CSCs to investigate how IL-6 regulates the conversion of non-CSCs to CSCs. MDA-MB-231 and MDA-MB-453 CSC-like cells formed mammospheres well, whereas most of non-stem cells died by anoikis and only part of the remaining non-stem cells produced viable mammospheres. Similar results were observed in xenograft tumor formation. Data from cytokine array assay show that IL-6 was secreted from non-CSCs when cells were cultured in ultra-low attachment plates. IL-6 regulates CSC-associated OCT-4 gene expression through the IL-6-JAK1-STAT3 signal transduction pathway in non-CSCs. Inhibiting this pathway by treatment with anti-IL-6 antibody (1 μg/ml) or niclosamide (0.5–2 μM)/LLL12 (5–10 μM) effectively prevented OCT-4 gene expression. These results suggest that the IL-6-JAK1-STAT3 signal transduction pathway plays an important role in the conversion of non-CSCs into CSCs through regulation of OCT-4 gene expression.
Keywords: cancer stem cells, stochastic status, hierarchy status, conversion
1. INTRODUCTION
Breast cancer has become the most frequently diagnosed cancer among women in the United States [1, 2]. The past decades have seen advances in the diagnosis and therapeutic treatment of breast cancer. Despite this progress, breast cancer is still a leading cause of cancer-related deaths among women, with as many 40% relapsing with cancer recurrence and subsequent metastatic disease [2, 3]. The major cause of death is not from the primary tumor itself but from the cancer cells at the distant sites to which the cancer has migrated [4]. Recent research in the past few years shows that breast cancer stem cells may be the cause of the recurrence and metastasis, which results in the spread of cancer to other parts of the body such as the bones, liver, brain, and lungs [5–7]. The concept of cancer stem cells being responsible for tumor origin, maintenance, and resistance to treatment has gained prominence in the field of cancer research [3, 8–10]. These cancer stem cells represent a minor subpopulation of cells in the tumor and are also distinct from the more differentiated tumor cells. Traditional cancer treatments are effective at reducing tumor mass but often fail to produce long-term clinical complete remissions, possibly due to their inability to eliminate the cancer stem cell population [3]. Therefore, targeted therapy for cancer stem cells has been proposed to improve the efficacy of cancer treatments [11–13]. The therapeutic targeting of cancer stem cells may have the potential to remove residual disease and become an important component of a multimodality treatment.
For a long time, human cancers have been recognized as a morphologically heterogeneous population of cells [14]. At least two models of tumor growth have been used to explain the heterogeneous potential of tumor cells and the process of metastasis in general [15–18]. The stochastic model claims that tumors are originally biologically homogeneous. However, according to random or stochastic influences that alter the behavior of individual cells in tumor, tumors can gain functional heterogeneity. These influences can be intrinsic (levels of transcription factors, signaling pathways) or extrinsic (microenvironment, immune response) [19–21]. The hierarchical model assumes that only a very small subpopulation such as cancer stem cells within the tumor actually has the capacity to initiate and sustain tumor growth. In contrast, the bulk heterogeneous tumor population is not tumorigenic and only cancer stem cells are able to grow primary tumor and metastatic tumor [22–24]. Interestingly, several researchers proposed that differentiated cancer cells can convert to stem-like cells to maintain equilibrium [25, 26]. Iliopoulos et al. [26] reported the inducible formation of breast CSCs and their dynamic equilibrium with non-CSCs via IL-6 secretion.
In this study, we observed that breast CSC-like cells were the major contributors to tumorigenicity. However, our data showed that even non-CSCs can contribute to tumor formation. Our mammosphere formation data showed that secreted IL-6 from non-CSCs activates the JAK1-STAT signal transduction pathway and upregulates CSC-associated OCT-4 gene expression. These results support the proposal that part of the non-CSC population can convert to CSC-like cells to maintian an equilibrium state and subsequently these CSC-like cells can initiate tumor formation.
2. MATERIALS AND METHODS
2.1. Cell culture
Permanently blocked cancer stem cell (CSC)-like MDA-MB-231 human breast adenocarcinoma and MDA-MB-453 human breast carcinoma cell lines, which can proliferate without differentiation and have characteristics of tumor-initiating cells, were generated in Dr. Prochownik’s laboratory as previously described following stable transfection with a human Oct3/4 promoter driving the expression of green fluorescent protein (GFP) [27] and their corresponding non-CSC cell lines were generated by stable transfection of DsRed-Monomer N1 (cat. 632465, Clontech, CA, USA) using Lipofectamine 2000 reagent (Invitrogen, NY, USA). Stably transfected clones were selected, examined for expression of tumor markers (CD44, CD24, and Oct-4), pooled, and maintained with G418 (800~1000 μg/ml, Cellgro, VA, USA). The cells were cultured in Roswell Park Memorial Institute medium (RPMI) 1640 or Dulbecco’s modi ed Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and 26 mM sodium bicarbonate for the monolayer cell culture. Petri dishes containing cells were kept in a 37°C humidified incubator with a mixture of 95% air and 5% CO2.
2.2. Drug treatment
Niclosamide (5-chloro-N-(2-chloro-4-nitrophenyl)-2-hydroxybenzamide) and LLL12 (5-hydroxy-9,10-dioxo-9,10-dihydroanthracene-1-sulfonamide) were purchased from Biovision (Milpitas, CA). These drugs were dissolved in dimethylsulfoxide (DMSO) and applied to cells. Treatment of cells with drugs was accomplished by aspirating the medium and replacing it with medium containing these drugs.
2.3. Fluorescence microscopy
The morphological features and fluorescence signals for CSC-like and non-CSC cells were confirmed with phase contrast and fluorescence microscopy (Axiovert 40 CFL, Carl Zeiss Microimaging, NY, USA). The data were analyzed by microscope imaging processing software AxioVision from Zeiss.
2.4. Protein extracts and PAGE
Cells were scraped with 1 × Laemmli lysis buffer (including 2.4 M glycerol, 0.14 M Tris (pH 6.8), 0.21 M SDS, and 0.3 mM bromophenol blue) and boiled for 5 minutes. Protein concentrations were measured with BCA protein assay reagent (Pierce, Rockford, IL, USA). The samples were diluted with 1 × lysis buffer containing 1.28 M β-mercaptoethanol, and an equal amount of protein was loaded on 8–12% SDS-polyacrylamide gels. SDS-PAGE analysis was performed using a Hoefer gel apparatus.
2.5. Immunoblot analysis
Proteins were separated by SDS-PAGE, electrophoretically transferred to mitrocellulose membranes and blocked with 5% skim milk in TBS-Tween 20 (0.05%, v/v) for 30 minutes. The membrane was incubated with antibodies against anti-JAK-1, anti-phospho-JAK1, anti-STAT3, anti-phospho-STAT3, anti-Oct 4 (Cell Signaling, Danvers, MA, USA), or β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG was used as the secondary antibody. Immunoreactive protein was visualized by the enhanced chemiluminescence protocol.
2.6. Mammosphere formation assay
Prior to mammosphere formation, CSC-like and non-CSC breast cancer cells were grown as a monolayer culture as described above. One thousand cells were trypsinized and plated to an Ultra-Low Attachment 24 well plate (Corning, Lowell, MA, USA). After 4–10 days, the mammosphere’s form was observed. For comparison of the mammosphere size for stem-like and non-stem cancer cells, we used the Adobe photoshop program (Adobe Photoshop CS3, San Jose, CA, USA).
2.7. ALDEFLUOR assay and flow cytometry
To measure and isolate cells with high aldehyde dehydrogenase (ALDH) activity, the Aldefluor assay was performed according to manufacturer’s guidelines (Stemcell Technologies, NC, USA). Dissociated single cells were suspended in Aldefluor assay buffer containing the ALDH substrate, bodipy aminoacetaldehyde (BAAA), at 1.5 mM and incubated for 40 minutes at 37°C. To distinguish between ALDH-positive and ALDH-negative cells, a fraction of cells was incubated under identical condition in the presence of a 10-fold molar excess of the ALDH inhibitor, diethylaminobenzaldehyde (DEAB), which resulted in a significant decrease in the fluorescence intensity of ALDH-positive cells and was used to compensate the flow cytometry. Analysis was performed using the FACScan flow cytometer, and results were analyzed with CellQuest software (both from Becton Dickinson Immunocytometry Systems, Franklin Lakes, NJ, USA).
2.8. Annexin V binding
Phosphatidylserine externalization, a marker of apoptotic events, was detected by binding of allophycocyanin (APC)-conjugated Annexin V. Non-stem cells were plated into ultra-low attachment plates, incubated various days, and stained with mouse anti-human Annexin V antibody. Cells were analyzed by flow cytometry. Typically, 100,000 events were collected using excitation/emission wavelengths of 488/525 nm.
2.9. Immunofluorescent staining
Mammospheres of CSC-like cells and non-stem cells were harvested by cytospin and fixed in 2% paraformaldehyde for 15 minutes. These slides were washed 3 times in 1 x PBS buffer, and then permeabilized in 0.5% Triton for 10 minutes. After further washing in PBS, using 1% BSA in PBS buffer, cells were blocked for 30 minutes. Cells were incubated with Oct-4 primary antibody (Abcam, MA, USA) during 1 hour at room temperature. Following a wash in PBS, the cells were incubated for 1 hour at room temperature in secondary antibody Alexa 647-conjugated goat anti-rabbit (Invitrogen, NY, USA) diluted 1:500 ratio in PBS. Cells were washed with PBS buffer, stained with 0.5 μg/ml DAPI (4′, 6-diamidino-2-phenylindole, Invitrogen) for 1 minute, washed again 3 times and then covered with mounting medium (Vector Laboratories). Immunofluorescent staining was observed and photographed using an FLUOVIEW FV1000 CONFOCAL MICROSCOPE (Filters- ALEXA 647 and DAPI) and software FV10-ASW version 02.01.01.04 interfaced to an Olympus (OLYMPUS, PA, USA).
2.10. Animal model
For xenograft tumor formation, MDA-MB-231 CSC-like cells or non-stem cancer cells (1 × 104 cells in 0.1 ml of sterile 0.9% NaCl and 0.1 ml of Matrigel) were injected into the upper mammary fat pads of six-week-old female nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (Jackson Laboratories, Bar Harbor, ME, USA). All animal experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh (Permit Number: 101904).
2.11. Cryosection and immunoblot analysis in tissue
Immediately after sacrifice the formed tumors were removed and cut in two burdens for the cryosection and western blot analysis. For the cryosection, one half was fixed in 2% paraformaldehyde overnight at 4°C and then soaked in 30% sucrose solution for an additional 4 hours at 4°C. The frozen tumors were cut to 8 μm thickness by MICROM cryostat (MICROM International, Walldorf, Germany) and examined by fluorescence microscope. The other half tumor was homogenized and dissolved in SDS lysis buffer. Lysates containing equal amounts of protein from tumor tissues were separated by SDS-PAGE and immunoblotted with anti-Oct-4 antibody.
2.12. Cytokine array assay
To detect the levels of cytokines and growth factors in MDA-MB-231 non-stem or stem-like cell-conditioned media, antibody-based cytokine array system (Raybio Human Cytokine Antibody Array 3, RayBiotech, Norcross, GA) was used following the manufacturer’s instructions. Briefly, cells were cultured by RPMI media including 10% FBS in 6 well plate to 60–70% confluence. Fresh media was placed on the cells for 36 hours and then collected. To remove any floating cells, collected media was centrifuged at 1,000 g for 5 minutes. The supernatant was collected again, and used in the array experiment. The assay membranes were incubated in blocking solution for 30 minutes at room temperature. Each conditioned media sample was added to the membrane and incubated with gentle rocking at 4°C overnight. After washing the membrane, biotin-conjugated anti-cytokine antibody was added to each membrane for 2 hours at room temperature. Again washing the membrane, HRP-conjugated streptavidin secondary antibody reaction was allowed to proceed for 2 hours at room temperature by rocking. Cytokines bound to membranes were evaluated by chemiluminescence assay. Signal quantification was measured by subtracting the background signal using the UN-SCAN-IT program (Silk Scientific, Orem, Utah).
2.17. Neutralization with anti-IL-6 antibody
MDA-MB-231 non-CSCs were plated into ultra-low attachment plates and treated with anti-IL-6 antibody (Biolegend, San Diego, CA).
2.18. Statistical analysis
Statistical analysis was carried out using GraphPad InStat 3 software (GraphPad Software, Inc., San Diego, CA, USA). Signi cance was set at values of p < .01 or p < .001.
3. RESULTS
3.1. Characterization of CSC-like cells and non-stem cells
MDA-MB 231 and MDA-MB-453 CSC-like cells can proliferate without further differentiation and have characteristics of tumor-initiating cells [27]. This property arises as the result of stable transfection of the cells with a human Oct-3/4 promotor driving the expression of GFP, although the mechanism of the block remains unclear. On the other hand, the corresponding non-stem cancer cells were isolated with a plasmid expressing RFP under the control of a CMV immediate early promoter. CSC-like cell and non-stem cell populations can be readily shown to express high levels of GFP and RFP, respectively (Fig. 1A). Figure 1B shows MDA-MB-231 and MDA-MB-453 CSC-like cells selectively expressed octamer binding transcription factor 3/4 (Oct-4), which is known to maintain CSC-like properties [28]. Figure 1C shows that unlike non-stem cancer cells, CSC-like cells were highly enriched with CD44+ and CD24− as previously reported [27].
Figure 1. Characterization of MDA-MB-231 and MDA-MB-453 cancer stem cell–like (CSC-like) and non-stem cancer (non-CSC) cells.

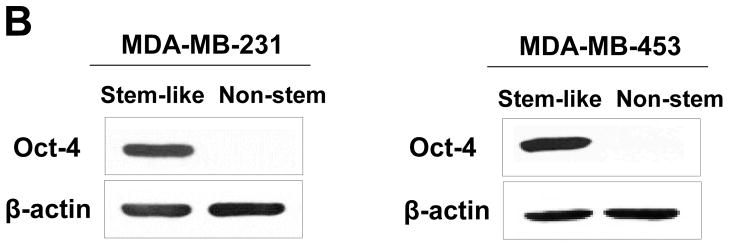

(A) Cells were transfected with a plasmid encoding GFP under the Oct-3/4 promoter for selection of stem-like cells or RFP encoding under the control of the CMV promoter for selection of non-stem cells. After selection using G418, positive colonies were verified by tumor markers (CD44, CD24, and Oct-4) expression and then pooled. Phase-contrast images or fluorescence images of Oct-3/4-GFP-transfected stem-like cells (left panels) and CMV-RFP-transfected non-stem cells (right panels) were visualized by light (phase-contrast) or UV (fluorescence) microscopy, respectively. (B) Stem-associated Oct-4 gene expression was examined in stem-like cells (left side) and non-stem cells (right side) by western blot analysis. Actin was shown as an internal standard. (C) Flow cytometry characterization of CSC-like or non-stem cells was performed by staining with surface marker antibodies (CD24, CD44) and evaluated by flow cytometry.
3.2. Mammosphere formation in CSC-like cells and non-stem cells
Stem cells have several unique properties which distinguishes them from differentiated cells. One of those is a self-renewal capacity. Gabriela Dontu et al. [29] reported that nonadherent mammospheres were enriched in cells with the functional characteristics of self-renewal potential of stem cells. They developed a strategy that allows for the cultivation of undifferentiated human mammary epithelial cells-mammospheres in suspension like neurospheres. For mammosphere formation assay, MDA-MB-231 and MDA-MB-453 CSC-like cells or non-stem cells were placed in ultra-low attachment 24 well culture plates. CSC-like cells formed mammospheres well, whereas most of the non-stem cells died and only part of the remaining non-stem cells produced viable mammospheres (Fig. 2A). Mammospheres of CSC-like cells grew faster than those of non-stem cells and the difference of mammosphere size between CSC-like cells and non-stem cells was about 3–4 times 9 days after plating (Fig. 2B). Data from cytometric assay clearly show that most of the non-stem cells died by detachment-induced apoptosis--anoikis (Fig. 2C).
Figure 2. Mammosphere formation of MDA-MB-231 and MDA-MB-453 CSC-like and non-stem cancer cells.


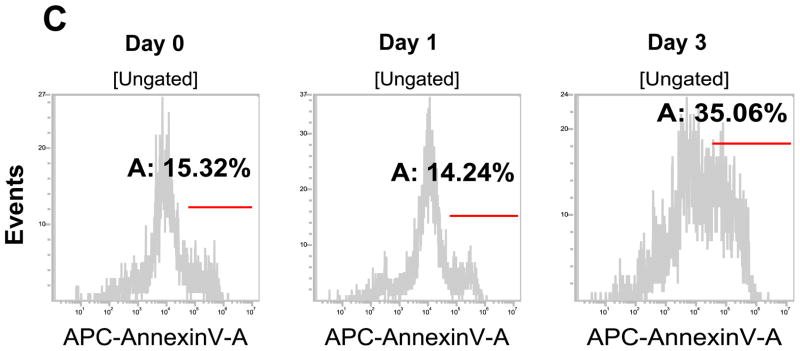
(A) Cells (1,000) were plated into ultra-low attachment plates and phase-contrast images (upper panels) or fluorescence images (lower panels) of mammospheres of stem-like (left panels) or non-stem (right panels) cells were obtained 4 days or 9 days later. (B) The numbers and sizes of mammospheres were determined and statistically analyzed. Error bars represent the SD from six samples (P<0.05). (C) Non-stem cells were plated into ultra-low attachment plates. After 1 or 3 days, cells were stained with APC-conjugated annexin V. Apoptosis was detected by the flow cytometric assay. Control: monolayer cultured cells.
3.3. Xenograft tumor formation in CSC-like cells and non-stem cells
We used a xenograft model of mammary fat pad (both sites of upper mammary fat pad) by subcutaneous injection of CSC-like cells and non-stem cells respectively to assess directly if distinct tumor formation differences were apparent between these cell lines (Fig. 3A). Our observation was that not only CSC-like cells but also non-stem cells formed tumors finally in all ten nude mice. However, the growth rate and final size of the tumors in each population was quite distinct. Figure 3B shows that the average tumor size from CSC-like cells was 323.5 mm3 and from non-stem cells was 70.4 mm3 (a difference of about 4.5 times). After the mice were sacrificed, we dissected the whole tumor mass and immediately confirmed the fluorescence derived from individual cell lines (Fig. 3C). From CSC-like cells, tumor mass showed green fluorescence, GFP, and from non-stem cells, tumor mass showed red fluorescence, RFP. To determine OCT-4 gene expression in the tumors, we used western blot analysis. As shown in Figure 3D, as well as the large amount of Oct-4 expressed by tumors of CSC-like cells, a small amount of Oct-4 was expressed by tumors of non-stem cells. From these observations, we hypothesize a dynamic equilibrium theory. We propose that removing cancer stem cells prompts part of the non-stem cell population to convert into CSC-like status during mammosphere formation, similar to what occurred in xenograft tumor.
Figure 3. Comparison of xenograft tumor formation in MDA-MB-231 stem-like cells and non-stem cells.
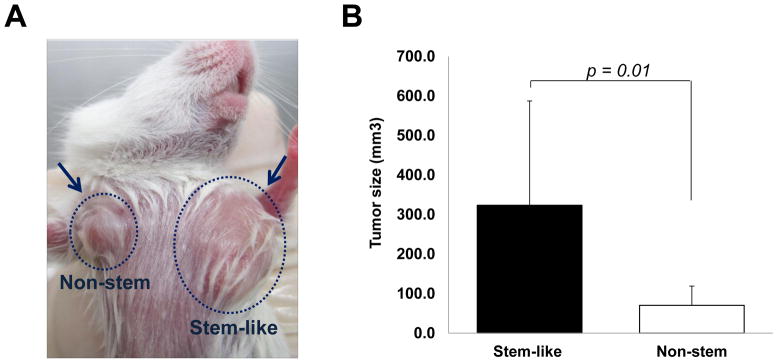

(A) Stem-like (1 × 10 4) cells and non-stem (1 × 10 4) cells were injected into the upper mammary fat pad of left and right side, respectively, in NOD/SCID mice (n=10). (B) Tumor volumes were measured 35 days after injection. The tumor volume showed significant difference (p=0.01). (C) Tumor was harvested 35 days later and cryosection was performed to detect cells containing GFP (stem-like cells) or RFP (non-stem cells). (D) Lysates containing equal amounts of protein from tumor tissues were separated by SDS-PAGE and immunoblotted with anti-Oct-4 antibody. Actin was shown as an internal standard.
3.4. Conversion of non-stem cells into stem-like cells
To test a dynamic equilibrium hypothesis, we examined whether non-stem cells can convert to stem-like cells during mammosphere formation. Ginestier et al. [30] reported that aldehyde dehydrogenase (ALDH) was increased in a subpopulation of normal and cancerous human mammary epithelial cells which exhibited stem/progenitor cell properties. Data from the Aldeflour assay revealed an increase in ALDH activity in non-stem cells during mammosphere formation. Figure 4A shows, compared to monolayer cells, a 10 to 11-fold increase in ALDH positive population in mammospheres. These data suggest the presence of CSC-like cell population in mammospheres. This possibility was examined by immunofluorescent staining assay (Fig. 4B). The expression of Oct-4 in the mammosphere was detected in most of the population from MDA-MB-231 and MDA-MB-453 CSC-like cells (both upper panels in Fig. 4B). The expression of Oct-4 was also detected in part of the non-stem cell population (both lower panels in Fig. 4B). These results suggest that part of the non-stem cells convert to CSC-like cells by inducing Oct-4 gene expression during mammosphere culture.
Figure 4. ALDEFLUOR assay, Oct-4 immunofluorescent staining, and Oct-4 gene expression in mammosphere of non-stem cells.


(A) MDA-MB-231 cells from non-stem monolayer culture or duplicate mammosphere cultures (30 days) were labeled with the Aldefluor (BAAA) with and without the ALDH inhibitor DEAB and analyzed with flow cytometry. The numbers shown in each panel reflect the percentage of ALDH+ cells in each corresponding group. (B) Mammospheres from MDA-MB-231 and MDA-MB-453 stem-like cells or non-stem cells were cultured for 30 days (left panels), harvested by cytospin, and stained with DAPI or anti-Oct-4 antibody, and then displayed in overlay images.
3.5. Cytokine profiles in CSC-like cells and non-stem cells
Recent studies have revealed that several cytokines such as interleukin-4 (IL-4), interleukin-6 (IL-6), interleukin-8 (IL-8) and interleukin-32 (IL-32) are involved in the interaction between cancer stem cells and their tumor microenvironment [26, 31–35]. We examined whether cytokines are involved in the conversion of non-stem cells into CSC-like cells. Cytokine production was compared by using cytokine array assay during monolayer culture and mammosphere culture. As shown in Figure 5, there is no significant change in CSC-like cells. Unlike in CSC-like cells, secretion of growth-related oncogene (Gro) chemokines (CXCL1, 2, 3) and IL-6 was detected during mammosphere culture in non-stem cells.
Figure 5. Comparison of cytokine production in MDA-MB-231 stem-like cells and non-stem cells.


(A) Cells were plated in 60-mm Petri dishes or ultra-low attachment plates. Conditioned media from monolayer culture or mammosphere culture were harvested and subjected to cytokine antibody arrays. (B) The cytokine array image was analyzed with densitometer and average of area integration of optical intensities for each pair of cytokine spots was plotted.
3.6. Role of the IL-6-JAK1-STAT3 signal transduction pathway in Oct-4 gene expression in non-stem cells
Previous studies have shown that IL-6 exerts its effects through the JAK1-STAT3 signal transduction pathway [36–40]. We postulated that Oct-4 gene expression is promoted through the IL-6-JAK1-STAT3 signal transduction pathway. To examine this possibility, non-stem cells were treated with anti-IL-6 antibody during mammosphere culture and the IL-6-JAK1-STAT3-Oct-4 signal transduction pathway was analyzed. Data from immunoblotting assay show that activation (phosphorylation) of JAK1 and STAT3 and increase in Oct-4 expression occurred in non-stem cells during mammosphere culture (Fig. 6A). Treatment with anti-IL-6 antibody inhibited the JAK1 and STAT3 activation as well as OCT-4 gene expression (Fig. 6B). These data suggest that the IL-6-JAK1-STAT3 signal transduction pathway is involved in the action of OCT-4 gene expression during mammosphere culture of non-stem cells. These results were confirmed by using STAT3 inhibitors, niclosamide and LLL12. These drugs inhibited STAT3 phosphorylation as well as OCT-4 gene expression during mammosphere culture (Fig. 7).
Figure 6. Effect of anti-IL-6 antibody on the IL-6-JAK1-STAT3-Oct-4 signal transduction pathway in MDA-MB-231 non-stem cells.

(A) Cells were cultured in regular plate (monolayer culture) or ultra-low attachment plate (mammosphere culture). After 1 or 3 days, cells were harvested and lysates containing equal amounts of protein were separated by SDS-PAGE and immunoblotted with anti-phospho-JAK1, anti-JAK1, anti-phospho-STAT3, anti-STAT3, or anti-Oct4 antibody. Actin was shown as an internal standard. (B) Cells were incubated with or without 1 μg/ml anti-IgG as a negative control or 1 μg/ml anti-IL-6 antibody for 1 day in the ultra-low attachment plate for mammosphere culture. Lysates from these cells were assessed by immunoblot analysis.
Figure 7. Effect of STAT3 inhibitors on STAT activation and OCT-4 gene expression in MDA-MB-231 non-stem cells.
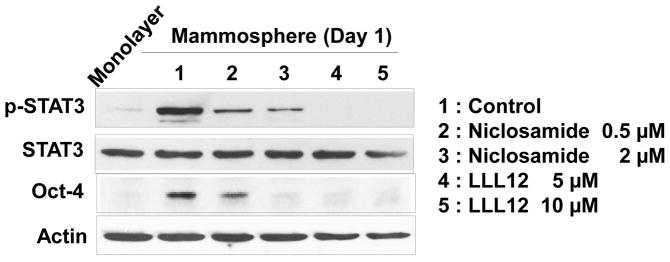
Cells were treated with niclosamide (0.5, 2 μM) or LLL12 (5, 10 μM) for 1 day. Lysates containing equal amounts of protein were separated by SDS-PAGE and immunoblotted with anti-phospho-STAT3, anti-STAT3, or anti-Oct-4 antibody. Actin was shown as an internal standard.
4. DISCUSSION
In this study, we observe that part of the non-stem cell population converted to CSC-like status during tumor formation by promoting OCT-4 gene expression. Non-stem cells, but not CSC-like cells, produced IL-6 which activated the JAK1-STAT3 signal transduction pathway. This autocrine signaling pathway plays an important role in the conversion of non-stem cells into stem-like cells through upregulation of Oct-4.
Cytokines exert their effects through specific receptors. Various signal transduction pathways are activated through distinct regions of each receptor’s cytoplasmic domain. Such pathways include those mediated by the Src (cellular homolog of the Src oncoprotein of Rous sarcoma virus) and JAK (Janus-activated kinase) tyrosine kinase families, STAT (signal transducer and activator of transcription), Smad (Sma and Mad Related Family), MAPK (mitogen-activated protein kinase), and PI3K (phosphatidyl inositol 3 kinase) [41–51]. Among these signaling molecules, STAT proteins play a central role in transmitting cytokine signals [42, 52]. In this study, we investigated the IL-6 signal transduction pathway which is known to be activated through the IL-6 receptor. Figure 6 and previous studies have shown that IL-6 activates the JAK1-STAT3 signal transduction pathway [36–40].
STAT3 is a transcription factor which is encoded by the STAT3 gene [53]. STAT3 is activated through phosphorylation of tyrosine residue 705, which induces homodimerization or heterodimerization with other STAT proteins and results in nuclear translocation and activation of the STAT3 transcriptional regulatory function [54, 55]. Phosphorylation of STAT3 can be induced by various cytokines including interferons, IL-5 and IL-6 (Fig. 6 and ref. 56) and also by receptor and nonreceptor tyrosine kinases such as epidermal growth factor receptor (EGFR) [57, 58] and Src [59]. Activation by IL-6 is mediated by members of the JAK kinase family; the tyrosine kinases EGFR and Src can directly phosphorylate STAT3 [60]. STAT3 mediates the expression of a variety of genes including autotaxin, twist, snail, tenascin-C, IL-8, vascular endothelial growth factor (VEGF), survivin, and matrix metalloproteinase-9 (MMP-9) in response to cell stimuli [61–64]. In this study, we observed that STAT3 mediates the expression of Oct-4 (Fig. 6). STAT3-mediated stem cell marker OCT-4 gene expression was effectively suppressed by treatment with STAT3 inhibitors, niclosamide or LLL12 (Fig. 7).
Our studies suggest that secreted IL-6 from non-stem cells plays an important role in the conversion of non-CSCs to CSCs through activation of the JAK1-STAT3-Oct-4 signal transduction pathway. An unanswered question is how non-stem cells produce IL-6 during mammosphere culture. At the present time, we can only speculate on the production of IL-6 in non-stem cells. One possibility is that different types of integrin-associated signal transduction pathways such as Notch and Wnt signaling [65] or different levels of integrin-associated proteins such as ILK (integrin-linked kinase), PINCH (Particularly Interesting Cys-His-rich Protein), parvin, and migfilin may exist in CSCs and non-CSCs. Previous studies have shown that among these integrin-associated proteins, migfilin regulates anoikis by influencing Src activation [66]. During mammosphere culture, migfilin is degraded and degradation of migfilin causes Src inactivation which leads to anoikis [66]. Cells of the portion of the non-CSC cells which develops resistance and survives during anoikis may activate Src-p38 MAPK-mediated IL-6 production [67, 68]. Obviously, this possibility needs to be examined to understand the mechanism of the conversion of non-CSCs into CSCs.
Acknowledgments
This work was supported by the following grants: NCI grant CA140554 (Y.J.L.) and HL083365 (Y.T.K.), DOD Breast Cancer Program BC103217 (Y.J.L.), and World Class University R31-2008-000-10103-0 (Y.T.K.). This project used the UPCI Core Facility and was supported in part by award P30CA047904.
Abbreviations used in this paper
- ALDH
aldehyde dehydrogenase
- BSA
bovine serum albumin
- CMV
cytomegalovirus
- EtOH
ethanol
- G418
Geneticin
- GFP
green fluorescent protein
- HRP
horseradish peroxidase
- JAK
Janus-activated kinase
- ILK
integrin-linked kinase
- MAPK
mitogen-activated protein kinas
- MMP-9
matrix metalloproteinase-9
- Oct-4
octamer binding transcription factor 4
- PAGE
polyacrylamide gel electrophoresis
- PBS
phosphate buffered saline
- PI3K
phosphatidyl inositol 3 kinase
- PINCH
particularly interesting Cys-His-rich protein
- RFP
red fluorescence protein
- SDS
sodium dodecyl sulfate
- STAT
signal transducer and activator of transcription
- VEGF
vascular endothelial growth factor
Footnotes
Author Contribution
Seog-Young Kim: Conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing
Jin Wook Kang: Collection and assembly of data, data analysis and interpretation
Xinxin Song: Collection and assembly of data
Bo Kyoung Kim: Collection and assembly of data
Young Dong Yoo: Collection and assembly of data
Yong Tae Kwon: Conception and design, financial support
Yong J. Lee: Conception and design, financial support, data analysis and interpretation, manuscript writing, final approval of manuscript
References
- 1.Siegel R, Naishadham D, Jemal A. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Desantis C, Siegel R, Bandi P, Jemal A. CA Cancer J Clin. 2011;61:409–418. doi: 10.3322/caac.20134. [DOI] [PubMed] [Google Scholar]
- 3.Morrison BJ, Schmidt CW, Lakhani SR, Reynolds BA, Lope JA. Breast Cancer Res. 2008;10:210–224. doi: 10.1186/bcr2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehlen P, Puisieux A. Nat Rev Cancer. 2006;6:449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 5.Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Breast Dis. 2006;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- 6.Wicha MS. Clin Cancer Res. 2006;12:5606–5607. doi: 10.1158/1078-0432.CCR-06-1537. [DOI] [PubMed] [Google Scholar]
- 7.Li F, Tiede B, Massague J, Kang Y. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 8.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, et al. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 10.O’Brien CA, Pollett A, Gallinger S, Dick JE. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 11.Lou H, Dean M. Oncogene. 2007;26:1357–1360. doi: 10.1038/sj.onc.1210200. [DOI] [PubMed] [Google Scholar]
- 12.Todaro M, Francipane MG, Medema JP, Stassi G. Gastroenterology. 2010;138:2151–2162. doi: 10.1053/j.gastro.2009.12.063. [DOI] [PubMed] [Google Scholar]
- 13.Oishi N, Wang XW. Int J Biol Sci. 2011;7:517–535. doi: 10.7150/ijbs.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hope KJ, Jin L, Dick JE. Nat Immunol. 2004;5:738–743. doi: 10.1038/ni1080. [DOI] [PubMed] [Google Scholar]
- 15.Gudjonsson T, Magnusson MK. APMIS. 2005;113:922–929. doi: 10.1111/j.1600-0463.2005.apm_371.x. [DOI] [PubMed] [Google Scholar]
- 16.Ward RJ, Dirks PB. AnnuRev Pathol Mech Dis. 2007;2:175–189. doi: 10.1146/annurev.pathol.2.010506.091847. [DOI] [PubMed] [Google Scholar]
- 17.Visvader JE, Lindeman GJ. Nature Reviews Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 18.Dick JE. Nature Biotechnology. 2009;27:44–46. doi: 10.1038/nbt0109-44. [DOI] [PubMed] [Google Scholar]
- 19.Eric Ft, Fearon, Vogelstein Bert. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 20.Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, et al. Curr Probl Cancer. 1997;21:233–300. doi: 10.1016/s0147-0272(97)80003-7. [DOI] [PubMed] [Google Scholar]
- 21.Karakosta A, Golias Ch, Charalabopoulos A, Peschos D, Batistatou A, et al. J Exp Clin Cancer Res. 2005;24:505–514. [PubMed] [Google Scholar]
- 22.Dalerba P, Cho RW, Clarke MF. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 23.Cho RW, Clarke MF. Current Opinion in Genetics & Development. 2008;18:48–53. doi: 10.1016/j.gde.2008.01.017. [DOI] [PubMed] [Google Scholar]
- 24.Liu S, Wicha MS. J Clin Oncol. 2010;28:4006–4012. doi: 10.1200/JCO.2009.27.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, et al. Cell. 2011;146:633–644. doi: 10.1016/j.cell.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 26.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Proc Natl Acad Sci USA. 2011;108:1397–1402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sajithlal GB, Rothermund K, Zhang F, Dabbs DJ, Latimer JJ, et al. Stem Cells. 2010;28:1008–1018. doi: 10.1002/stem.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen YC, Hsu HS, Chen YW, Tsai TH, How CK, et al. PLoS One. 2008;3:e2637. doi: 10.1371/journal.pone.0002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Cell Prolif. 2003;36(Suppl 1):59–72. doi: 10.1046/j.1365-2184.36.s.1.6.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, et al. Cell Stem Cell. 2007;1:555–67. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korkaya H, Liu S, Wicha MS. J Clin Invest. 2011;121:3804–3809. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Todaro M, Alea MP, Scopelliti A, Medema JP, Stassi G. Cell Cycle. 2008;7:309–313. doi: 10.4161/cc.7.3.5389. [DOI] [PubMed] [Google Scholar]
- 33.Francipane MG, Alea MP, Lombardo Y, Todaro M, Medema JP, et al. Cancer Res. 2008;68:4022–4025. doi: 10.1158/0008-5472.CAN-07-6874. [DOI] [PubMed] [Google Scholar]
- 34.Sansone P, Storci G, Tavolari S, Guarnieri T, Giovannini C, et al. J Clin Invest. 2007;117:3988–4002. doi: 10.1172/JCI32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang CJ, Chien Y, Lu KH, Chang SC, Chou YC, et al. Biochem Biophys Res Commun. 2011;415:245–251. doi: 10.1016/j.bbrc.2011.10.024. [DOI] [PubMed] [Google Scholar]
- 36.Kim NH, Lee MY, Park SJ, Choi JS, Oh MK, et al. Immunology. 2007;122:607–614. doi: 10.1111/j.1365-2567.2007.02679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Slinger E, Maussang D, Schreiber A, Siderius M, Rahbar A, et al. Sci Signal. 2010;3:ra58. doi: 10.1126/scisignal.2001180. [DOI] [PubMed] [Google Scholar]
- 38.Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, et al. Breast Cancer Res. 2007;9:R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niemand C, Nimmesgern A, Haan S, Fischer P, Schaper F, et al. J Immunol. 2003;170:3263–3272. doi: 10.4049/jimmunol.170.6.3263. [DOI] [PubMed] [Google Scholar]
- 40.Murray PJ. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 41.Birge RB, Knudsen BS, Besser D, Hanafusa H. Genes Cells. 1996;1:595–613. doi: 10.1046/j.1365-2443.1996.00258.x. [DOI] [PubMed] [Google Scholar]
- 42.Darnell JE., Jr Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 43.Heldin CH. Cell. 1995;80:213–223. doi: 10.1016/0092-8674(95)90404-2. [DOI] [PubMed] [Google Scholar]
- 44.Heldin CH, Miyazono K, ten Dijke P. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi-Tezuka M, Hibi M, Fujitani Y, Fukada T, Yamaguchi T, et al. Oncogene. 1997;14:2273–2282. doi: 10.1038/sj.onc.1201071. [DOI] [PubMed] [Google Scholar]
- 46.Winston LA, Hunter T. Curr Biol. 1996;6:668–671. doi: 10.1016/s0960-9822(09)00445-x. [DOI] [PubMed] [Google Scholar]
- 47.Ihle JN. Philos Trans R Soc Lond B Biol Sci. 1996;351:159–166. doi: 10.1098/rstb.1996.0012. [DOI] [PubMed] [Google Scholar]
- 48.Massagué J, Weis-Garcia F. Cancer Surv. 1996;27:41–64. [PubMed] [Google Scholar]
- 49.Pawson T, Scott JD. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 50.Schlessinger J, Bar-Sagi D. Cold Spring Harb Symp Quant Biol. 1994;59:173–179. doi: 10.1101/sqb.1994.059.01.021. [DOI] [PubMed] [Google Scholar]
- 51.Taniguchi Science. 1995;268:251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- 52.Darnell JE, Jr, Kerr IM, Stark GR. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- 53.Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, et al. Cell. 1994;77:63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 54.Sasse J, Hemmann U, Schwartz C, Schniertshauer U, Heesel B, et al. Mol Cell Biol. 1997;17:4677–4686. doi: 10.1128/mcb.17.8.4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Duan Z, Bradner JE, Greenberg E, Levine R, Foster R, et al. Clin Cancer Res. 2006;12:6844–6852. doi: 10.1158/1078-0432.CCR-06-1330. [DOI] [PubMed] [Google Scholar]
- 56.Dimberg LY, Dimberg A, Ivarsson K, Fryknäs M, Rickardson L, et al. BMC Cancer. 2012;12:318. doi: 10.1186/1471-2407-12-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou W, Grandis JB, Wells A. Br J Cancer. 2006;95:164–171. doi: 10.1038/sj.bjc.6603234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grandis JR, Drenning SD, Chakraborty A, Zhou MY, Zeng Q, et al. J Clin Invest. 1998;102:1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cao X, Tay A, Guy GR, Tan YH. Mol Cell Biol. 1996;16:1595–1603. doi: 10.1128/mcb.16.4.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vogt PK, Hart JR. Cancer Discov. 2011;1:481–486. doi: 10.1158/2159-8290.CD-11-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azare J, Doane A, Leslie K, Chang Q, Berishaj M, et al. PLos One. 2011;6:e27851. doi: 10.1371/journal.pone.0027851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang CH, Xu GL, Jia WD, Li JS, Ma JL, et al. J Surg Res. 2012;174:120–129. doi: 10.1016/j.jss.2010.10.030. [DOI] [PubMed] [Google Scholar]
- 63.Su L, Rao K, Guo F, Li X, Ahmed AA, et al. Domest Anim Endocrinol. 2012;43:26–36. doi: 10.1016/j.domaniend.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 64.Schröer N, Pahne J, Walch B, Wickenhauser C, Smola S. Cancer Res. 2011;71:87–97. doi: 10.1158/0008-5472.CAN-10-2193. [DOI] [PubMed] [Google Scholar]
- 65.Rallis C, Pinchin SM, Ish-Horowicz D. Development. 2010;137:3591–3601. doi: 10.1242/dev.050070. [DOI] [PubMed] [Google Scholar]
- 66.Zhao J, Zhang Y, Ithychanda SS, Tu Y, Chen K, et al. J Biol Chem. 2009;284:34308–34320. doi: 10.1074/jbc.M109.045021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tobe B, Frink M, Choudhry MA, Schwacha MG, Bland KI, et al. Am J Physiol Cell Physiol. 2006;291:C476–C482. doi: 10.1152/ajpcell.00076.2006. [DOI] [PubMed] [Google Scholar]
- 68.Hou CH, Fong YC, Tang CH. J Cell Physiol. 2011;226:2006–2015. doi: 10.1002/jcp.22541. [DOI] [PubMed] [Google Scholar]