

Abstract
Purpose
To identify a robust prognostic gene expression signature as an independent predictor of survival of patients with acute myeloid leukemia (AML) and use it to improve established risk classification.
Patients and Methods
Four independent sets totaling 499 patients with AML carrying various cytogenetic and molecular abnormalities were used as training sets. Two independent patient sets composed of 825 patients were used as validation sets. Notably, patients from different sets were treated with different protocols, and their gene expression profiles were derived using different microarray platforms. Cox regression and Kaplan-Meier methods were used for survival analyses.
Results
A prognostic signature composed of 24 genes was derived from a meta-analysis of Cox regression values of each gene across the four training sets. In multivariable models, a higher sum value of the 24-gene signature was an independent predictor of shorter overall (OS) and event-free survival (EFS) in both training and validation sets (P < .01). Moreover, this signature could substantially improve the European LeukemiaNet (ELN) risk classification of AML, and patients in three new risk groups classified by the integrated risk classification showed significantly (P < .001) distinct OS and EFS.
Conclusion
Despite different treatment protocols applied to patients and use of different microarray platforms for expression profiling, a common prognostic gene signature was identified as an independent predictor of survival of patients with AML. The integrated risk classification incorporating this gene signature provides a better framework for risk stratification and outcome prediction than the ELN classification.
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous group of hematopoietic malignancies with diverse cytogenetic and molecular abnormalities and variable responses to treatment.1–5 Many gene expression profiling studies have been performed in AML using DNA microarrays,6–21 and the results have proven to be valuable in diagnosis, classification, and prognosis.22–24 However, gene signatures from different laboratories are not always consistent.23 Indeed, because of the lack of consistency between different patient sets, no gene signature has been incorporated into well-established risk classifications so far.
To identify genes that are consistently associated with prognosis of patients with AML, we used four independent patient sets comprising 499 patients as training sets. We performed Cox regression to assess the association of gene expression values with overall survival (OS) of patients in each training set and then conducted a meta-analysis of Cox regression values of each individual gene across the four training sets to identify the genes with the most consistent and robust prognostic impact. We identified 24 such genes and then derived a prognostic gene signature. The prognostic impact of this gene signature was confirmed in two independent validation sets totaling 825 patients in both OS and event-free survival (EFS) models. More importantly, we showed that this gene signature could substantially improve the widely accepted European LeukemiaNet (ELN) risk classification25 of AML. Thus, we developed an integrated risk classification, which provided better risk stratification and outcome prediction than the ELN risk classification in both test and validation sets.
PATIENTS AND METHODS
Additional information about methods is provided in the Data Supplement. Figure 1 illustrates the entire study design.
Fig 1.

Overview of research design and work flow. (A) Identification and validation of 24-gene signature; (B) development and validation of integrated risk classification scheme. AML, acute myeloid leukemia; APL, acute promyelocytic leukemia; CALGB, Cancer and Leukemia Group B; EFS, event-free survival; ELN, European LeukemiaNet; FDR, false discovery rate; HOVON, Hemato-Oncologie voor Volwassenen Nederland; OS, overall survival.
Patient Samples
All samples of patients with AML were obtained at time of diagnosis and with informed consent at corresponding hospitals, and study protocols were approved by the institutional review boards of corresponding institutes and hospitals. All patients were treated according to the protocols of corresponding institutes and hospitals. The clinical and molecular characteristics of these patients are provided in the Data Supplement.
Gene Expression Profiling
Microarray assays of Germany Set 1 (n = 106),10 Netherlands Set 1 (n = 241),11 and Netherlands Set 2 (n = 277)17 were conducted using Stanford cDNA (Stanford University, Stanford, CA), Affymetrix U133A (Affymetrix, Santa Clara, CA), and Affymetrix U133 Plus2.0 arrays (Affymetrix), respectively. USA Sets 1 (n = 65; data for 35 patients were reported previously26) and 2 (n = 87)26 were analyzed using an Agilent custom-designed microarray (Agilent Technologies, Santa Clara, CA) and Affymetrix GeneChip Human Exon 1.0 ST array (Affymetrix), respectively. Germany Set 2 (n = 548) was generated on Affymetrix U133A+B and Affymetrix U133 Plus2.0 arrays. Data analyses are described in the Data Supplement. The microarray data have been deposited in the Gene Expression Omnibus, and the accession numbers include GSE425, GSE1159, GSE14468, GSE30258, GSE30285, and GSE37642.
Statistical Analyses
OS was measured from the date the patient was enrolled onto the study until the date of death, and patients alive at last follow-up were censored. EFS was measured from the date of entry into a study to the date of induction treatment failure, relapse from complete remission, or death resulting from any cause; patients not known to have any of these events at last follow-up were censored. Survival was estimated according to the Kaplan-Meier method. The log-rank test was used to assess statistical significance. Cox regression was used to assess the association of a given variable with OS or EFS. Multivariable testing was performed using Cox proportional hazards models. P values < .05 were considered statistically significant.
Identification of 24 Consistently Prognostic Genes From Four Training Sets via Meta-Analysis
Cox regression was used to estimate the prognostication of OS for each gene in each training set. A meta-analysis was then conducted on the Cox-regression P values of OS from the four training sets using the Stouffer method27:
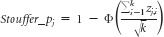 , where Zi=Φ−1(1−pi), pi is a P value for the ith study of k studies in total, and Φ and Φ−1 denote the standard normal cumulative distribution function and its inverse. A total of 24 genes (Data Supplement) remained significant predictors after false discovery rate (FDR) adjustment for multiple comparisons of meta-analysis of P values (FDR < 0.05; R Stats Package (http://www.r-project.org); Hommel adjustment28). For genes with multiple probe sets (transcripts), the average expression value for a given gene was used in all analyses. Notably, our meta-analysis did not require cross-platform normalization to combine the four individual training sets into a single set, thus avoiding potential biases resulting from cross-platform normalization.
, where Zi=Φ−1(1−pi), pi is a P value for the ith study of k studies in total, and Φ and Φ−1 denote the standard normal cumulative distribution function and its inverse. A total of 24 genes (Data Supplement) remained significant predictors after false discovery rate (FDR) adjustment for multiple comparisons of meta-analysis of P values (FDR < 0.05; R Stats Package (http://www.r-project.org); Hommel adjustment28). For genes with multiple probe sets (transcripts), the average expression value for a given gene was used in all analyses. Notably, our meta-analysis did not require cross-platform normalization to combine the four individual training sets into a single set, thus avoiding potential biases resulting from cross-platform normalization.
Derivation of the 24-Gene Signature
As described previously,26,29 a compound covariate30 was derived for each sample by computing a linear combination of expression values of the 24 genes. The value of the compound covariate for patient i was ci = Σ wj xij, where xij is the log-transformed expression value for probe set j in patient i, and wj is the weight assigned to probe set j (here wj was set equal to the average Cox regression correlation coefficient from the four training sets for each gene; Data Supplement). The sum was over all 24 genes.
RESULTS
Identification of a Common Prognostic 24-Gene Signature in AML via Meta-Analysis of Four Independent AML Sets
We collected four independent cohorts for a total of 499 patients with AML, including USA Set 1 (n = 65), USA Set 2 (n = 87), Netherlands Set 1 (n = 241), and Germany Set 1 (n = 106). All cohorts contained at least four different cytogenetic subtypes of AML, and except for USA Set 2, all contained cytogenetically normal AML (Data Supplement). Notably, these four data sets were generated by four different array platforms; patients were collected from different institutions and countries, and importantly, they were treated with distinct protocols (Patients and Methods; Fig 1). We performed univariable Cox regression analysis to assess the association of expression levels of each gene with OS of patients in each training set, followed by a meta-analysis of Cox regression P values. We identified 24 genes (including ALS2CR8, ANGEL1, ARL6IP5, BSPRY, BTBD3, C1RL, CPT1A, DAPK1, ETFB, FGFR1, HEATR6, LAPTM4B, MAP7, NDFIP1, PBX3, PLA2G4A, PLOD3, PTP4A3, SLC25A12, SLC2A5, TMEM159, TRIM44, TRPS1, and VAV3), the increased expression levels of which were significantly associated with worse (22 genes) or favorable (two genes: FGFR1 and PLOD3) OS after FDR adjustment for multiple tests across four training sets (FDR < 0.05; Hommel adjustment28; Data Supplement). Interestingly, based on the shortest-paths method of GeneGO (Carlsbad, CA), we show that among the 24 genes, five genes (FGFR1, LAPTM4B, PLOD3, TRPS1, and VAV3) form a mini-network, as do nine other genes (ARL6IP5/JWA, DAPK1, CPT1A, MAP7, PBX3, PLA2G4A, PTP4A3, SLC25A12, and TRIM44; Data Supplement), highlighting the frequent interactions among the genes within this signature.
We then derived a gene sum-value signature, which is a linear combination26,30 of the expression values of the 24 genes weighted by the average correlation coefficient (Patients and Methods). Kaplan-Meier and univariable Cox regression analyses demonstrated that patients with AML with a higher sum value experienced significantly (P < .01) shorter OS in all four training sets (Data Supplement). Through multivariable testing, we revealed that the increased sum value of the 24-gene signature remained significantly (P < .05) associated with poor OS in all four training sets (Data Supplement), after adjusting for all other variables that have a P < .1 in univariable analyses (Data Supplement).
Increased Sum Value of 24-Gene Signature Is Independent Predictor of Poor Survival in Two Independent Validation Sets
We then validated the prognostic impact of this gene signature in two independent AML sets, namely Netherlands (n = 277) and Germany Sets 2 (n = 548). As expected, Kaplan-Meier and univariable Cox regression analyses demonstrated that increased sum value of the signature was significantly (P < .01) associated with shorter OS and EFS (Fig 2; Data Supplement). Our gene signature performed well for both younger (age < 60 years) and older patients (Data Supplement) and patients with de novo and non–de novo AML (Data Supplement). Through multivariable testing, we showed that the increased sum value of the 24-gene signature remained significantly associated with worse OS and EFS in both validation sets (Table 1), after adjusting for all other variables [including age, WBC count, CEBPA mutation, NPM1 mutation, KRAS mutation, FLT3 internal tandem duplication (ITD), t(9;11), t(11q23), t(8;21), inv(16), t(15;17), +8, complex, del(3q)/inv(3)/t(3;3), −7/del(7q), and/or other AML types] that had P < .1 in univariable analyses (Data Supplement).
Fig 2.
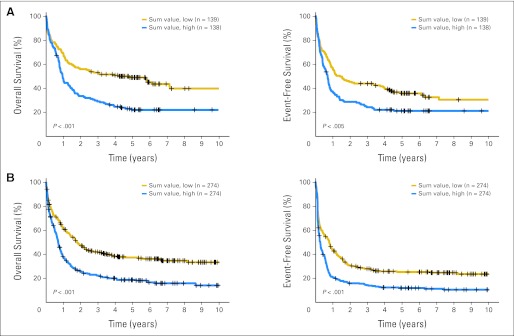
Survival curves of patients with acute myeloid leukemia (AML) in the two validation sets ([A] Netherlands Set 2, n = 277; [B] Germany Set 2, n = 548) predicted by the sum-value signature of 24 genes. Patients in each set were dichotomized into two groups based on median value of the sum-value signature, and Kaplan-Meier curves were generated to depict outcomes. P values were determined by log-rank test. Plus signs indicate censored patients. Patients with AML with higher sum values experienced significantly shorter (P < .005) overall (left panels) and event-free survival (right panel) rates than those with lower sum values in (A) Netherlands Set 2 and (B) Germany Set 2.
Table 1.
Multivariable Analyses of OS and EFS of Patients With AML From Validation Sets*
| Variable | HR | 95% CI | P |
|---|---|---|---|
| OS | |||
| Netherlands Set 2 (n = 277) | |||
| 24-gene signature: high v low | 1.9 | 1.4 to 2.9 | < .001 |
| KRAS mutation v wild type | 92.2 | 23.9 to 356.3 | < .001 |
| Complex v others | 3.8 | 2.2 to 6.4 | < .001 |
| FLT3-ITD v others | 1.4 | 1.0 to 2.1 | .04 |
| t(8;21) v others | 0.4 | 0.1 to 1.0 | .04 |
| Age group: ≥ 60 v < 60 years | 2.1 | 1.5 to 2.9 | < .001 |
| Germany Set 2 (n = 548) | |||
| 24-gene signature: high v low | 1.4 | 1.1 to 1.8 | .002 |
| FLT3-ITD v others | 1.5 | 1.2 to 1.9 | < .001 |
| inv(16) v others | 0.3 | 0.2 to 0.6 | < .001 |
| t(15;17) v others | 0.4 | 0.2 to 0.6 | < .001 |
| Complex v others | 2.1 | 1.5 to 2.9 | < .001 |
| Age group: ≥ 60 v < 60 years | 1.7 | 1.4 to 2.1 | < .001 |
| t-AML v others | 2.8 | 1.3 to 6.1 | .01 |
| EFS | |||
| Netherlands Set 2 (n = 277) | |||
| 24-gene signature: high v low | 1.7 | 1.2 to 2.2 | .001 |
| KRAS mutation v wild type | 87.3 | 23.2 to 328.3 | < .001 |
| Complex v others | 3.0 | 1.8 to 4.8 | < .001 |
| t(15;17) v others | 3.1 | 1.6 to 5.9 | < .001 |
| t(8;21) v others | 0.4 | 0.2 to 0.9 | .04 |
| del(3q) v others | 2.1 | 1.1 to 4.0 | .03 |
| Age group: ≥ 60 v < 60 years | 1.8 | 1.3 to 2.5 | < .001 |
| Germany Set 2 (n = 548) | |||
| 24-gene signature: high v low | 1.4 | 1.1 to 1.7 | .005 |
| NPM1 mutation v wild type | 0.5 | 0.4 to 0.7 | < .001 |
| FLT3-ITD v others | 1.6 | 1.3 to 2.1 | < .001 |
| inv(16) v others | 0.4 | 0.2 to 0.7 | .002 |
| −7/del(7q) v others | 2.1 | 1.2 to 3.6 | .01 |
| inv(3)/t(3;3) v others | 3.0 | 1.6 to 5.4 | < .001 |
| Complex v others | 2.2 | 1.6 to 3.0 | < .001 |
| Age group: ≥ 60 v < 60 years | 1.5 | 1.3 to 1.9 | < .001 |
| t-AML v others | 2.9 | 1.4 to 6.0 | .005 |
NOTE. The following variables were evaluated in univariable Cox regression models for outcome: sum-value signature, age, sex, platelet count, WBC, percentage of blood blasts, percentage of bone marrow blasts, presence or absence of various chromosomal translocations [ie, inv(16), t(8;21), t(15;17), t(9;11), t(11q23), t(6;9), t(9;22)] or other abnormalities [+8, −3/inv(3q)/t(3;3), −7/del(7q)], and presence or absence of gene mutations (ie, FLT3-ITD, FLT3-TKD, NRAS, KRAS, NPM1, CEBPA, MLL-PTD). Variables for which P < .1 in univariable models (Data Supplement) were included in multivariable analysis for sum-value signature. Only variables for which P < .05 in multivariable models are shown in Table. HRs > 1 or < 1 indicate, respectively, higher or lower risk of event for higher values of continuous variables and for first category listed for categorical variables in OS or EFS models.
Abbreviations: AML, acute myeloid leukemia; EFS, event-free survival; HR, hazard ratio; ITD, internal tandem duplication; OS, overall survival; PTD, partial tandem duplication; t-AML, therapy-related AML; TKD, tyrosine kinase domain.
24-Gene Signature Provides Additional Prognostic Power to ELN Risk Classification of AML
We then sought to investigate whether this gene signature provides additional prognostic value to established risk stratification systems. Recently, an international expert panel on behalf of the ELN recommended a risk classification scheme for adult AML (acute promyelocytic leukemia was excluded) based on major cytogenetic and molecular genetic data (Data Supplement).25 All patient samples studied in Netherlands Set 111 (n = 241) had been reanalyzed using Affymetrix U133 Plus2.0 arrays (Affymetrix) along with Netherlands Set 217 (n = 277; Fig 1). Thus, we had Affymetrix U133 Plus2.0 platform–derived expression data for all 518 patients, of whom 480 (termed Netherlands Set 2+; Data Supplement) could be classified into either the ELN favorable (n = 161), intermediate-I (n = 133), intermediate-II (n = 108), or adverse (n = 78) risk group (Data Supplement). An increased sum value of the 24-gene signature remained an independent predictor of shorter OS and EFS in Netherlands Set 2+ after adjusting for other variables (Data Supplement).
As shown in the Data Supplement, in both OS and EFS models, there was no significant (P > .1) difference between intermediate-I and intermediate-II risk groups in either the entire set of patients or those younger or older than age 60 years, suggesting that the ELN risk classification needs to be improved. We then incorporated the sum-value information (ie, low or high) of the 24-gene signature into the ELN risk classification and found that within each ELN risk group, patients with a higher sum value of 24-gene signature experienced much shorter OS or EFS (Data Supplement). Thus, the 24-gene prognostic signature could provide additional prognostic power to the ELN risk classification of AML.
Development of an Integrated Risk Classification Scheme
On the basis of the results shown in the Data Supplement, we developed an integrated risk classification scheme through integration of the ELN risk classification and the 24-gene signature classification (Fig 3A): favorable, ELN favorable-risk patients with a low sum value of the 24-gene signature; intermediate, ELN favorable-risk patients with a high sum value and ELN intermediate-I– or intermediate-II–risk patients with a low sum value; and adverse, ELN intermediate-I– or intermediate-II–risk patients with a high sum value and all ELN adverse-risk patients. We then applied this new risk classification to the test set (ie, Netherlands Set 2+, n = 480; Fig 3B). Clearly, this new integrated risk classification separated the patients into three distinct risk groups, with significant (P < .05) differences in both OS and EFS of patients in Netherlands Set 2+ (Fig 3C). Similar patterns were observed when either the younger or older patients were considered separately (Figs 3D, 3E).
Fig 3.

Distribution and survival of patients with acute myeloid leukemia (AML) in the test set (Netherlands Set 2+, n = 480) according to or predicted by integrated risk categories. (A) Scheme of reclassification of the four European LeukemiaNet (ELN) risk groups into three new integrated risk groups by integrating the 24-gene signature classification (ie, low or high) with ELN risk classification. (B) Distribution of the whole set of patients, those age < 60 years, and those age > 60 years according to integrated risk class criteria. (C, D, E) Overall (left panels) and event-free survival (right panels) rates of (C) the whole set of patients, (D) those age < 60 years, and (E) those age > 60 years predicted by integrated risk categories.
Validation and Evaluation of the Integrated Risk Classification
A total of 484 patients from Germany Set 2 could be classified into four ELN risk groups (Fig 4A); this subset was termed Germany Set 2′ (Data Supplement) and was used as the validation set. The 484 patients of Germany Set 2′ were then reclassified into three risk groups based on our new integrated risk classification (Fig 4A). On the basis of the ELN risk classification, the intermediate-I, intermediate-II, and adverse groups had fairly similar OS and EFS (Table 2; Fig 4A), indicating that ELN risk classification did not clearly distinguish the risk stratification of those patients. In contrast, on the basis of our new integrated risk classification, the favorable risk group experienced significantly longer OS (P< .001) and EFS (P = .01) than the intermediate risk group, and the latter experienced significantly longer OS (P< .001) and EFS (P< .001) than the adverse risk group (Fig 4A). Median OS for the favorable, intermediate, and adverse risk groups were 3.8 (95% CI, 3.1 to 5.5), 1.2 (95% CI, 0.9 to 1.8), and 0.6 years (95% CI, 0.5 to 0.7), respectively (Table 2). A similar trend was observed in younger (age < 60 years; Data Supplement) and older (age ≥ 60 years; Data Supplement) patients (Table 2). As expected, the higher risk grade of the integrated risk class (favorable, 1; intermediate, 2; adverse, 3) remained an independent predictor of shorter OS and EFS after adjusting other variables (Data Supplement). The statistics of patients with risk classification changes between the ELN risk and our integrated risk classifications is summarized in the Data Supplement.
Fig 4.

Validation and further evaluation of new integrated risk classification. (A) Distribution (upper panels) and overall survival (OS; middle panels) or event-free survival (lower panels) of patients with acute myeloid leukemia in validation set (Germany Set 2′, n = 484) according to or predicted by the European LeukemiaNet (ELN; left panels) or integrated (right panels) risk categories. Additional predictive value displayed by prediction error curves for OS in (B) Netherlands Set 2+ and (C) Germany Set 2′. Lower curve (ie, lower prediction error) indicates better predictive value. Reference line indicates Kaplan-Meier estimation without additional variables; integrated risk class indicates combined prediction model based on ELN and 24-gene signature; 24 gene signature (categorial) indicates model based on 24-gene signature categorized by median (ie, as high or low); ELN indicates model based on ELN risk classification; 24-gene signature (numerical) indicates model based on numerical 24-gene signature.
Table 2.
CR Rate and Median OS and EFS of Distinct Risk Groups in Germany Set 2′
| Risk Group | Whole Set (n = 484) |
Patients Age < 60 Years (n = 255) |
Patients Age ≥ 60 Years (n = 229) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CR (%) | OS |
EFS |
CR (%) | OS |
EFS |
CR (%) | OS |
EFS |
|||||||
| Median (years) | 95% CI | Median (years) | 95% CI | Median (years) | 95% CI | Median (years) | 95% CI | Median (years) | 95% CI | Median (years) | 95% CI | ||||
| ELN risk classification | |||||||||||||||
| Favorable | 72 | 3.8 | 2.7 to 4.8 | 1.8 | 1.2 to 3.2 | 76 | 4.4 | 3.3 to 5.8 | 2.5 | 1.6 to 4.8 | 63 | 1.7 | 0.6 to 3.9 | 0.9 | 0.3 to 2.0 |
| Intermediate-I | 60 | 0.7 | 0.6 to 0.9 | 0.4 | 0.2 to 0.6 | 62 | 0.7 | 0.6 to 1.1 | 0.5 | 0.2 to 0.7 | 59 | 0.7 | 0.5 to 0.8 | 0.3 | 0.2 to 0.5 |
| Intermediate-II | 53 | 1.0 | 0.8 to 1.5 | 0.3 | 0.2 to 0.7 | 62 | 1.4 | 0.8 to 2.3 | 0.7 | 0.2 to 1.4 | 44 | 0.6 | 0.1 to 1.3 | 0.1 | 0.1 to 0.7 |
| Adverse | 34 | 0.5 | 0.4 to 0.7 | 0.2 | 0.1 to 0.2 | 36 | 0.6 | 0.3 to 1.0 | 0.2 | 0.1 to 0.3 | 33 | 0.4 | 0.3 to 0.6 | 0.2 | 0.1 to 0.2 |
| Integrated risk classification | |||||||||||||||
| Favorable | 75 | 3.8 | 3.1 to 5.5 | 1.8 | 1.3 to 3.3 | 80 | 4.2 | 3.1 to 6.4 | 2.5 | 1.6 to 4.3 | 66 | 3.5 | 1.1 to 6.5 | 1.2 | 0.2 to 3.3 |
| Intermediate | 60 | 1.2 | 0.9 to 1.8 | 0.7 | 0.3 to 0.9 | 64 | 2.1 | 1.0 to 4.2 | 1.0 | 0.4 to 1.9 | 54 | 0.9 | 0.5 to 1.3 | 0.4 | 0.1 to 0.8 |
| Adverse | 45 | 0.6 | 0.5 to 0.7 | 0.2 | 0.2 to 0.3 | 48 | 0.8 | 0.6 to 1.0 | 0.2 | 0.2 to 0.3 | 42 | 0.5 | 0.4 to 0.6 | 0.2 | 0.1 to 0.2 |
Abbreviations: CR, complete remission; EFS, event-free survival; ELN, European LeukemiaNet; OS, overall survival.
Moreover, we performed a Brier score analysis31 and calculated prediction error curves32 in Netherlands Set 2+ (n = 480) and Germany Set 2′ (n = 484). We showed that the 24-gene signature provided additional prognostic value to the ELN risk classification, and the integrated risk classification provided higher prediction accuracy than the ELN risk classification (Figs 4B and 4C).
DISCUSSION
Despite different treatment regimens used in the various patient groups, and different microarray platforms and normalization strategies employed in data generation (Patients and Methods; Fig 1), we identified a common prognostic gene signature composed of 24 genes (Data Supplement) from four independent training sets (Fig 1; Data Supplement). The prognostic impact of this signature was subsequently confirmed in two independent validation sets (Figs 1, 2; Data Supplement). Thus, our study indicates that despite the heterogeneous genetic lesions found in AML, a common prognostic gene signature can be derived, and the differential expression of the genes contained in this signature likely contributes to the distinct outcome among various subtypes of AML. Our strategy to identify the most robust and consistent gene signature demonstrated that directly comparing different data sets obtained by different groups using different microarray platforms and normalization methods can substantially minimize false-positive discoveries caused by potential systematic bias of any particular platform or normalization method. Therefore, this strategy might also be applicable in identifying such gene signatures in other types of cancers.
More importantly, we showed that this 24-gene signature could substantially improve the ELN risk classification (Table 2; Figs 1, 3, 4; Data Supplement). In both testing and validation sets, no significant difference in OS or EFS was observed between the ELN intermediate-I and intermediate-II groups (Data Supplement). A similar pattern was previously observed in elderly patients with AML (age > 60 years).33 To improve the ELN risk classification, we developed a new integrated risk classification, which provided a better risk stratification and outcome prediction in both the test and validation sets (Table 2; Figs 3, 4; Data Supplement).
In the clinical setting, it is important to identify high-risk patients with poor prognosis, for whom improved/novel treatment strategies are needed. In fact, in Netherlands Set 2+ (n = 480), median OS times for the ELN favorable, intermediate-I, intermediate-II, and adverse risk groups were 5.1, 1.1, 1.6, and 0.8 years, respectively; in Germany Set 2′ (n = 484), median OS times were 3.8, 0.7, 1.0, and 0.5 years, respectively; similarly, in younger patients (age ≤ 60 years) in the data set of Rollig et al,33 median OS times were 5.3, 1.1, 1.6, and 0.5 years, respectively. It seems the outcomes of many patients in the ELN intermediate risk groups were indeed poor; thus, it is critical to reclassify them into the adverse risk group. With our integrated risk classification, median OS times for patients in the favorable, intermediate, and adverse risk groups in Netherlands Set 2+ became 6.1, 2.6, and 0.8 years, respectively; in Germany Set 2′, median OS times were 3.8, 1.2, and 0.6 years, respectively. Therefore, our integrated risk classification seems to be more clinically useful than the ELN risk classification in identification of high-risk patients.
Recently, extensive efforts have been devoted to identifying prognostic gene mutations.34 In addition to CEBPA mutations,17,35–39 FLT3-ITD,35,40–44 and NPM1 mutations,45–49 which have been incorporated into the ELN risk classification, mutations with prognostic implications in a number of other genes (eg, TET2,50 ASXL1,51 DNMT3A,52–54 p53,55–58 and KIT59) have also been identified.25 TET2 and ASXL1 mutations have also been shown to be able to improve the risk stratification of the ELN favorable-risk patient group.50,51 We expect that there are some correlations between gene mutations and expression levels of the 24-gene signature. Indeed, we found that CEBPA mutations were significantly associated with low levels of expression of the 24-gene signature in both Netherlands Set 2+ and Germany Set 2′; the opposite was true for FLT3-ITD, whereas the association of NPM1 mutations with high-level signature expression was likely the result of frequently coexisting FLT3-ITD (Data Supplement). In Netherlands Set 2+, DNMT3A mutations were significantly correlated with high levels of expression of the 24-gene signature in the whole data set as well as the ELN favorable risk group, whereas no significant correlation was observed for ASXL1 mutations (Data Supplement). Furthermore, several recent studies have shown that the favorable prognostic impact of CEBPA mutations is dependent on biallelic, not monoallelic, mutations.17,37 On the basis of our integrated risk classification, 90% of the patients with biallelic CEBPA mutations (nine of 10 patients), in contrast to approximately only half of those with monoallelic mutations (six of 11 patients), remained in the favorable risk group; the others were classified as belonging to the intermediate risk group. Thus, our integrated risk classification also accurately reflected the difference in prognosis between biallelic and monoallelic CEBPA mutations. Collectively, our gene signature may reflect the sum of the prognostic impacts of many gene mutations.
Given the large number of prognostic gene mutations that have been identified (and more will be identified) in AML,34 it will become increasingly difficult to integrate all prognostic mutations into a risk classification framework. Instead, our integrated risk classification, by relying on a limited number of cytogenetic and molecular abnormalities recommended by ELN plus the 24-gene signature classification, could be easily used in routine clinical practice. A combination of cytogenetic, molecular (using direct sequencing or RNA sequencing to detect gene mutations), and gene expression (using custom-design microarrays, real-time quantitative polymerase chain reaction, or RNA sequencing to assess expression of the 24 genes) assays can be used to classify all patients with AML into one of the three new risk groups, which will provide accurate information for prognostic and therapeutic decisions. Thus, although a large-scale independent, prospective validation trial is needed to prove its robustness, this new integrated risk classification does hold great potential for clinical application in risk stratification and outcome prediction in AML.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: None Stock Ownership: Peter J.M. Valk, Skyline Diagnostics Honoraria: None Research Funding: None Expert Testimony: None Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Stefan K. Bohlander, Jianjun Chen
Financial support: Zejuan Li, Miao Sun, Xinan Yang, Hao Huang, Xi Jiang, Lars Bullinger, Paul P. Liu, Ruud Delwel, Guido Marcucci, Bob Lowenberg, Clara D. Bloomfield, Janet D. Rowley, Stefan K. Bohlander, Jianjun Chen
Administrative support: Zejuan Li, Tobias Herold, Peter J.M. Valk, Maria-Cristina Sauerland, Thomas Büchner, Wolfgang Hiddemann, Richard A. Larson, Michael A. Caligiuri, Konstanze Döhner, Lars Bullinger, Paul P. Liu, Ruud Delwel, Guido Marcucci, Bob Lowenberg, Clara D. Bloomfield, Janet D. Rowley, Stefan K. Bohlander, Jianjun Chen
Provision of study materials or patients: Zejuan Li, Tobias Herold, Peter J.M. Valk, Maria-Cristina Sauerland, Thomas Büchner, Wolfgang Hiddemann, Yanming Zhang, Richard A. Larson, Michelle M. Le Beau, Michael A. Caligiuri, Konstanze Döhner, Lars Bullinger, Paul P. Liu, Ruud Delwel, Guido Marcucci, Bob Lowenberg, Clara D. Bloomfield, Janet D. Rowley, Stefan K. Bohlander, Jianjun Chen
Collection and assembly of data: Zejuan Li, Tobias Herold, Chunjiang He, Peter J.M. Valk, Ping Chen, Michael D. Radmacher, Kati S. Maharry, Miao Sun, Xinan Yang, Hao Huang, Xi Jiang, Maria-Cristina Sauerland, Thomas Büchner, Wolfgang Hiddemann, Abdel Elkahloun, Mary Beth Neilly, Yanming Zhang, Richard A. Larson, Michelle M. Le Beau, Michael A. Caligiuri, Konstanze Döhner, Lars Bullinger, Paul P. Liu, Ruud Delwel, Guido Marcucci, Bob Lowenberg, Clara D. Bloomfield, Janet D. Rowley, Stefan K. Bohlander, Jianjun Chen
Data analysis and interpretation: Zejuan Li, Tobias Herold, Chunjiang He, Vindi Jurinovic, Ulrich Mansmann, Michael D. Radmacher, Kati S. Maharry, Miao Sun, Xinan Yang, Maria-Cristina Sauerland, Thomas Büchner, Wolfgang Hiddemann, Lars Bullinger, Paul P. Liu, Guido Marcucci, Bob Lowenberg, Clara D. Bloomfield, Janet D. Rowley, Stefan K. Bohlander, Jianjun Chen
Manuscript writing: All authors
Final approval of manuscript: All authors
Supplementary Material
Acknowledgment
We thank Masha Kocherginsky, PhD, Theodore Karrison, PhD, and Dezheng Huo, MD, PhD, for data coordination and advice on statistical analyses.
Footnotes
Z.L., T.H., and C.H. contributed equally to this work; S.K.B. and J.C. contributed equally to this work.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Supported in part by the Leukemia and Lymphoma Society (LLS) Translational Research Grant No. 6199-09 (J.D.R., J.C.), Cancer and Leukemia Group B (CALGB) Pilot Grant No. 20801 (J.D.R., J.C.), German Ministry of Education and Research National Genome Research Network Grant No. 01G0876 (S.K.B.), the Dutch Cancer Foundation Queen Wilhelmina Fund (P.J.M.V., R.D., B.L.), National Institutes of Health (NIH) Grant No. R01 CA127277 (J.C.), American Cancer Society Research Scholar Grant No. 120993-RSG-11-156-01-LIB (J.C.), an LLS Special Fellowship (Z.L.), the G. Harold and Leila Y. Mathers Charitable Foundation (J.C.), Gabrielle's Angel Foundation for Cancer Research (Z.L., X.J., H.H., J.C.), CALGB Leukemia Correlative Science Committee Grant No. U10 CA101140 (G.M.), Ohio State University Leukemia Specialized Program of Research Excellence Grant No. P50CA140158 Project 2 (G.M., C.D.B.), Leukemia Clinical Research Foundation grant (G.M., C.D.B.), the NIH Intramural Research Program of the National Human Genome Research Institute (A.E., P.P.L.), Department of Defense Predoctoral Traineeship Award No. W81XWH-10-1-0396 (M.S.), National Natural Science Foundation of China Grant No. 60971099 (X.Y.), Deutsche José Carreras Leukämie Stiftung Grant No. R 06/41v (L.B.), Deutsche Forschungsgemeinschaft Heisenberg-Stipendium Grant No. BU 1339/3-1 (L.B.), NIH Grant No. P01 CA40046 (M.M.L.B.), National Cancer Institute Cancer Center Support Grant No. P30 CA014599 (M.M.L.B.), the University of Chicago Committee on Cancer Biology Fellowship Program (X.J.), the Spastic Paralysis Foundation of the Illinois/Eastern Iowa Branch of Kiwanis International (J.D.R.), and the Fidelity Foundation (J.D.R., J.C.).
REFERENCES
- 1.Löwenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 2.Byrd JC, Mrózek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461) Blood. 2002;100:4325–4336. doi: 10.1182/blood-2002-03-0772. [DOI] [PubMed] [Google Scholar]
- 3.Marcucci G, Mrózek K, Bloomfield CD. Molecular heterogeneity and prognostic biomarkers in adults with acute myeloid leukemia and normal cytogenetics. Curr Opin Hematol. 2005;12:68–75. doi: 10.1097/01.moh.0000149608.29685.d1. [DOI] [PubMed] [Google Scholar]
- 4.Grimwade D, Mrózek K. Diagnostic and prognostic value of cytogenetics in acute myeloid leukemia. Hematol Oncol Clin North Am. 2011;25:1135–1161. doi: 10.1016/j.hoc.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 5.Smith ML, Hills RK, Grimwade D. Independent prognostic variables in acute myeloid leukaemia. Blood Rev. 2011;25:39–51. doi: 10.1016/j.blre.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 7.Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 8.Schoch C, Kohlmann A, Schnittger S, et al. Acute myeloid leukemias with reciprocal rearrangements can be distinguished by specific gene expression profiles. Proc Natl Acad Sci U S A. 2002;99:10008–10013. doi: 10.1073/pnas.142103599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yagi T, Morimoto A, Eguchi M, et al. Identification of a gene expression signature associated with pediatric AML prognosis. Blood. 2003;102:1849–1856. doi: 10.1182/blood-2003-02-0578. [DOI] [PubMed] [Google Scholar]
- 10.Bullinger L, Döhner K, Bair E, et al. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N Engl J Med. 2004;350:1605–1616. doi: 10.1056/NEJMoa031046. [DOI] [PubMed] [Google Scholar]
- 11.Valk PJ, Verhaak RG, Beijen MA, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med. 2004;350:1617–1628. doi: 10.1056/NEJMoa040465. [DOI] [PubMed] [Google Scholar]
- 12.Ross ME, Mahfouz R, Onciu M, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104:3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 13.Haferlach T, Kohlmann A, Schnittger S, et al. Global approach to the diagnosis of leukemia using gene expression profiling. Blood. 2005;106:1189–1198. doi: 10.1182/blood-2004-12-4938. [DOI] [PubMed] [Google Scholar]
- 14.Radmacher MD, Marcucci G, Ruppert AS, et al. Independent confirmation of a prognostic gene-expression signature in adult acute myeloid leukemia with a normal karyotype: A Cancer and Leukemia Group B study. Blood. 2006;108:1677–1683. doi: 10.1182/blood-2006-02-005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bullinger L, Rücker FG, Kurz S, et al. Gene-expression profiling identifies distinct subclasses of core binding factor acute myeloid leukemia. Blood. 2007;110:1291–1300. doi: 10.1182/blood-2006-10-049783. [DOI] [PubMed] [Google Scholar]
- 16.Metzeler KH, Hummel M, Bloomfield CD, et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood. 2008;112:4193–4201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wouters BJ, Löwenberg B, Erpelinck-Verschueren CA, et al. Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood. 2009;113:3088–3091. doi: 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verhaak RG, Wouters BJ, Erpelinck CA, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009;94:131–134. doi: 10.3324/haematol.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rockova V, Abbas S, Wouters BJ, et al. Risk stratification of intermediate-risk acute myeloid leukemia: Integrative analysis of a multitude of gene mutation and gene expression markers. Blood. 2011;118:1069–1076. doi: 10.1182/blood-2011-02-334748. [DOI] [PubMed] [Google Scholar]
- 20.Li Z, Huang H, Chen P, et al. MiR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat Commun. 2012;3:688. doi: 10.1038/ncomms1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X, Huang H, Li Z, et al. Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell. 2012;22:524–535. doi: 10.1016/j.ccr.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Downing J. Profiling in acute leukemia. Clin Adv Hematol Oncol. 2003;1:522–524. [PubMed] [Google Scholar]
- 23.Bullinger L, Valk PJ. Gene expression profiling in acute myeloid leukemia. J Clin Oncol. 2005;23:6296–6305. doi: 10.1200/JCO.2005.05.020. [DOI] [PubMed] [Google Scholar]
- 24.Theilgaard-Mönch K, Boultwood J, Ferrari S, et al. Gene expression profiling in MDS and AML: Potential and future avenues. Leukemia. 2011;25:909–920. doi: 10.1038/leu.2011.48. [DOI] [PubMed] [Google Scholar]
- 25.Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Huang H, Li Y, et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically-abnormal AML. Blood. 2012;119:2314–2324. doi: 10.1182/blood-2011-10-386235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stouffer SA, Suchman EA, DeVinney LC, et al. Adjustment During Army Life. Princeton, NJ: Princeton University Press; 1949. [Google Scholar]
- 28.Hommel G. A stagewise rejective multiple test procedure based on a modified Bonferroni test. Biometrika. 1988;75:383–386. [Google Scholar]
- 29.Marcucci G, Radmacher MD, Maharry K, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1919–1928. doi: 10.1056/NEJMoa074256. [DOI] [PubMed] [Google Scholar]
- 30.Radmacher MD, McShane LM, Simon R. A paradigm for class prediction using gene expression profiles. J Comput Biol. 2002;9:505–511. doi: 10.1089/106652702760138592. [DOI] [PubMed] [Google Scholar]
- 31.Brier GW. Verification of forecasts expressed in terms of probability. Mon Wea Rev. 1950;78:1–3. [Google Scholar]
- 32.Schumacher M, Binder H, Gerds T. Assessment of survival prediction models based on microarray data. Bioinformatics. 2007;23:1768–1774. doi: 10.1093/bioinformatics/btm232. [DOI] [PubMed] [Google Scholar]
- 33.Röllig C, Bornhäuser M, Thiede C, et al. Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: Evaluation of the proposed reporting system. J Clin Oncol. 2011;29:2758–2765. doi: 10.1200/JCO.2010.32.8500. [DOI] [PubMed] [Google Scholar]
- 34.Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–486. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 35.Schlenk RF, Döhner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 36.Preudhomme C, Sagot C, Boissel N, et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: A study from the Acute Leukemia French Association (ALFA) Blood. 2002;100:2717–2723. doi: 10.1182/blood-2002-03-0990. [DOI] [PubMed] [Google Scholar]
- 37.Dufour A, Schneider F, Metzeler KH, et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol. 2010;28:570–577. doi: 10.1200/JCO.2008.21.6010. [DOI] [PubMed] [Google Scholar]
- 38.Fröhling S, Schlenk RF, Stolze I, et al. CEBPA mutations in younger adults with acute myeloid leukemia and normal cytogenetics: Prognostic relevance and analysis of cooperating mutations. J Clin Oncol. 2004;22:624–633. doi: 10.1200/JCO.2004.06.060. [DOI] [PubMed] [Google Scholar]
- 39.Marcucci G, Maharry K, Radmacher MD, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: A Cancer and Leukemia Group B Study. J Clin Oncol. 2008;26:5078–5087. doi: 10.1200/JCO.2008.17.5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Whitman SP, Archer KJ, Feng L, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: A Cancer and Leukemia Group B study. Cancer Res. 2001;61:7233–7239. [PubMed] [Google Scholar]
- 41.Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: Analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 42.Fröhling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML Study Group Ulm. Blood. 2002;100:4372–4380. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 43.Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–2784. doi: 10.1182/blood-2007-08-109090. [DOI] [PubMed] [Google Scholar]
- 44.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 45.Döhner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: Interaction with other gene mutations. Blood. 2005;106:3740–3746. doi: 10.1182/blood-2005-05-2164. [DOI] [PubMed] [Google Scholar]
- 46.Verhaak RG, Goudswaard CS, van Putten W, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005;106:3747–3754. doi: 10.1182/blood-2005-05-2168. [DOI] [PubMed] [Google Scholar]
- 47.Schnittger S, Schoch C, Kern W, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–3739. doi: 10.1182/blood-2005-06-2248. [DOI] [PubMed] [Google Scholar]
- 48.Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML) Blood. 2006;107:4011–4020. doi: 10.1182/blood-2005-08-3167. [DOI] [PubMed] [Google Scholar]
- 49.Schlenk RF, Döhner K, Kneba M, et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica. 2009;94:54–60. doi: 10.3324/haematol.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metzeler KH, Maharry K, Radmacher MD, et al. TET2 mutations improve the new European LeukemiaNet risk classification of acute myeloid leukemia: A Cancer and Leukemia Group B study. J Clin Oncol. 2011;29:1373–1381. doi: 10.1200/JCO.2010.32.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Metzeler KH, Becker H, Maharry K, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011;118:6920–6929. doi: 10.1182/blood-2011-08-368225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marcucci G, Metzeler KH, Schwind S, et al. Age-related prognostic impact of different types of DNMT3A mutations in adults with primary cytogenetically normal acute myeloid leukemia. J Clin Oncol. 2012;30:742–750. doi: 10.1200/JCO.2011.39.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Renneville A, Boissel N, Nibourel O, et al. Prognostic significance of DNA methyltransferase 3A mutations in cytogenetically normal acute myeloid leukemia: A study by the Acute Leukemia French Association. Leukemia. 2012;26:1247–1254. doi: 10.1038/leu.2011.382. [DOI] [PubMed] [Google Scholar]
- 54.Ribeiro AF, Pratcorona M, Erpelinck-Verschueren C, et al. Mutant DNMT3A: A new marker of poor prognosis in acute myeloid leukemia. Blood. 2012;119:5824–5831. doi: 10.1182/blood-2011-07-367961. [DOI] [PubMed] [Google Scholar]
- 55.Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–2121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 56.Tavor S, Rothman R, Golan T, et al. Predictive value of TP53 fluorescence in situ hybridization in cytogenetic subgroups of acute myeloid leukemia. Leuk Lymphoma. 2011;52:642–647. doi: 10.3109/10428194.2010.551571. [DOI] [PubMed] [Google Scholar]
- 57.Seifert H, Mohr B, Thiede C, et al. The prognostic impact of 17p (p53) deletion in 2272 adults with acute myeloid leukemia. Leukemia. 2009;23:656–663. doi: 10.1038/leu.2008.375. [DOI] [PubMed] [Google Scholar]
- 58.Bowen D, Groves MJ, Burnett AK, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23:203–206. doi: 10.1038/leu.2008.173. [DOI] [PubMed] [Google Scholar]
- 59.Paschka P, Marcucci G, Ruppert AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol. 2006;24:3904–3911. doi: 10.1200/JCO.2006.06.9500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.