

Abstract
The International Diabetes Federation estimates that there are currently 336 million people worldwide who have type 2 diabetes (T2DM), and the global prevalence of diabetes has more than doubled since 1980. The rapid rise in rates of T2DM echoes a similar rise in rates of obesity, which causes insulin resistance and places an increased insulin secretory demand on pancreatic β cells. While diabetes is diagnosed clinically by elevated plasma glucose levels, loss of β-cell function is progressive over time and β-cell dysfunction is far advanced by the time diabetes is diagnosed. Methods for preserving or restoring β-cell function are important for the prevention and treatment of T2DM. Interventions that reduce body fat or that change fat biology provide the best evidence for slowing or arresting the deterioration of β-cell function that causes T2DM. These interventions should form the basis of interventions to prevent and treat T2DM, particularly early in its course.
Keywords: Diabetes, Prevention, β-cell function, Glucose, Type 2 diabetes
Introduction
The global prevalence of diabetes mellitus has more than doubled since 1980 and is expected to continue to rise at alarming rates [1]. An estimated 336 million people worldwide now have T2DM [2]. T2DM results from an interaction between genetic and environmental factors that impair β-cell function and insulin action. Diabetes is diagnosed clinically by elevated plasma glucose levels, however, loss of β-cell function is progressive over time and β-cell dysfunction is far advanced by the time diabetes is diagnosed clinically [3, 4]. Patients with impaired glucose tolerance have <50 % of normal β-cell function [5–7] and patients with T2DM have <15 % of normal β-cell function for their degree of insulin resistance [8], demonstrating the progressive nature of β-cell dysfunction in the course of T2DM. Therefore, methods for preserving or restoring β-cell function are important in our attempts to prevent and treat T2DM. In this review, we discuss current evidence for causes of the progressive loss of β-cell function in T2DM, and the effects of current therapeutic strategies on preservation of β-cell function and the prevention and treatment of T2DM.
Pathogenesis of Type 2 Diabetes
β-cell Compensation for Insulin Resistance
Diabetes is defined clinically as an increase in plasma glucose levels. Plasma glucose levels are determined by the sensitivity of tissues to insulin and by the amount of insulin secreted by the pancreatic β cells. A number of factors, including lack of exercise, obesity, and visceral fat are major determinants of insulin resistance [4].
Normally, increases in insulin resistance are matched by a compensatory increase in insulin secretion by the β cells, and the relationship between insulin resistance and insulin secretion is defined by a hyperbola [9]. Based on this hyperbolic relationship, β-cell compensation can be determined by the disposition index, defined as the product of insulin secretion and insulin sensitivity [9] (Fig. 1). As long as the product of insulin secretion times insulin sensitivity remains constant, glucose tolerance is preserved. For example, in a lean, insulin sensitive individual, less insulin secretion is required to maintain normal glucose levels. An obese, insulin resistant individual requires a compensatory increase in insulin secretion in order to maintain normal glucose levels. Inadequate β-cell compensation for insulin resistance results in impaired glucose homeostasis and eventually to T2DM. Longitudinal studies have shown that reduced β-cell function as reflected in the disposition index is a powerful predictor of conversion from normal glucose tolerance to T2DM in at-risk populations [10, 11].
Fig 1.
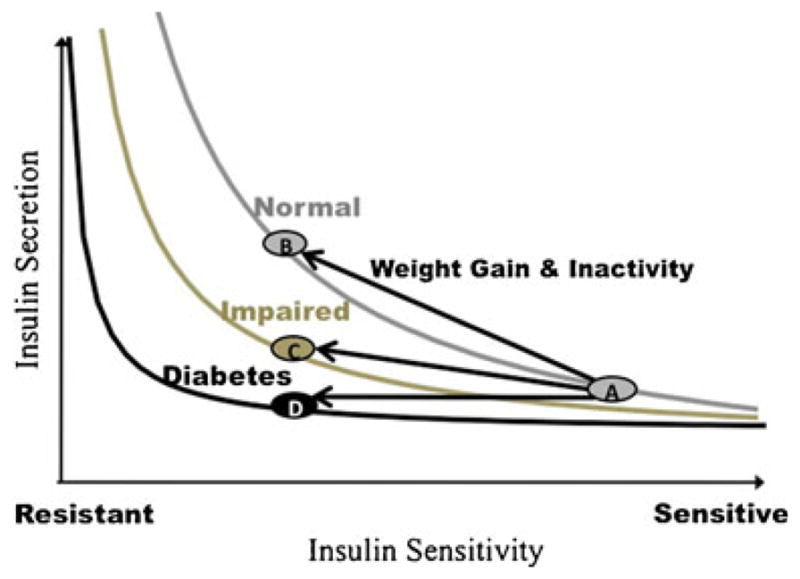
The Disposition Index (DI) is the product of insulin secretion and insulin sensitivity. Normally, increases in insulin resistance (due to factors such as weight gain and inactivity) are matched by a compensatory increase in insulin secretion in a hyperbolic relationship (A to B) and DI remains constant. Inadequate insulin secretion to compensate for insulin resistance results in a reduction in DI and impaired glucose homeostasis (A to C) and eventually to type 2 diabetes (A to D). (Data adapted from: Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68(6):1456–67) [9].
Multiple factors, including genetic predisposition, glucotoxicity, lipotoxicity, and decreased β-cell mass and function are thought to play a role in the pathogenesis of T2DM [2, 4].
Glucotoxicity and Lipotoxicity
Glucotoxicity refers to irreversible damage to pancreatic β cells caused by chronically elevated glucose levels and has been demonstrated with in vitro and in vivo studies [12–14]. Similarly to chronically elevated glucose levels, chronically elevated levels of free fatty acids (FFA) are known to cause β-cell dysfunction, a concept referred to as lipotoxicity [12]. Obesity, especially abdominal adiposity, results in increased FFA levels, and has been shown to correlate with decreased insulin gene expression and β-cell death [12]. In vitro and in vivo studies using lipid infusions have shown that chronic exposure to FFA results in decreased glucose stimulated insulin secretion, decreased insulin gene expression, and increased β-cell apoptosis in β cell lines and isolated human islets [15–17]. Recently, the concept of “glucolipotoxicity” has been introduced because of evidence suggesting that lipotoxicity is dependent on the simultaneous presence of hyperglycemia, and that elevated glucose and FFA act synergistically to impair β-cell function [12, 15, 18].
Two proposed mechanisms for glucotoxicity are endoplasmic reticulum (ER) stress and oxidative stress [15, 19].
ER Stress
The ER is the organelle responsible for folding, modification, and trafficking of proteins [20]. β cells are particularly rich in ER, given their secretory function. ER stress occurs when the ER’s folding capacity cannot match the protein load, and unfolded or misfolded proteins accumulate in the ER’s lumen [12]. Chronic hyperglycemia increases the production demand on β cells, which puts them at risk for ER stress. As the β cells of hyperglycemic individuals increase their preproinsulin production to account for elevated glucose levels, misfolded proinsulin may result in ER stress [12].
It is important to note that the processing of proinsulin into mature insulin is a critical step in insulin production and secretion, and high proinsulin levels relative to circulating levels of mature insulin can indicate β-cell stress. Recent genome-wide association studies (GWAS) have identified 9 genetic variants associated with fasting proinsulin that may contribute to β-cell function and susceptibility for T2DM [21].
When ER stress occurs, a number of different stress responses collectively referred to as the unfolded protein response (UPR) are activated in an attempt to restore cell function [20]. This is accomplished by (a) decreased translation, (b) increased ER folding capacity, (c) ER stress associated protein degradation and, ultimately (d) cell apoptosis if a, b, and c are unsuccessful [12].
ER stress also results in increased production of molecular chaperones, which help with protein folding, such as 78 kDa glucose-regulated protein (GRP78) and 4-phenylbutyrate (PBA) [19]. A recent study reported that glucose infusion to rats for 48 hours increased ER stress markers and induced β-cell dysfunction, but co-infusion with the chaperone molecule PBA prevented glucose-induced β-cell dysfunction [19].
Oxidative Stress
In vitro and in vivo studies implicate oxidative stress in glucose-induced β-cell dysfunction [19, 22, 23]. As glucose is metabolized in the mitochondria via oxidative phosphorylation, reactive oxygen species (ROS) are produced. As studies involving rat islets have shown, ROS reduce the ability of the mitochondria to produce ATP, thereby decreasing glucose-stimulated insulin secretion [24]. Isolated islets from T2DM patients have increased markers of oxidative stress compared with islets from controls without T2DM, and levels of oxidative stress correlate with the degree of impairment in glucose-stimulated insulin secretion [23]. Moreover, exposure to the antioxidant, glutathione, for 24 hours was shown to significantly improve glucose-stimulated insulin release and resulted in decreased markers of oxidative stress suggesting that reducing islet cell oxidative stress may improve the functional impairment of T2DM islets [23].
Reduced β-Cell Mass
A number of studies have demonstrated that β-cell mass and/or volume is reduced in patients with T2DM [25–29]. In a study examining pancreatic tissue from 124 autopsies, relative β-cell volume was found to be increased by 50 % in obese individuals without T2DM when compared with lean individuals without T2DM. In contrast, in obese individuals with impaired fasting glucose (IFG) there was a 40 % deficit in β-cell volume and in obese individuals with T2DM there was a 63 % deficit in β-cell volume when compared with obese individuals without T2DM [27]. The presence of decreased β-cell volume in individuals with IFG suggests that this process occurs early on in the process of developing T2DM [27].
Effects of Type 2 Diabetes Therapies on β-Cell Function
Clinical management of T2DM is currently based on achieving plasma glucose levels that are associated with a low risk of developing long-term microvascular complications [30]. There are a number of therapies that are effective in reducing plasma glucose levels through various mechanisms, however current therapeutic strategies have different effects on β-cell function. Interventions that reduce the load on β-cells by decreasing insulin demand have better durability on glycemic control and are more effective in preventing T2DM in high risk patients. Below, we discuss current therapies used in the treatment of T2DM and their effects on β-cell function, long-term durability, and effectiveness in preventing progression to T2DM in high risk individuals.
Intensive Lifestyle Modification
Obesity is a major risk factor for T2DM, and the increased prevalence of obesity is largely responsible for the concomitant increase in T2DM [1]. Obesity and lack of physical activity cause insulin resistance and increase the workload on β cells [31]. Weight loss and exercise interventions increase insulin sensitivity and unload the secretory demand on β-cells.
The United States Diabetes Prevention Program (DPP) showed that implementing a program that achieved at least a 7 % reduction in body weight through diet and exercise reduced the incidence of T2DM by 58 % in patients with impaired glucose tolerance (IGT) [32]. The effect of lifestyle intervention on reducing the incidence of T2DM was related to overall improvements in β-cell function driven by its gains in insulin sensitivity, such that the hyperbola describing the relationship between insulin secretion and in insulin sensitivity was shifted to the right [33]. The Finnish Diabetes Prevention Study (DPS) also showed a 58 % reduction in the incidence of T2DM in individuals with IGT who were assigned to a lifestyle intervention that included weight loss and increased physical activity [34] (Table 1). A recent analysis of the DPS indicated that lifestyle intervention helps to preserve β-cell function and prevent the development of T2DM through improvements in insulin sensitivity [35].
Table 1.
Summary of diabetes prevention trials
| Study | Participants at high-risk for diabetes | Intervention | Relative reduction in risk of diabetesa |
|---|---|---|---|
| DPP | IGT | Lifestyle | 58 % |
| Finnish DPS | IGT | Lifestyle | 58 % |
| XENDOS | IGT | Orlistat + lifestyle | 45%b |
| TRIPOD | Prior GDM | Troglitazone | 55 % |
| DPP | IGT | Troglitazone | 75 % |
| DREAM | IGT | Rosiglitazone | 60 % |
| ACT NOW | IGT | Pioglitazone | 72 % |
| DPP | IGT | Metformin | 31 % |
| Stop-NIDDM | IGT | Acarbose | 25 % |
DPP Diabetes Prevention Program, DPS Diabetes Prevention Study, TRIPOD troglitazone in prevention of diabetes, DREAM diabetes reduction assessment with ramipril and rosiglitazone medication, ACT NOW Actos now, IGT impaired glucose tolerance, GDM gestational diabetes mellitus
vs placebo and/or usual care
vs lifestyle intervention
Pharmacological Weight Loss
The Xenical in the Prevention of Diabetes in Obese Subjects (XENDOS) Study reported that orlistat, a gastrointestinal lipase inhibitor, added to lifestyle intervention resulted in greater weight loss and a 37 % reduced relative risk for diabetes compared with lifestyle intervention alone in obese adults with normal and impaired glucose tolerance [36]. In obese subjects with IGT at baseline, orlistat plus lifestyle intervention resulted in a 45 % reduced relative risk for diabetes vs lifestyle intervention alone (Table 1).
These randomized controlled trials demonstrate the importance of lifestyle modifications and pharmacological weight loss in T2DM prevention, but it is important to note that weight regain is common [37], even with pharmacological treatment. For example, in the XENDOS trial, mean weight loss after the first year of treatment was 10.6 kg with orlistat plus lifestyle intervention and 6.2 kg after lifestyle intervention alone, but after 4 years of treatment the mean weight loss was only 5.8 kg in orlistat plus lifestyle and 3.0 kg in lifestyle intervention alone [36]. Therefore, early intervention measures, including public health initiatives aimed at preventing obesity by promoting physical activity and healthier diets early in life may be particularly important to prevent the upward trends in prevalence of T2DM. For obese individuals with impaired glucose levels, interventions aimed at more robust and sustained weight loss, including modestly invasive bariatric surgery, deserve exploration as an approach towards β-cell preservation for the prevention of T2DM [38] (see “On the Horizon; Newer Approaches” below).
Sulfonylureas
Sulfonylureas are oral medications that stimulate insulin secretion by binding to the sulfonylurea receptor 1, resulting in membrane depolarization and calcium influx, which triggers exocytosis of insulin containing secretory granules [39]. While sulfonylureas are widely used clinically in the treatment of T2DM, evidence from the Diabetes Outcome Progression Trial (ADOPT) study demonstrates a more rapid deterioration of glycemic control with the sulfonylurea, glyburide, compared with treatment with metformin and the thiazolidinedione (TZD), rosiglitazone in patients with recently diagnosed T2DM [40]. In vitro studies have suggested that the lack of durability of sulfonylurea treatment observed in clinical studies may be due to its potentially damaging effects on pancreatic β-cells [41, 42]. Studies in isolated human islets indicate that the sulfonylurea, glibenclamide, decreases insulin content, and induces β-cell apoptosis [41, 42]; however, 1 study suggested that the sulfonylurea, gliclazide, may protect β cells from apoptosis potentially through antioxidant effects [42]. No clinical studies have demonstrated a beneficial effect of sulfonylureas in the prevention of T2DM, and as mentioned above, sulfonylurea monotherapy has less durable effects on glycemic control compared with a TZD or metformin in patients recently diagnosed with T2DM [40].
Metformin
Metformin is effective at reducing hyperglycemia primarily by inhibiting hepatic glucose production and by increasing insulin sensitivity [43]. It is currently recommended as a first-line drug for the treatment of T2DM [30]. In vitro studies demonstrated that metformin could protect isolated human islets from glucotoxicity and lipotoxicity suggesting that metformin may have beneficial effects on β-cell health [44, 45]. In clinical studies, the DPP showed that metformin reduced the conversion from IGT to T2DM by 31 % [32] suggesting that it has modest effects on slowing the progression of T2DM (Table 1). The U.K. Prospective Diabetes Study (UKPDS) showed similar rates of deterioration of β-cell function (assessed with HOMA-B index) and loss of glycemic control with metformin treatment compared with sulfonylureas or insulin treatment in patients with recently diagnosed T2DM [46, 47]. The ADOPT study showed that the durability of metformin monotherapy was better than glyburide, but it still resulted in a 21 % failure rate at 5 years in patients with recently diagnosed T2DM [40].
Acarbose
Acarbose is an α-glucosidase inhibitor that improves post-prandial hyperglycemia by inhibiting the activity of enzymes in the small intestine resulting in reduced glucose absorption. The Study to Prevent NIDDM (STOP-NIDDM) found a 25 % relative risk reduction in the development of T2DM over 3.3 years in patients with impaired glucose levels treated with acarbose compared with placebo [48] (Table 1). However, in the 3-month observation period after acarbose was discontinued, the incidence of diabetes in patients who had not converted was higher in the group initially assigned to acarbose (15 %) compared with group first randomized to placebo (10 %) suggesting that the benefit of acarbose is lost after discontinuation of active treatment [48].
Thiazolidinediones (TZDs)
TZDs are potent insulin sensitizers that improve glycemic control in patients with T2DM [43, 49]. TZDs are ligands for the nuclear transcription factor peroxisome-proliferator-activated-receptor-γ, and they have a wide spectrum of action [49]. Studies have shown that TZDs reduce lipotoxicity [50, 51], prevent β-cell apoptosis [52], increase serum adiponectin levels [53], and improve β-cell function [54–57]. Prevention trials have consistently shown that TZDs are effective in preventing the onset of type 2 diabetes in high-risk patients by ~50 %–75 % (DPP, Troglitazone in Prevention of Diabetes (TRIPOD), Pioglitazone in Prevention of Diabetes (PIPOD), Diabetes Reduction Assessment with Ramipril and Rosiglitazone Medication (DREAM), and Actos now (ACT-NOW) [55, 56, 58–60] (see Table 1). The TRIPOD study was the first to carefully examine changes in β-cell function through treatment of insulin resistance as a way to reduce T2DM risk. Notably, the TRIPOD study showed that protection from diabetes in women with previous gestational diabetes persisted 8 months after T2DM treatment stopped, and patients who were protected from diabetes during TZD treatment had stable β-cell function and insulin resistance for almost 5 years demonstrating that TZDs slow the natural progression of the disease [55, 56]. The ability of TZDs to slow or stop progression to T2DM was supported by DREAM and DPP, in which the protection from diabetes that was achieved during treatment persisted after treatment was stopped [61].
The ADOPT Study demonstrated that TZDs had significantly greater durability for glycemic control in patients with recently diagnosed T2DM when compared with both metformin and sulfonylureas [40]. Subsequent analysis of the ADOPT data showed that the reduction in treatment failure with rosiglitazone corresponded with improved β-cell function and increased insulin sensitivity [62] supporting results from the TRIPOD prevention study and suggesting that TZDs may preserve β-cell function by improving insulin sensitivity and unloading the secretory demand on β-cells [55].
The clinical use of TZDs for the prevention of T2DM is limited due to adverse side effects, including fluid retention and weight gain, and recent safety concerns including reports of an association with increased risk for bone fractures and bladder cancer [49] The development of safer compounds with selective PPARγ modulation offers the potential for targeted insulin sensitization in the absence of adverse side effects [49].
GLP-1 Receptor Agonists
Glucagon-like polypeptide-1 (GLP-1) is an incretin hormone secreted by the L cells in the intestines in response to nutrient stimulation. GLP-1 possesses a number of properties that make it an ideal agent for the treatment of T2DM. GLP-1 potentiates glucose stimulated insulin secretion, suppresses glucagon secretion, delays gastric emptying and suppresses appetite [43]. GLP-1 is cleaved by the enzyme dipeptidyl peptidase-4 (DPP-4) leading to its rapid inactivation, but DPP-4 resistant GLP-1 receptor agonists, including exenatide and liraglutide, and DPP-4 inhibitors have been developed for the treatment of T2DM [43]. Studies in animals have shown the GLP-1 analogues decrease β-cell apoptosis and increase β-cell mass [63–65], and human studies indicate that exenatide exerts potent anti-inflammatory effects that are independent of weight loss [66]. Findings from recent randomized controlled trials demonstrate that exenatide improves β-cell function [67, 68, 69•, 70••]. Bunck and colleagues compared the effects of exenatide with the insulin, glargine, on β-cell function over 3 years in metformin treated patients with T2DM [68, 69•]. They found that exenatide significantly improved β-cell function during 52 weeks of active treatment compared with glargine, but after stopping the treatments for 4 weeks, β-cell function returned to pre-treatment values in both groups [68]. In contrast, in the 3-year extension study, β-cell function was sustained in the exenatide group after a 4-week off-drug period whereas the glargine treated patients had a reduction in β-cell function suggesting that at least 3 years of exenatide treatment may be necessary to delineate a significant, prolonged benefit on β-cell function [69•].
A recent study in obese adults showed that a 20-week treatment with liraglutide (in doses ranging from 1.8 to 3 mg per day) resulted in greater weight loss and an 84 %–96 % reduction in the prevalence of prediabetes compared with placebo [71••]. Another clinical trial showed that a 24-week treatment with exenatide plus lifestyle modification resulted in greater weight loss and normalization of glucose tolerance in 77 % of obese participants with impaired glucose homeostasis (IGT or IFG) compared with 56 % in the placebo group [72•]. Longer term prevention trials in high-risk patients are needed to determine whether GLP-1 agonists can modify the progressive course of T2DM.
A recent randomized controlled trial suggested that the GLP-1 receptor agonist, exenatide, may have durable effects on glycemic control in patients with T2DM [70••]. The EUREXA study compared exenatide with glimepiride as add-on to metformin for durability of glycemic control in patients with T2DM who were inadequately controlled with metformin alone [70••]. Results showed that glycemic control was maintained for longer (180 weeks with exenatide vs 142 weeks with glimepiride) and treatment failure was lower in the exenatide (41 %) compared with glimepiride (54 %) group over the 48 month follow up period. The exenatide group also had a significantly greater increase in disposition index compared with the glimepiride group demonstrating beneficial effects of exenatide on β-cell function [70••].
On the Horizon; Newer Approaches
Early Intensive Treatment with Insulin and/or Multiple Agents
In vitro studies investigating the effects of glucose toxicity on a pancreatic β-cell line showed that shortening the duration of antecedent glucose toxicity increases the likelihood of recovering β-cell function [73]. Thus, early and more aggressive treatment strategies that quickly normalize glucose levels in patients with newly diagnosed T2DM may preserve residual β-cell function by reducing glucotoxicity. Clinical studies have shown beneficial effects of early short-term (2–3 weeks) intensive insulin therapy in patients with newly diagnosed T2DM [74–78]. A multi-center randomized trial showed that early, short-term intensive insulin treatment at the time of T2DM diagnosis led to improved β-cell function and greater diabetes remission rates at 1 year compared with early, short-term treatment with oral hypoglycemic agents [75]. A recent randomized control trial evaluated β-cell function preservation after 3.5 years of intensive therapy with insulin plus metformin compared with triple oral therapy with metformin, glyburide, and pioglitazone after an initial 3-month insulin treatment period and found that both approaches were effective in preserving β-cell function [77]. Based on these results, the authors suggested that patients with newly diagnosed T2DM should be treated with an initial period of intensive insulin therapy to maximize β-cell recovery and then continued either on insulin therapy or switched to a combination of oral agents with complementary mechanisms of action [77].
The Outcome Reduction with an Initial Glargine Intervention (ORIGIN) trial recently reported results of the comparison between treatment with insulin glargine to normalize fasting plasma glucose levels compared with standard care on cardiovascular outcomes, cancer, and incident diabetes in over 12,000 patients with cardiovascular disease risk factors plus IFG or T2DM who were followed for a median of 6.2 years [79]. They found that glargine had a neutral effect on cardiovascular outcomes and cancers, but that it reduced new-onset diabetes among the participants without diabetes at randomization providing support for the idea that the early use of insulin may preserve β-cell function in patients with impaired glucose homeostasis.
There is a growing interest in the early use of combination therapies for the treatment of patients with IGT and T2DM [67, 80]. A recent clinical study examined the effects of combination therapy with exenatide and rosiglitazone vs each therapy alone on insulin sensitivity and β-cell function in patients with T2DM already on metformin [67]. The 20-week combination therapy with exenatide and rosiglitazone resulted in greater improvements in insulin sensitivity and β-cell function and better glycemic control compared with either treatment alone suggesting a beneficial effect of the combination therapy [67]. However, randomized controlled studies are needed to determine whether early use of a combination of oral agents is effective in preventing T2DM in high risk patients and the long-term durability of this treatment strategy on glycemic control.
Bariatric Surgery
Bariatric surgery is an effective and durable treatment for obesity and provides substantial improvements in glycemic control in obese patients with T2DM [81]. Four bariatric surgical procedures are used conventionally, including Roux-en-Y gastric bypass (RYGB), laparoscopic adjustable gastric banding (LAGB), biliopancreatic diversion (BPD), and laparoscopic sleeve gastrectomy (LSG), and RYGB and LAGB are the most widely used surgical procedures [81]. A meta-analysis indicated that bariatric surgery results in resolution of diabetes in 78 % of patients with T2DM demonstrating its significant impact on T2DM remission [82]. The effects of bariatric surgery on β-cell function were recently summarized in a review by Bradley et al [83], and indicate that LAGB surgery increases the disposition index following modest weight loss, RYGB surgery increases the disposition index after significant weight loss, BPD surgery increases early insulin secretion and disposition index in patients with T2DM, and LSG surgery increases early insulin secretion in patients with T2DM, but its effects on disposition index are currently unknown [83].
Three randomized controlled trials have compared the effects of bariatric surgery to medical therapy on remission of T2DM, and all 3 studies showed that bariatric surgery was significantly more effective than medical therapy in achieving remission of T2DM within 1 to 2 years following surgery [84, 85••, 86••]. The first randomized controlled trial showed that LAGB surgery resulted in T2DM remission in 73 % of patients vs only 13 % who received standard medical treatment [84]. In a recent randomized controlled trial, 42 % of obese patients with T2DM who underwent RYGB surgery and 37 % of those who underwent LSG surgery achieved remission of T2DM compared with 12 % of patients treated with conventional medical therapy [85••]. In another recent randomized controlled trial, 95 % of individuals with T2DM who underwent BPD and 75 % of those who underwent RYGB surgery, but none of those who received standard medical therapy achieved remission of T2DM [86••].
The long- term effects of bariatric surgery on T2DM remission were examined in the Swedish Obese Subjects Study (SOS), a nonrandomized prospective case-control study in over 4,000 obese patients who underwent bariatric surgery [87]. They found that 72 % of T2DM patients achieved remission at 2 years after surgery, and 36 % had maintained T2DM remission at 10 years after surgery [87]. Similar results were found in another smaller study which showed durable remission of T2DM (>5 years) in over half of the 89 % of patients who had achieved early following RYBG remission [88], and a meta-analysis which reported that 62 % of patients with T2DM remained free of diabetes for more than 2 years following bariatric surgery [82].
The effect of bariatric surgery (LAGB, VBG, RYGB) on the prevention of T2DM in obese adults was recently examined in the SOS study which followed surgically treated and matched controls for 15 years [89••]. Bariatric surgery compared with standard care reduced the long-term relative risk of T2DM by 78 % in obese adults, and among those with IFG it reduced the relative risk of T2DM by 82 %. The postoperative mortality was 0.2 %, and 2.8 % of patients had complications that required a reoperation [89••]. These findings indicate that bariatric surgery has effective and durable effects on the prevention of T2DM in obese adults, particularly among those with IFG. Randomized controlled trials are needed to confirm whether bariatric surgery is an effective and safe approach for preventing T2DM in high-risk individuals.
Conclusions
T2DM is characterized by a progressive loss of β-cell function that occurs against a background of chronic insulin resistance. The rapid rise in rates of T2DM echoes a similar rise in rates of obesity, which causes insulin resistance and may have additional effects on β-cell health. Interventions that reduce body fat (such as diet and exercise, GLP-1 receptor agonists, or bariatric surgery) or that change fat biology (TZDs) provide the best evidence for slowing or arresting the deterioration of β-cell function that causes T2DM. These interventions should form the basis of interventions to prevent and treat T2DM, particularly early in its course.
Footnotes
Disclosure No potential conflicts of interest relevant to this article were reported.
Contributor Information
Kathleen A. Page, Email: kpage@usc.edu, Division of Endocrinology and Diabetes, Department of Internal Medicine, Keck School of Medicine, University of Southern California, 1333 San Pablo Street; BMT-B11, Los Angeles, CA 90033, USA
Tamar Reisman, Email: tamar.reisman@gmail.com, Department of Internal Medicine, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA.
References
Papers of particular interest that have been published recently have been highlighted as:
• Of importance
•• Of major importance
- 1.Chen L, Magliano DJ, Zimmet PZ. The worldwide epidemiology of type 2 diabetes mellitus–present and future perspectives. Nat Rev Endocrinol. 2012;8:228–36. doi: 10.1038/nrendo.2011.183. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–71. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchanan TA. Pancreatic beta-cell loss and preservation in type 2 diabetes. Clin Ther. 2003;25(Suppl B):B32–46. doi: 10.1016/s0149-2918(03)80241-2. [DOI] [PubMed] [Google Scholar]
- 4.Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46:3–19. doi: 10.1007/s00125-002-1009-0. [DOI] [PubMed] [Google Scholar]
- 5.Abdul-Ghani MA, Tripathy D, DeFronzo RA. Contributions of beta-cell dysfunction and insulin resistance to the pathogenesis of impaired glucose tolerance and impaired fasting glucose. Diabetes Care. 2006;29:1130–9. doi: 10.2337/diacare.2951130. [DOI] [PubMed] [Google Scholar]
- 6.Buchanan TA, Xiang AH, Peters RK, Kjos SL, Berkowitz K, Marroquin A, et al. Response of pancreatic beta-cells to improved insulin sensitivity in women at high risk for type 2 diabetes. Diabetes. 2000;49:782–8. doi: 10.2337/diabetes.49.5.782. [DOI] [PubMed] [Google Scholar]
- 7.Bergman RN, Ader M, Huecking K, Van Citters G. Accurate assessment of beta-cell function: the hyperbolic correction. Diabetes. 2002;51 (Suppl 1):S212–20. doi: 10.2337/diabetes.51.2007.s212. [DOI] [PubMed] [Google Scholar]
- 8.Porte D, Jr, Kahn SE. beta-cell dysfunction and failure in type 2 diabetes: potential mechanisms. Diabetes. 2001;50 (Suppl 1):S160–3. doi: 10.2337/diabetes.50.2007.s160. [DOI] [PubMed] [Google Scholar]
- 9.Bergman RN, Phillips LS, Cobelli C. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J Clin Invest. 1981;68:1456–67. doi: 10.1172/JCI110398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes. 2000;49:2094–101. doi: 10.2337/diabetes.49.12.2094. [DOI] [PubMed] [Google Scholar]
- 11.Lyssenko V, Almgren P, Anevski D, Perfekt R, Lahti K, Nissen M, et al. Predictors of and longitudinal changes in insulin sensitivity and secretion preceding onset of type 2 diabetes. Diabetes. 2005;54:166–74. doi: 10.2337/diabetes.54.1.166. [DOI] [PubMed] [Google Scholar]
- 12.Kim MK, Kim HS, Lee IK, Park KG. Endoplasmic reticulum stress and insulin biosynthesis: a review. Exp Diabetes Res. 2012;2012:509437. doi: 10.1155/2012/509437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonner-Weir S, Trent DF, Weir GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest. 1983;71:1544–53. doi: 10.1172/JCI110910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leahy JL, Bonner-Weir S, Weir GC. Beta-cell dysfunction induced by chronic hyperglycemia. Current ideas on mechanism of impaired glucose-induced insulin secretion. Diabetes Care. 1992;15:442–55. doi: 10.2337/diacare.15.3.442. [DOI] [PubMed] [Google Scholar]
- 15.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev. 2008;29:351–66. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes. 2002;51:1437–42. doi: 10.2337/diabetes.51.5.1437. [DOI] [PubMed] [Google Scholar]
- 17.Oprescu AI, Bikopoulos G, Naassan A, Allister EM, Tang C, Park E, et al. Free fatty acid-induced reduction in glucose-stimulated insulin secretion: evidence for a role of oxidative stress in vitro and in vivo. Diabetes. 2007;56:2927–37. doi: 10.2337/db07-0075. [DOI] [PubMed] [Google Scholar]
- 18.Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002;51 (Suppl 3):S405–13. doi: 10.2337/diabetes.51.2007.s405. [DOI] [PubMed] [Google Scholar]
- 19.Tang C, Koulajian K, Schuiki I, Zhang L, Desai T, Ivovic A, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia. 2012;55:1366–79. doi: 10.1007/s00125-012-2474-8. [DOI] [PubMed] [Google Scholar]
- 20.Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767–93. doi: 10.1146/annurev-biochem-072909-095555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes. 2011;60:2624–34. doi: 10.2337/db11-0415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–4. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 23.Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes. 2005;54:727–35. doi: 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- 24.Li N, Brun T, Cnop M, Cunha DA, Eizirik DL, Maechler P. Transient oxidative stress damages mitochondrial machinery inducing persistent beta-cell dysfunction. J Biol Chem. 2009;284:23602–12. doi: 10.1074/jbc.M109.024323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakuraba H, Mizukami H, Yagihashi N, Wada R, Hanyu C, Yagihashi S. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45:85–96. doi: 10.1007/s125-002-8248-z. [DOI] [PubMed] [Google Scholar]
- 26.Yoon KH, Ko SH, Cho JH, Lee JM, Ahn YB, Song KH, et al. Selective beta-cell loss and alpha-cell expansion in patients with type 2 diabetes mellitus in Korea. J Clin Endocrinol Metab. 2003;88:2300–8. doi: 10.1210/jc.2002-020735. [DOI] [PubMed] [Google Scholar]
- 27.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–10. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 28.Marchetti P, Del Guerra S, Marselli L, Lupi R, Masini M, Pollera M, et al. Pancreatic islets from type 2 diabetic patients have functional defects and increased apoptosis that are ameliorated by metformin. J Clin Endocrinol Metab. 2004;89:5535–41. doi: 10.1210/jc.2004-0150. [DOI] [PubMed] [Google Scholar]
- 29.Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care. 2006;29:717–8. doi: 10.2337/diacare.29.03.06.dc05-1538. [DOI] [PubMed] [Google Scholar]
- 30.Standards of medical care in diabetes-2011. Diabetes Care. 2011;34(Suppl 1):S11–61. doi: 10.2337/dc11-S011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergman RN. Minimal model: perspective from 2005. Horm Res. 2005;64 (Suppl 3):8–15. doi: 10.1159/000089312. [DOI] [PubMed] [Google Scholar]
- 32.Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitabchi AE, Temprosa M, Knowler WC, Kahn SE, Fowler SE, Haffner SM, et al. Role of insulin secretion and sensitivity in the evolution of type 2 diabetes in the diabetes prevention program: effects of lifestyle intervention and metformin. Diabetes. 2005;54:2404–14. doi: 10.2337/diabetes.54.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–50. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 35.de Mello VD, Lindstrom J, Eriksson J, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Sundvall J, et al. Insulin secretion and its determinants in the progression of impaired glucose tolerance to type 2 diabetes in impaired glucose-tolerant individuals: the Finnish Diabetes Prevention Study. Diabetes Care. 2012;35:211–7. doi: 10.2337/dc11-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torgerson JS, Hauptman J, Boldrin MN, Sjostrom L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27:155–61. doi: 10.2337/diacare.27.1.155. [DOI] [PubMed] [Google Scholar]
- 37.Venditti EM, Bray GA, Carrion-Petersen ML, Delahanty LM, Edelstein SL, Hamman RF, et al. First versus repeat treatment with a lifestyle intervention program: attendance and weight loss outcomes. Int J Obes (Lond) 2008;32:1537–44. doi: 10.1038/ijo.2008.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchanan TA. Stemming the tide of type 2 diabetes: bring on the “big guns”. Obesity (Silver Spring) 2010;18:1065–7. doi: 10.1038/oby.2010.111. [DOI] [PubMed] [Google Scholar]
- 39.Lang V, Youssef N, Light PE. The molecular genetics of sulfonyl-urea receptors in the pathogenesis and treatment of insulin secretory disorders and type 2 diabetes. Curr Diab Rep. 2011;11:543–51. doi: 10.1007/s11892-011-0233-8. [DOI] [PubMed] [Google Scholar]
- 40.Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355:2427–43. doi: 10.1056/NEJMoa066224. [DOI] [PubMed] [Google Scholar]
- 41.Maedler K, Carr RD, Bosco D, Zuellig RA, Berney T, Donath MY. Sulfonylurea induced beta-cell apoptosis in cultured human islets. J Clin Endocrinol Metab. 2005;90:501–6. doi: 10.1210/jc.2004-0699. [DOI] [PubMed] [Google Scholar]
- 42.Del Guerra S, Marselli L, Lupi R, Boggi U, Mosca F, Benzi L, et al. Effects of prolonged in vitro exposure to sulphonylureas on the function and survival of human islets. J Diabetes Complications. 2005;19:60–4. doi: 10.1016/j.jdiacomp.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 43.Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378:182–97. doi: 10.1016/S0140-6736(11)60207-9. [DOI] [PubMed] [Google Scholar]
- 44.Lupi R, Del Guerra S, Fierabracci V, Marselli L, Novelli M, Patane G, et al. Lipotoxicity in human pancreatic islets and the protective effect of metformin. Diabetes. 2002;51 (Suppl 1):S134–7. doi: 10.2337/diabetes.51.2007.s134. [DOI] [PubMed] [Google Scholar]
- 45.Patane G, Piro S, Rabuazzo AM, Anello M, Vigneri R, Purrello F. Metformin restores insulin secretion altered by chronic exposure to free fatty acids or high glucose: a direct metformin effect on pancreatic beta-cells. Diabetes. 2000;49:735–40. doi: 10.2337/diabetes.49.5.735. [DOI] [PubMed] [Google Scholar]
- 46.UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- 47.UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- 48.Chiasson JL, Josse RG, Gomis R, Hanefeld M, Karasik A, Laakso M. Acarbose for prevention of type 2 diabetes mellitus: the STOP-NIDDM randomised trial. Lancet. 2002;359:2072–7. doi: 10.1016/S0140-6736(02)08905-5. [DOI] [PubMed] [Google Scholar]
- 49.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARgamma agonists: time for a reassessment. Trends in endocrinology and metabolism: TEM. 2012;23:205–15. doi: 10.1016/j.tem.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Lupi R, Del Guerra S, Marselli L, Bugliani M, Boggi U, Mosca F, et al. Rosiglitazone prevents the impairment of human islet function induced by fatty acids: evidence for a role of PPARgamma2 in the modulation of insulin secretion. Am J Physiol Endocrinol Metab. 2004;286:E560–7. doi: 10.1152/ajpendo.00561.2002. [DOI] [PubMed] [Google Scholar]
- 51.Prieur X, Mok CY, Velagapudi VR, Nunez V, Fuentes L, Montaner D, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. 2011;60:797–809. doi: 10.2337/db10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akiyama M, Hatanaka M, Ohta Y, Ueda K, Yanai A, Uehara Y, et al. Increased insulin demand promotes while pioglitazone prevents pancreatic beta cell apoptosis in Wfs1 knockout mice. Diabetologia. 2009;52:653–63. doi: 10.1007/s00125-009-1270-6. [DOI] [PubMed] [Google Scholar]
- 53.Yu JG, Javorschi S, Hevener AL, Kruszynska YT, Norman RA, Sinha M, et al. The effect of thiazolidinediones on plasma adiponectin levels in normal, obese, and type 2 diabetic subjects. Diabetes. 2002;51:2968–74. doi: 10.2337/diabetes.51.10.2968. [DOI] [PubMed] [Google Scholar]
- 54.Gastaldelli A, Ferrannini E, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. Thiazolidinediones improve beta-cell function in type 2 diabetic patients. Am J Physiol Endocrinol Metab. 2007;292:E871–83. doi: 10.1152/ajpendo.00551.2006. [DOI] [PubMed] [Google Scholar]
- 55.Buchanan TA, Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, et al. Preservation of pancreatic beta-cell function and prevention of type 2 diabetes by pharmacological treatment of insulin resistance in high-risk hispanic women. Diabetes. 2002;51:2796–803. doi: 10.2337/diabetes.51.9.2796. [DOI] [PubMed] [Google Scholar]
- 56.Xiang AH, Peters RK, Kjos SL, Marroquin A, Goico J, Ochoa C, et al. Effect of pioglitazone on pancreatic beta-cell function and diabetes risk in Hispanic women with prior gestational diabetes. Diabetes. 2006;55:517–22. doi: 10.2337/diabetes.55.02.06.db05-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hanley AJ, Zinman B, Sheridan P, Yusuf S, Gerstein HC. Effect of Rosiglitazone and Ramipril on {beta}-cell function in people with impaired glucose tolerance or impaired fasting glucose: the DREAM trial. Diabetes Care. 2010;33:608–13. doi: 10.2337/dc09-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerstein HC, Yusuf S, Bosch J, Pogue J, Sheridan P, Dinccag N, et al. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]
- 59.DeFronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, et al. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med. 2011;364:1104–15. doi: 10.1056/NEJMoa1010949. [DOI] [PubMed] [Google Scholar]
- 60.Knowler WC, Hamman RF, Edelstein SL, Barrett-Connor E, Ehrmann DA, Walker EA, et al. Prevention of type 2 diabetes with troglitazone in the Diabetes Prevention Program. Diabetes. 2005;54:1150–6. doi: 10.2337/diabetes.54.4.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buchanan TA. (How) can we prevent type 2 diabetes? Diabetes. 2007;56:1502–7. doi: 10.2337/db07-0140. [DOI] [PubMed] [Google Scholar]
- 62.Kahn SE, Lachin JM, Zinman B, Haffner SM, Aftring RP, Paul G, et al. Effects of rosiglitazone, glyburide, and metformin on beta-cell function and insulin sensitivity in ADOPT. Diabetes. 2011;60:1552–60. doi: 10.2337/db10-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rolin B, Larsen MO, Gotfredsen CF, Deacon CF, Carr RD, Wilken M, et al. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am J Physiol Endocrinol Metab. 2002;283:E745–52. doi: 10.1152/ajpendo.00030.2002. [DOI] [PubMed] [Google Scholar]
- 64.Drucker DJ. Glucagon-like peptide-1 and the islet beta-cell: augmentation of cell proliferation and inhibition of apoptosis. Endocrinology. 2003;144:5145–8. doi: 10.1210/en.2003-1147. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Hansotia T, Yusta B, Ris F, Halban PA, Drucker DJ. Glucagon-like peptide-1 receptor signaling modulates beta cell apoptosis. J Biol Chem. 2003;278:471–8. doi: 10.1074/jbc.M209423200. [DOI] [PubMed] [Google Scholar]
- 66.Chaudhuri A, Ghanim H, Vora M, Sia CL, Korzeniewski K, Dhindsa S, et al. Exenatide exerts a potent antiinflammatory effect. J Clin Endocrinol Metab. 2012;97:198–207. doi: 10.1210/jc.2011-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DeFronzo RA, Triplitt C, Qu Y, Lewis MS, Maggs D, Glass LC. Effects of exenatide plus rosiglitazone on beta-cell function and insulin sensitivity in subjects with type 2 diabetes on metformin. Diabetes Care. 2010;33:951–7. doi: 10.2337/dc09-1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bunck MC, Diamant M, Corner A, Eliasson B, Malloy JL, Shaginian RM, et al. One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: a randomized, controlled trial. Diabetes Care. 2009;32:762–8. doi: 10.2337/dc08-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69•.Bunck MC, Corner A, Eliasson B, Heine RJ, Shaginian RM, Taskinen MR, et al. Effects of exenatide on measures of beta-cell function after 3 years in metformin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2041–7. doi: 10.2337/dc11-0291. This randomized clinical trial showed that following 3 years of treatment with exenatide, β-cell function was sustained after a 4-week off-drug period suggesting a benefical effect of exenatide on β-cell health in metformin-treated patients with T2DM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70••.Gallwitz B, Guzman J, Dotta F, Guerci B, Simo R, Basson BR, et al. Exenatide twice daily versus glimepiride for prevention of glycaemic deterioration in patients with type 2 diabetes with metformin failure (EUREXA): an open-label, randomised controlled trial. Lancet. 2012;379:2270–8. doi: 10.1016/S0140-6736(12)60479-6. A large multicenter open label randomized controlled trial showing that exenatide had more durable effects on glycemic control compared to glimepiride over a 48 month follow up period in patients with T2DM inadequately controlled with metformin alone. [DOI] [PubMed] [Google Scholar]
- 71••.Astrup A, Rossner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374:1606–16. doi: 10.1016/S0140-6736(09)61375-1. A large multicenter randomized controlled trial showing that treatment with liraglutide resulted in signicantly greater weight loss and a reduction in the prevalence of prediabetes in obese adults. [DOI] [PubMed] [Google Scholar]
- 72•.Rosenstock J, Klaff LJ, Schwartz S, Northrup J, Holcombe JH, Wilhelm K, et al. Effects of exenatide and lifestyle modification on body weight and glucose tolerance in obese subjects with and without pre-diabetes. Diabetes Care. 2010;33:1173–5. doi: 10.2337/dc09-1203. Randomized trial showing that a 24-week treatment with exenatide plus lifestyle modification resulted in greater weight loss and normalization of glucose tolerance in 77 % of obese participants with impaired glucose homeostasis compared to 56 % in the placebo group. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gleason CE, Gonzalez M, Harmon JS, Robertson RP. Determinants of glucose toxicity and its reversibility in the pancreatic islet beta-cell line, HIT-T15. Am J Physiol Endocrinol Metab. 2000;279:E997–1002. doi: 10.1152/ajpendo.2000.279.5.E997. [DOI] [PubMed] [Google Scholar]
- 74.Ilkova H, Glaser B, Tunckale A, Bagriacik N, Cerasi E. Induction of long-term glycemic control in newly diagnosed type 2 diabetic patients by transient intensive insulin treatment. Diabetes Care. 1997;20:1353–6. doi: 10.2337/diacare.20.9.1353. [DOI] [PubMed] [Google Scholar]
- 75.Weng J, Li Y, Xu W, Shi L, Zhang Q, Zhu D, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multi-centre randomised parallel-group trial. Lancet. 2008;371:1753–60. doi: 10.1016/S0140-6736(08)60762-X. [DOI] [PubMed] [Google Scholar]
- 76.Chen HS, Wu TE, Jap TS, Hsiao LC, Lee SH, Lin HD. Beneficial effects of insulin on glycemic control and beta-cell function in newly diagnosed type 2 diabetes with severe hyperglycemia after short-term intensive insulin therapy. Diabetes Care. 2008;31:1927–32. doi: 10.2337/dc08-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harrison LB, Adams-Huet B, Raskin P, Lingvay I. beta-cell function preservation after 3.5 years of intensive diabetes therapy. Diabetes Care. 2012;35:1406–12. doi: 10.2337/dc11-2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu Y, Li L, Xu Y, Yu T, Tong G, Huang H, et al. Short-term intensive therapy in newly diagnosed type 2 diabetes partially restores both insulin sensitivity and beta-cell function in subjects with long-term remission. Diabetes Care. 2011;34:1848–53. doi: 10.2337/dc10-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gerstein HC, Bosch J, Dagenais GR, Diaz R, Jung H, Maggioni AP, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. 2012;367:319–28. doi: 10.1056/NEJMoa1203858. [DOI] [PubMed] [Google Scholar]
- 80.DeFronzo RA, Abdul-Ghani MA. Preservation of beta-cell function: the key to diabetes prevention. J Clin Endocrinol Metab. 2011;96:2354–66. doi: 10.1210/jc.2011-0246. [DOI] [PubMed] [Google Scholar]
- 81.Dixon JB, le Roux CW, Rubino F, Zimmet P. Bariatric surgery for type 2 diabetes. Lancet. 2012;379:2300–11. doi: 10.1016/S0140-6736(12)60401-2. [DOI] [PubMed] [Google Scholar]
- 82.Buchwald H, Estok R, Fahrbach K, Banel D, Jensen MD, Pories WJ, et al. Weight and type 2 diabetes after bariatric surgery: systematic review and meta-analysis. Am J Med. 2009;122:248–56e5. doi: 10.1016/j.amjmed.2008.09.041. [DOI] [PubMed] [Google Scholar]
- 83.Bradley D, Magkos F, Klein S. Effects of bariatric surgery on glucose homeostasis and type 2 diabetes. Gastroenterology. 2012;143:897–912. doi: 10.1053/j.gastro.2012.07.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dixon JB, O’Brien PE, Playfair J, Chapman L, Schachter LM, Skinner S, et al. Adjustable gastric banding and conventional therapy for type 2 diabetes: a randomized controlled trial. JAMA. 2008;299:316–23. doi: 10.1001/jama.299.3.316. [DOI] [PubMed] [Google Scholar]
- 85••.Schauer PR, Kashyap SR, Wolski K, Brethauer SA, Kirwan JP, Pothier CE, et al. Bariatric surgery versus intensive medical therapy in obese patients with diabetes. N Engl J Med. 2012;366:1567–76. doi: 10.1056/NEJMoa1200225. Randomized controlled single center trial showing that RYGB surgery was more effective than intensive medical therapy in achieving remission of diabetes at 12 months of follow up in obese adults. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86••.Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Leccesi L, et al. Bariatric surgery versus conventional medical therapy for type 2 diabetes. N Engl J Med. 2012;366:1577–85. doi: 10.1056/NEJMoa1200111. Randomized controlled single center trial that showed at 2 years of follow up diabetes remission had occurred in no patients in the medical therapy group versus 75 % in the RYGB surgery group and 95 % in the biliopancreatic-diversion group. [DOI] [PubMed] [Google Scholar]
- 87.Sjostrom L, Lindroos AK, Peltonen M, Torgerson J, Bouchard C, Carlsson B, et al. Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med. 2004;351:2683–93. doi: 10.1056/NEJMoa035622. [DOI] [PubMed] [Google Scholar]
- 88.Chikunguwo SM, Wolfe LG, Dodson P, Meador JG, Baugh N, Clore JN, et al. Analysis of factors associated with durable remission of diabetes after Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2010;6:254–9. doi: 10.1016/j.soard.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 89••.Carlsson LM, Peltonen M, Ahlin S, Anveden A, Bouchard C, Carlsson B, et al. Bariatric surgery and prevention of type 2 diabetes in Swedish obese subjects. N Engl J Med. 2012;367:695–704. doi: 10.1056/NEJMoa1112082. Large prospective case–control study showing that bariatric surgery as compared to usual care significanlty reduced the incidence of diabetes in obese patients, particularly among obese patients with impaired fasting glucose. [DOI] [PubMed] [Google Scholar]