

Abstract
Apoptosis is critical for organismal homeostasis and a wide variety of diseases. Caspases are the ultimate executors of the apoptotic programmed cell death pathway. As caspases play such a central role in apoptosis, there is significant demand for technologies to monitor caspase function. We recently developed a caspase activatable-GFP (CA-GFP) reporter. CA-GFP is unique due to its “dark” state, where chromophore maturation of the GFP is inhibited by the presence of a C-terminal peptide. Here we show that chromophore maturation is prevented because CA-GFP does not fold into the robust β-barrel of GFP until the peptide has been cleaved by active caspase. Both CA-GFP and GFP1-10, a split form of GFP lacking the 11th strand, have similar secondary structure, different from mature GFP. A similar susceptibility to proteolytic digestion indicates that this shared structure is not the robust, fully formed GFP β-barrel. We have developed a model that suggests that as CA-GFP is translated in vivo it follows the same folding path as wild-type GFP; however, the presence of the appended peptide does not allow CA-GFP to form the barrel of the fully matured GFP. CA-GFP is therefore held in a “pro-folding” intermediate state until the peptide is released, allowing it to continue folding into the mature barrel geometry. This new understanding of the structural basis of the dark state of the CA-GFP reporter will enable manipulation of this mechanism in the development of reporter systems for any number of cellular processes involving proteases and potentially other enzymes.
Keywords: apoptosis, NMR, soluble aggregate, caspase reporter, dark state of GFP
Introduction
Caspases are critical cysteine proteases that mediate apoptotic cell death. Caspases also function in a variety of biological cascades including inflammation and neurodegeneration. Due to the centrality of apoptosis in organismal development and homeostasis as well as many disease states, a full understanding of caspase activity is essential. In particular, there is a demonstrated need for a genetically-encoded reporter of caspase activity that has properties compatible with real-time, longitudinal monitoring of apoptosis in living organisms. Monitoring caspases contributes to our ability to observe and understand apoptosis in the context of organismal development and enables other important pursuits such as predicting drug toxicity mediated by apoptosis. Although a number of caspase reporters have been developed previously, none of them have optimal properties for in depth studies of caspases in many relevant contexts.
We recently reported the development of a genetically-encoded reporter of caspase activity called caspase activatable-GFP (CA-GFP)1 that has many advantageous properties. The genetically-encoded characteristic of CA-GFP makes it amenable to a number of applications that cannot be fulfilled by existing reporters. This reporter is composed of the green fluorescent protein (GFP), the caspase recognition sequence, DEVD, and a quenching peptide derived from the tetramerization domain of influenza M2 protein [Fig. 1(A)]. The presence of the quenching peptide on the C-terminus of GFP changes the oligomeric properties of the protein. Full-length CA-GFP is a mixture of tetramers and higher order oligomers. This oligomerization appears to be related to the function of CA-GFP. When CA-GFP is oligomeric, the chromophore, which provides GFP with fluorescent properties, is not formed and CA-GFP remains in a dark, non-fluorescent state. When CA-GFP is expressed under conditions where active caspases are present, CA-GFP is cleaved and the quenching peptide is released. GFP can then undergo some conformational rearrangement allowing maturation of the chromophore and gain of fluorescence. Thus, response time is dictated by the kinetics of chromophore maturation.
Figure 1.
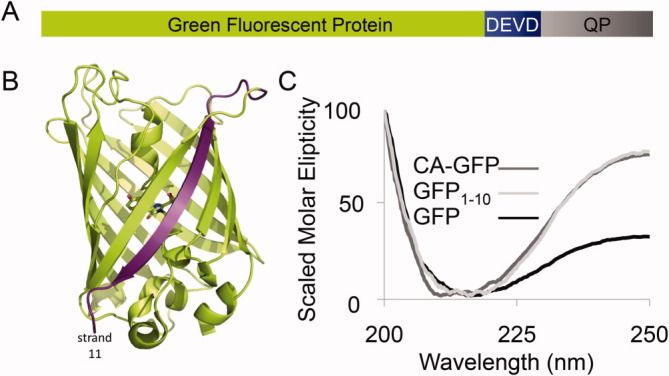
A. CA-GFP is constructed from the Green Fluorescent Protein (S65T) GFP fused to a linker containing the caspase-3 and -7 recognition sequence DEVD to the 27 amino acid quenching peptide derived from the transmembrane domain of influenza M2. B. The structure of the GFP β-barrel (PDB ID:2YOG, green) with the chromophore residues shown as sticks. The 11th strand of the barrel, removed in the GFP1-10 construct is highlighted in purple. C. The CD spectra of GFP, CA-GFP and GFP1-10 are superimposed to allow assessment of the overall shape of the three spectra. The maximum intensity of the three spectra have been scaled for the relative concentration of each protein (Supporting Information Fig. S1). The non-scaled molar intensities of CD spectra for these proteins can be seen in Figure 2.
A number of studies have reported other versions of GFP in which maturation of the chromophore is prevented.2–4 GFP is a β-barrel protein [Fig. 1(B)] in which all the 11 β-strands must be properly assembled to attain the fluorescent state. In each of these darkened states the GFP chromophore is not mature and maturation of the chromophore only occurs when the full GFP barrel assembles. Only when the chromophore-forming residues are in precisely the correct structural environment can the reaction to form the chromophore occur. For all of the split GFP variants a substantial number of amino acids (10–50% of the protein) are missing, so it is not surprising that these proteins do not attain the properly folded state. In contrast, CA-GFP is composed of all the amino acids necessary for GFP to fold. We hypothesize that some aspect of CA-GFP folding may be sub-optimal as chromophore maturation does not occur. Thus we expect that structural changes within the GFP portion of CA-GFP must be present, preventing the chromophore from maturing. These structural perturbations are reflected by alterations to the chromophore maturation properties: ranging from extremely slow (similar to the R96A or R96M mutants which mature in a matter of months5) to entirely prevented (like split versions of GFP).
Due to its response to caspases and the dark state prior to cleavage, CA-GFP has been a robust reporter of caspase activity. In bacteria heterologously expressing caspases, we observe a 50-fold increase in fluorescence after caspase cleavage. In mammalian systems, where low levels of active caspases are present constitutively, the fluorescent background is higher, and we observe a threefold increase in fluorescence upon induction of apoptosis. Although this increase in fluorescence is lower than in bacterial systems, it is still superior to any other genetically-encoded caspase reporter systems. Perhaps the most striking characteristic of CA-GFP is its profoundly dark state prior to caspase cleavage. Because the chromophore is immature, CA-GFP provides nearly no fluorescent background. As a consequence, the signal-to-noise ratio for CA-GFP is strikingly improved compared to other previously reported caspase reporters. While CA- GFP is robust at reporting global apoptotic events, a dark reporter that housed a more rapidly maturing chromophore would enable us to capture more rapid kinetic details of the apoptotic cascade. The goal of this project is to understand, from a structural perspective, how chromophore maturation is prevented in CA-GFP. Understanding the conformational state of CA-GFP would enable us to improve the response time of CA-GFP, optimize the fluorescent increase upon cleavage of CA-GFP, and finally enable us to design reporters related to CA-GFP for other enzymatic processes.
Results
The most broadly applicable attribute of CA-GFP is the fully quenched nature of the uncleaved state, which results from structural perturbations that maintain an immature chromophore. We have previously shown that the appendage of the 27-amino acid quenching peptide causes CA-GFP to oligomerize into a mixed population that is predominantly tetramer and higher order oligomers.1 We hypothesize that this oligomerization is at the root of the CA-GFP quenching mechanism. To fully harness the potential of CA-GFP for other uses, it is essential to understand the structural basis of the immature chromophore at a mechanistic level. The goal of this work is to assess the overall folded state of CA-GFP to determine what structural factors prevent maturation of the GFP chromophore.
CA-GFP has secondary structure similar to GFP1-10
Both mature GFP and CA-GFP have a circular dichroism (CD) signal that is consistent with a predominantly β-stranded structure [Fig. 1(C)]. The fact that GFP is known to be a stable and well-folded β-barrel protein suggests that CA-GFP is likewise a folded protein despite the fact that the CD spectra are not superimposable, particularly at wavelengths greater than 225 nm. Several other factors also suggest that CA-GFP is in a folded state. CA-GFP is soluble in solution for an extended period of time and is not prone to aggregation-induced insolubility. CA-GFP can also be cleaved rapidly by active caspases, suggesting that it is not massively aggregated and therefore resistant to proteolytic cleavage. On the other hand, the fact that CA-GFP does form some high-order oligomers suggests that the stability of the protein has been compromised by the appendage of the quenching peptide. Thus, we sought to determine whether CA-GFP is folded into the β-barrel observed in mature GFP or whether CA-GFP forms a stable, partially folded intermediate.
The folding pathway for GFP has been studied previously by a number of groups.6–12 It is now clear that folding of GFP occurs co-translationally and that proper folding into the GFP β-barrel only occurs once the entire protein (strands 1 through 11) has been translated.13, 14 Despite the fact that all 11 strands must be available for proper folding of GFP, it is possible to cut GFP following the 10th strand to make split GFP.4 In the split-GFP system, co-expression of GFP strands 1–10 (GFP1-10) with the 11th strand allows reassociation of the 11th strand with GFP1-10 and folding of the GFP β-barrel. Once the full GFP β-barrel is formed the chromophore is competent to mature, and split GFP becomes fluorescent. Chromophore maturation in split GFP occurs with similar kinetics to CA-GFP.4, 15 We hypothesized that CA-GFP might adopt a stable, partially folded structure similar to that of GFP1-10 [Fig. 1(C)]. Indeed, the overall shape of the GFP1-10 spectrum was also indicative of a predominantly β-strand structure. More importantly, the CD spectra for GFP1-10 and CA-GFP are more similar to each other than to mature GFP. This suggests a higher degree of structural similarity between GFP1-10 and CA-GFP than mature GFP.
In addition to the shape of the CD spectra, the thermal stability of CA-GFP also provides insight into the structure relative to mature GFP and GFP1-10. To assess stability, we collected spectra from 200 to 250 nm of each protein every 5° as the temperature was raised from 20 to 90°C [Fig. 2(A–C)]. As was the case with the CD spectra, the denaturation properties of CA-GFP are more similar to GFP1-10 than to GFP. As the temperature was increased for mature GFP the molar ellipticity decreased. Previous work has suggested that an increase in CD signal is a sign of aggregation. Thus it appears that as GFP becomes unfolded it is prone to aggregation. In the cases of both GFP1-10 and CA-GFP the signal very gradually increases as a function of temperature up to 85°C. After this temperature the signal precipitously drops, suggesting a significant aggregation occurring at the highest temperatures. The melting profile can be illustrated by plotting the molar ellipticity at a selected wavelength as a function of temperature. We observed the greatest temperature-dependent change in CD signal at 214 nm, so we have reported those data [Fig. 2(D–E)] but similar trends are seen at wavelengths > 210 nm. The measured melting temperature (Tm) for GFP of 76°C, closely agrees with published value of 78°C based on loss of fluorescence.15, 16 The curves for both GFP1-10 and CA-GFP cannot be fit to a two-state melting model but show similar stability profiles to one another. These data suggest that CA-GFP adopts an ensemble of structures that have more similar thermodynamic properties to GFP1-10 than to mature GFP.
Figure 2.
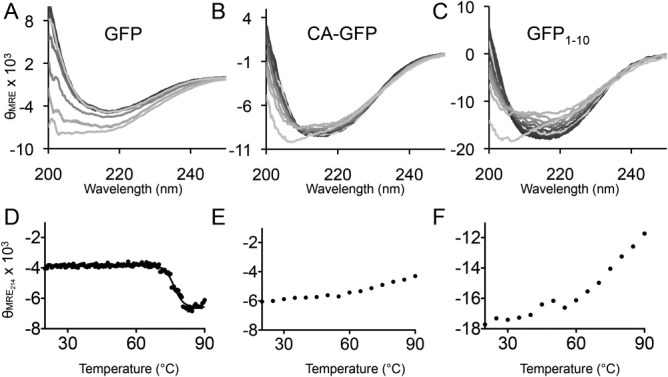
Circular dichroism spectra. A. The CD spectra of GFP from 200 to 250 nm collected at 5° increments from 20 to 90°C. The 20°C spectra is shown in the darkest shading; the 90°C spectra in the lightest shading and intermediate temperatures are shown in decreasing color intensity as a function of temperature. Spectra are plotted as the mean residual ellipticity (MRE × 103: degree cm2 dmol–1 number of residues–1) as a function of wavelength B. The CD spectra of CA-GFP collected and shown as in A. C. The CD spectra of GFP1-10 collected and shown as in A. D. Profile of thermal denaturation of GFP as monitored by CD signal at 214 nm plotted as a function of temperature. E. Profile of thermal denaturation of CA-GFP. F. Profile of thermal denaturation of GFP1-10.
We have previously shown that CA-GFP adopts an oligomeric state greater than a monomer and consistent with a trimer or tetramer by size exclusion chromatography. We aimed to compare the oligomeric state of CA-GFP to mature GFP and GFP1-10 as further insight into the organization of CA-GFP. As expected, GFP elutes as a monomer. Approximately 50% of CA-GFP elutes as a lower-order oligomer (trimer/tetramer) and 50% higher-order oligomer. In contrast, GFP1-10 elutes in a single peak at the void volume of the Superdex 200 column (Fig. 3), indicating a highly aggregated state. The size exclusion chromatography analysis corroborates the observations from CD, suggesting that GFP1-10 exists in a higher oligomeric state than CA-GFP, which is more oligomeric than GFP. Thus, CA-GFP appears to have oligomerization properties that are intermediate between mature GFP and GFP1-10.
Figure 3.
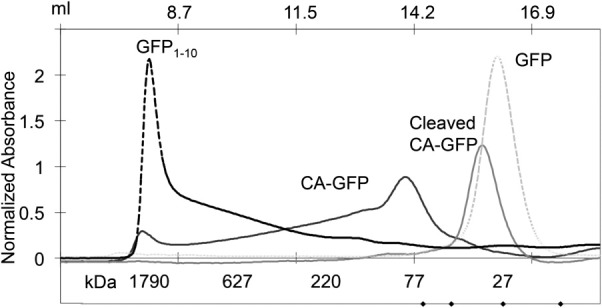
The size exclusion chromatogram of GFP, CA-GFP, cleaved CA-GFP, and GFP1-10. GFP and cleaved CA-GFP elute at a retention volume consistent with a monomeric form of the protein. The monomer molecular weights are GFP: 27.7 kDa, CA-GFP: 32 kDa, cleaved CA-GFP: 28.2 kDa, and GFP1-10: 20.5 kDa. The dark CA-GFP elutes as two peaks with retention volumes consistent with a population that is 50% lower order oligomer (putatively trimer or tetramer) and 50% higher oligomer. GFP1-10 elutes in the void volume indicating a largely aggregated population.
We reasoned that structural studies of CA-GFP would be dramatically eased if we attained oligomerically homogeneous preparations of CA-GFP. We also isolated the low-order oligomeric form of CA-GFP following size exclusion chromatography and allowed it to re-equilibrate at room temperature for 1 or 18 h. Following incubation, CA-GFP had re-equilibrated to a mixture that contained both low- and higher-order oligomers. This occurred in a time-dependent manner (data not shown). We tried extensively to identify detergent-containing conditions that would shift CA-GFP entirely to a homogenous low oligomeric state, but were unsuccessful in this pursuit. Despite this we performed extensive crystallization trials, reasoning that perhaps crystallization solely of a trimeric/tetrameric state could occur. These crystallization trials were also uniformly unsuccessful (data not shown). Thus, we think it likely that CA-GFP does not exist in a single homogenous conformation in solution but is indeed a mixture of oligomeric species as observed by size exclusion chromatography.
β-Barrel of CA-GFP not fully formed
To further investigate whether the solution structure of CA-GFP more closely resembles the mature GFP or GFP1-10, we subjected all three proteins to limited proteolysis by proteinase K [Fig. 4(A)]. Proteinase K cleaves proteins at sites containing aromatic and aliphatic amino acids and is thus a useful means of achieving nearly full proteolysis of most protein substrates. The propensity of GFP1-10 toward aggregation has been suggested to indicate the presence of a less ordered folding intermediate of GFP. We expect that such a folding intermediate should be relatively more susceptible to proteolysis. In contrast, fully folded GFP is notoriously resistant to proteolytic cleavage due to the stability of the β-barrel.17 β-barrels are exquisitely stable, cooperatively folding structures because all of the hydrogen bond donor and acceptors in each of the β-strands are fully satisfied. As predicted, GFP was resistant to digestion up to 10 min while both CA-GFP and GFP1-10 were nearly fully digested within one minute [Fig. 4(A)]. This strong susceptibility to digestion suggests that the β-barrel structure, which renders GFP resistant to the proteinase K treatment is altered. These alterations could range from subtle, such as displacement of a single strand, to catastrophic with the entire β-barrel disrupted in CA-GFP.
Figure 4.
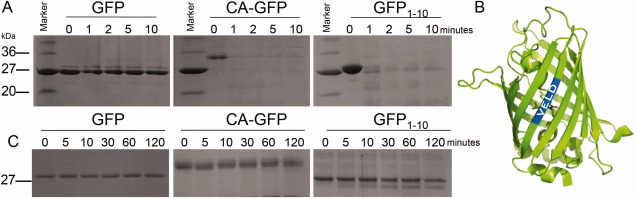
Protease susceptibility of GFP, CA-GFP and GFP1-10. A. GFP, GFP1-10, and CA-GFP were subjected to digestion by proteinase K. Samples were collected and analyzed at 0, 1, 2, 5, and 10 min. GFP is highly resistant to digestion while GFP1-10 and CA-GFP are nearly completely degraded after 1 min. B. The GFP β-barrel (green) is drawn with the predicted caspase-6 cleavage sequence (VELD, blue) highlighted. The sequence falls in the center of the first β-strand, however, caspases are predicted to cleave in loop regions. C. GFP, GFP1-10, and CA-GFP were subjected to digestion by caspase-6. Caspase-6 is capable of cleaving GFP at a single site near the N-terminus. GFP is resistant to digestion while GFP1-10 and CA-GFP undergo partial cleavage after a two-hour incubation. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Whereas Proteinase K has broad specificity and cleaves most proteins at multiple sites we sought to cleave the barrel region with a more structurally specific protease. Though we considered several potential candidate proteases we found only one, caspase-6, that would cleave the GFP barrel in only one location, in a structure-dependant manner. Caspases are very specific proteases, recognizing particular aspartate-terminated tetra peptide motifs and have a strong preference for loop regions over those with more ordered secondary structure in a substrate protein. Sequence analysis indicated that a caspase-6-cleavage site exists independent of the designed caspase-7 cleavage site in the linker. We have tested CA-GFP against other caspases including caspase-7, -8, and -9 and have not observed cleavage at any site other than the recognition-site-containing linker (data not shown). Caspase-6 has a preference for cleaving protein substrates within loop regions at the recognition sequence VEXD.18 Thus, caspase-6 cleavage serves as a useful indicator of the ordered structure of a particular region of a protein containing a recognition motif.
A caspase-6 compatible sequence, VELD (residues 16–19), is present natively in the N-terminal region of GFP, CA-GFP, and GFP1-10. This VELD sequence is present in a fully folded GFP in the center of the first β-strand [Fig. 4(B)]. We hypothesized that if this region of CA-GFP and GFP1-10 is organized into a protected β-strand conformation like that in mature GFP, caspase-6 would be unable to access the site and cleave the protein. As anticipated, GFP is resistant to cleavage by caspase-6 after incubation for 2 h at room temperature [Fig. 4(C)]. In contrast, caspase-6 is able to partially cleave both CA-GFP and GFP1-10 within 5 min [Fig. 4(C)]. This suggests the first strand region of CA-GFP and GFP1-10 is not resting in a β-strand conformation and likely exists in a disordered or loop conformation.
Together, limited proteolysis by proteinase K and caspase-6 indicate that GFP1-10 and CA-GFP lack the stable β-barrel structure present in mature GFP. If prevention of chromophore maturation is caused by the presence of a less-folded, stable folding intermediate or by small perturbations in a mostly formed β-barrel geometry then TROSY-HSQC NMR spectroscopy with 15N-labeled GFP and CA-GFP would indicate a spectral shift for specific residues. A CA-GFP spectrum with chemical shifts similar to those of GFP could indicate which residues were being perturbed if the differences were subtle. The spectrum of GFP [Fig. 5(A)] showed well-defined and well-dispersed peaks, consistent with a well-folded protein.19 The spectra of CA-GFP [Fig. 5(B)] looked much different than the control spectra and the signal resembled that of an unfolded protein with poorly dispersed peaks.20 In sum, these experiments suggest that CA-GFP does not fold into a stable β-barrel conformation and is more similar to GFP1-10 than to the mature GFP.
Figure 5.
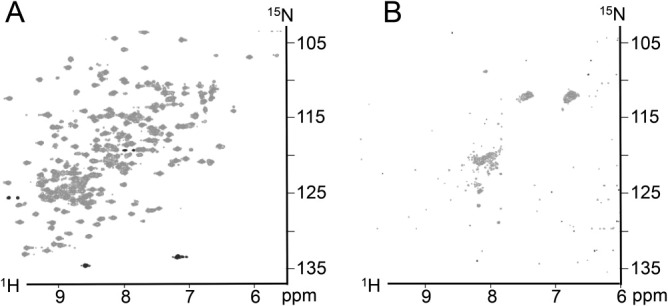
TROSY NMR HSQC spectra. A. Spectra of GFP collected at 37°C. B. Spectra of CA-GFP also at 37°C. The spectra of GFP is indicative of a well-folded protein while the spectra of CA-GFP resembles that of an unfolded protein.
CA-GFP exists in “pro-folding” conformation
The similarity of CA-GFP to the GFP1-10 conformation, as assessed by CD and proteolysis, suggests a number of potential models for how the quenching peptide prevents chromophore maturation. Due to the sequential proximity of the quenching peptide to the C-terminal 11th strand of GFP, we favor the model that fusing the quenching peptide to the GFP C-terminus may prevent proper folding of the final 11th strand in CA-GFP. With the 11th strand unable to complete the GFP barrel, the environment for chromophore maturation is never attained, so CA-GFP remains in the dark state with an immature chromophore until the quenching peptide is cleaved and released. Dissociation of the quenching peptide allows the GFP barrel to properly fold, enabling maturation of the chromophore. We reasoned that if this model were correct, fusion of the quenching peptide to other parts of GFP would likely have a differential effect on the behavior of the resulting “reporter.” We made a fusion protein with the quenching peptide on the GFP N-terminus (nCA-GFP) [Fig. 6(A)]. To compare the effectiveness of nCA-GFP with that of CA-GFP, we co-transformed E. coli with nCA-GFP and either active caspase-7 or an inactive caspase-7 variant where the active site cysteine has been mutated to alanine (C186A). In the absence of active caspase, nCA-GFP was equally “dark” as the C-terminal fusion, CA-GFP, suggesting that the quenching peptide was equally effective at disrupting assembly of the GFP barrel thereby preventing chromophore maturation. However, when nCA-GFP was co-expressed with active caspase-7, it showed less than a 3.8-fold increase in fluorescence in E. coli lysates [Fig. 6(B)]. This is in contrast to the C-terminal construct, CA-GFP, which shows a greater than 40-fold increase in fluorescence when cleaved by active caspase. Western blot analysis using an anti-GFP antibody shows that nCA-GFP is only partially processed in comparison to CA-GFP [Fig. 6(C)], indicating the linker region is less accessible to proteolytic cleavage than in the C-terminal fusion. Partial digestion of nCA-GFP when co-expressed with active caspase indicates that the linker region may be less accessible to the protease. The low fluorescence recovery is then due to a smaller fraction of nCA-GFP being cleaved and a smaller portion of the cleaved protein attaining the mature barrel geometry to allow for chromophore maturation. Thus, it is clear that the conformational state attained by CA-GFP is better poised to allow cleavage, refolding, chromophore maturation and regaining fluorescence than GFP1-10. Given the greater propensity for the C-terminal fusions of the quenching peptide to be cleaved and recover fluorescence, we can conclude that the dark state can be attained by a variety of unfolded and partially folded states. Nevertheless, the partially folded, partially aggregated state that CA-GFP obtains has more optimal properties for cleavage and refolding than that induced by adding the quenching peptide to other parts of GFP, such as the N-terminus.
Figure 6.
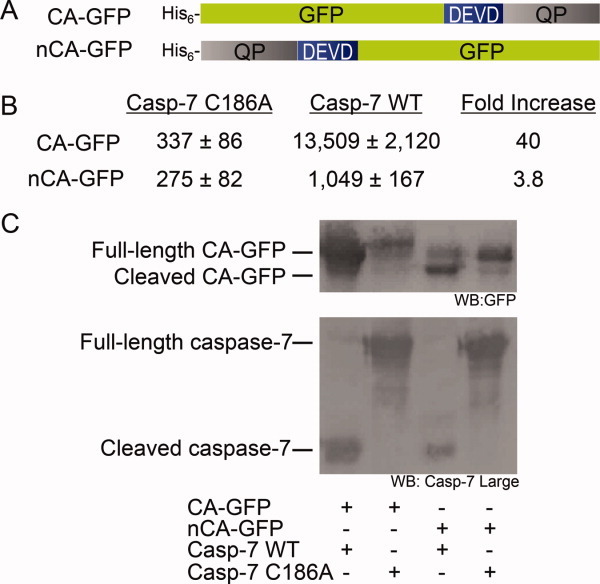
A. Constructs of CA-GFP and N-terminally tagged nCA-GFP with their relative fluorescence when co-expressed in E. coli with active caspase-7 or an inactive version caspase-7 C186A. CA-GFP shows a 40-fold increase in fluorescence in the presence of active caspase vs. nCA-GFP which shows less than 4-fold. B. Western blots of the CA-GFP and nCA-GFP co-transformations with active and inactive caspase. The top blot was probed with an anti-GFP primary antibody shows that CA-GFP is fully cleaved in the presence of active caspase while nCA-GFP is only partially cleaved. The bottom blot was probed with a primary antibody specific for the large sub-unit of caspase-7 shows that caspase-7 is cleaved and active in the case of the wild-type (WT) and in the full-length, unprocessed inactive form in the case of the C186A mutant. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The notion that folding of GFP is integral to the quenching mechanism is further supported by our observation that the quenching peptide is not capable of preventing fluorescence when appended to superfolder GFP (sfGFP). Superfolder GFP was developed by Waldo and coworkers to fold with faster kinetics and therefore is less prone to aggregation.21 When the quenching peptide is fused to the C-terminus the fluorescence of sfGFP is not quenched (data not shown). We predicted that a shorter linker would be a more stringent test of the quenching ability of the peptide. A CA-superfolder GFP (CA-sfGFP) construct was made with a shorter linker (DELD) rather than the longer linker in CA-GFP (DEVDFQGP, Supporting Information Fig. S2) but the resulting CA-sfGFP was still brightly fluorescent suggesting that the kinetics of folding is involved in the quenching mechanism that is at work in CA-GFP. Thus, we venture to state that CA-GFP exists in a relatively optimal folding competent or “pro-folding” state. Although we cannot firmly conclude that CA-GFP is quenched by prevention of the 11th strand from inserting into the partially formed barrel, this model is compatible with our observations.
Discussion
Our initial model of the structure of CA-GFP in the dark state was that of a well-formed β-barrel conformation similar to that of mature GFP, with the presence of the quenching peptide inducing subtle perturbations, potentially a result of the oligomeric state, inhibiting chromophore maturation. The fully formed β-barrel of GFP has been shown to be impressively robust. Once the barrel has folded and the chromophore is mature it requires fairly extreme conditions to disrupt the fluorescence and unfold the protein. Melnik et al. showed that even after partial digestion the fluorescence of the cycle-3 mutant remained nearly unchanged.22 They hypothesized that the proteases may cleave in loops but that the barrel remained formed and “sticks” to the chromophore to retain fluorescence. They also observed that GFP fluorescence was unaffected in concentrations of urea up to 4 M. For example, when a trypsin site was engineered into a loop to remove the 11th strand, the protein still had to be denatured and refolded to remove the cleaved strand from the barrel.23 As we observed throughout this study, CA-GFP lacks the remarkable stability of the mature fold, leading us to speculate that it does not exist in the fully formed β-barrel conformation but potentially exists as a stable intermediate along the native folding pathway.
Several recent protein-folding studies have pointed out that the canonical models of protein folding fail to take into account the kinetics and sterics of co-translational folding.14, 24–26 One recent study has shown that a stable folding intermediate consisting of the first 10 strands of GFP form prior to the 11th strand's release from the ribosome tunnel.14 It follows that if CA-GFP is folding co-translationally, as GFP does, it will sample the same folding intermediates as the native protein prior to release of the 11th strand. As the peptide is the last portion to be translated and released it is possible that the quenching peptide interferes with the folding of the final stave of the barrel. Since CA-GFP is in a “pro-folding” state, after the peptide is released a majority of the individual CA-GFP molecules can proceed to the native GFP fold.
This is slightly different from the case of nCA-GFP where the very hydrophobic peptide is being translated and released from the ribosome first, leading to a very different sampling of folding states as it is being translated. The grouping of long stretches of hydrophobic residues has been shown to lead to an increase in aggregation,27 which could lead to very different folding intermediates being sampled than the native GFP. This type of behavior has been harnessed to use GFP as a folding reporter. When peptides or proteins have been fused to the N-terminus of GFP, fluorescence is an indication of the stably-folded state of the fused peptide or protein, whereas unstably folded protein or peptide fusions prevent GFP fluorescence.28, 29 Our results suggest that the quenching peptide is not well-folded, which would therefore lead to a less stably folded intermediate of the GFP barrel in nCA-GFP.
The differences in the oligomeric states of CA-GFP and GFP1-10 suggest that CA-GFP is in a more advanced folding intermediate conformation than GFP1-10. CA-GFP has a SEC profile consistent with 50% of the protein being in a low order oligomeric state. The heterogeneous mixture of oligomeric states suggests that there may be several intermediate folding states present as these conformations tend towards self-aggregation24 with the majority being in a similar stable state that forms a low-order oligomer. This is in contrast to the largely aggregated GFP1-10. It is not surprising that GFP1-10 is in an aggregated state, even the robust mature GFP can be coaxed to aggregate upon the addition of 2,2,2-trifluoroethanol (TFE), a chemical commonly used to induce aggregation in proteins.30 The observation that CA-GFP is not as aggregated then supports the hypothesis that it is more ordered than GFP1-10.
If the dark state of CA-GFP is dictated by the co-translational folding of the GFP barrel, then faster folding variants of GFP should behave differently in the presence of the quenching peptide. The fact that superfolder GFP remains fluorescent in the presence of the quenching peptide indicates that folding kinetics control the dark or bright properties of this class of reporters. The improved folding kinetics of sfGFP prevent populating kinetic traps required for the quenching peptide to function. Thus, to develop the next generation CA-GFPs with faster response times, it will also be important to develop quenching peptides that can interact with GFP folding intermediates on a more rapid time scale.
Based on the structural insights we report above we propose a working model of the dark state of CA-GFP. We envision that as CA-GFP is being translated the N-terminus begins sampling the native GFP folding states. The presence of the quenching peptide on the C-terminus prevents it from attaining the fully mature β-barrel conformation and it is trapped in a stable, “pro-folding” intermediate. After the quenching peptide is cleaved and released the GFP barrel is then allowed to fold into the robust fold and the chromophore matures, yielding fluorescence. Controlling the folding path of CA-GFP to attain a rapidly maturing chromophore yet maintaining the dark state will be critical in further development of the next generation of CA-GFP reporters. Once a more detailed understanding of the “pro-folding” state is attained the ability to engineer further fluorescent protease reporters as well as expand the utility of the platform to respond to other enzymatic processes will be greatly eased.
Materials and Methods
Molecular cloning
CA-GFP was generated by amplification of GFP (S65T) by PCR using a reverse primer encoding the 27 amino acid transmembrane domain of the influenza A virus. After amplification of the new gene it was ligated into the XhoI and NdeI sites of pET21b. A linker sequence containing the caspase-3 and -7 cleavage recognition site DEVD was then inserted between GFP and the peptide using a site-directed mutagenesis using overlapping inverted primers and amplification of the entire plasmid similar to the QuikChange® (Agilent) approach for a final fusion sequence of DEVDFQGPCNDSSDPLVVAASIIGILHLILWILDRL at the C-terminus of GFP. The expression construct for GFP (S65T) GFP1-10 was generated by inserting a stop codon (UAA) after residue K214 using a similar site-directed mutagenesis using GFP (S65T) in pET21b as the template.
The N-terminal peptide version of CA-GFP (nCA-GFP) was generated by separately amplifying the peptide (M2) region from the GFP region of the gene using two primers to amplify M2 and two primers to amplify GFP. The first primer (P1) annealed to the N-terminal region of GFP with the linker sequence included N-terminally to the GFP. The second primer (P2) annealed to the C-terminal of GFP and included a stop codon (UAA) and the restriction site for XhoI. The third primer (P3) included an NdeI restriction enzyme site as well as a 6His sequence and annealed to the N-terminal region of the M2 portion of the CA-GFP gene. The last primer (P4) annealed to the C-terminus of M2 and included the same sequence for the linker as P1, giving primers P1 and P4 a 24 bp overlapping region. The GFP fragment was then amplified using primers P1 and P2 while the M2 portion was amplified using primers P3 and P4. After gel purification of the amplified fragments they were combined and allowed to anneal through the overlapping region for five PCR cycles before the addition of primers P2 and P3 which then amplified the full length gene. The gene was then ligated into pET21b into NdeI and XhoI sites. The final sequence of the N-terminal peptide and linker is MHHHHHHMCNDSSDPLVVAASIIGILHLILWILDRLDEVDFQGP.
Protein expression and purification
CA-GFP, CA-sfGFP, nCA-GFP, GFP1-10, and GFP were transformed into E. coli strain BL21(DE3) for expression. One liter cultures of 2×YT media were inoculated with 1 mL of dense overnight culture and grown at 37°C to an A600 of 0.6. The cultures were then induced with 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) at 25°C for 3 h. Cells were then harvested by centrifugation and disrupted by microfluidization. After centrifugation at 15,000g for 45 min. the proteins were purified from supernatant using Co2+ affinity chromatography (HiTrap Chelating HP, GE). The column was washed with a buffer of 50 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4 pH 8.0 and eluted in a buffer of 300 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4 pH 8.0. Protein purity was assessed by gel electrophoresis; proteins were estimated to be at lease 95% pure.
The caspase-6 E. coli codon-optimized sequence gene construct in pET11a was transformed into the BL21(DE3) T7 express strain of E. coli (NEB). The cultures were grown in 2×YT media with Amp (100 mg/mL, Sigma-Aldrich) at 37°C until they reached A600=0.6. The temperature was reduced to 20°C and cells were induced with 1 mM IPTG (Anatrace) to express soluble His-tagged protein. Cells were harvested after 18 h to ensure complete processing. Cell pellets stored at –20 °C were freeze-thawed and lysed in a microfluidizer (Microfluidics, Inc.) in 300 mM NaCl, 2 mM imidazole, and 50 mM Tris (pH 8.5). Lysed cells were centrifuged at 18,000 rpm to remove cellular debris. The filtered supernatant was loaded onto a 5-mL HiTrap Ni-affinity column (GE Healthcare). The column was washed with 300 mM NaCl, 50 mM imidazole, and 50 mM Tris (pH 8.5), and the protein was eluted with 300 mM NaCl, 250 mM imidazole, and 50 mM Tris (pH 8.5). The eluted fraction was diluted by fivefold into 2 mM DTT and 20 mM Tris (pH 8.5) buffer to reduce the salt concentration. This protein sample was loaded onto a 5-mL Macro-Prep High Q column (Bio-Rad Laboratories, Inc.). The column was developed with a linear NaCl gradient and eluted in 120 mM NaCl, 2 mM DTT, and 20 mM Tris (pH 8.5) buffer. The eluted protein was stored at –80 °C in the above buffer conditions. The identity and purity of the purified caspase-6 was analyzed by SDS-PAGE.
Proteolysis
Proteolysis of 20 μM CA-GFP, GFP1-10, and GFP by the addition of 500 nM Proteinase K (sigma) was performed in a buffer of 50 mM Tris pH 7.5, 5 mM CaCl2. Aliquots were taken from the reaction at 0, 1, 2, 5, and 10 min and added to protease inhibitor phenylmethylsulfonyl fluoride (PMSF) to stop the reaction. SDS loading buffer was then added to the samples, which were boiled for 10 min and analyzed by SDS-PAGE. The presence of the PMSF made the loading of samples onto the SDS-PAGE gel challenging but was aided by the chilling of the samples, gel and running buffer prior to loading. It was also helpful to load the gel while a low current (50 mV) was conducted over the gel.
Purified CA-GFP, GFP1-10, and GFP were incubated at a concentration of 5 μM in a 1:1 molar ratio with purified active caspase-6 at room temperature in a buffer of 100 mM HEPES pH 7.5, 10% sucrose, 0.1% CHAPS, 30 mM NaCl, and 5 mM DTT. Aliquots of each reaction were taken at 0, 5, 10, 30, 60, and 120 min time intervals and added to SDS loading buffer with DTT and immediately boiled for 10 min to stop cleavage. The samples were then assessed using SDS-PAGE to determine cleavage at each time point.
Circular dichroism (CD)
Purified proteins were buffer exchanged into a buffer containing 10 mM NaH2PO4, pH 7 using Millipore Ultra-free 10K NMWL membrane concentrators and diluted to 10 μM as determined by A280 nm using a Nanodrop 2000C Spectrophotometer. CD spectra were measured on a J-715 circular dichroism spectrometer at intervals of every 5° from 20 to 90°C at a rate of temperature increase of 1°C per minute in a quartz cuvette with a 0.1 cm path length.
TROSY NMR
15N-labeled CA-GFP and GFP were obtained by growth in M9 minimal media using 15N ammonium chloride as its sole nitrogen source (Cambridge isotopes).31 A 20 ml LB culture was inoculated from a dense overnight culture. After reaching an OD600 of 0.5, the cells were spun down and washed once with PBS. The cells were resuspended in 100 ml 15N-labeled M9 media. The culture was incubated about 3.5 h at 37°C until reaching an OD600 of 0.5. The doubling time of the cells in the M9 media was approximately 90 min in agreement with published values.32 The culture was then diluted into 200 ml of labeled M9. After incubation at 37°C until reaching an OD600 of 0.5 once more the culture was diluted to 1 L of labeled M9 and then induced with 1 mM isopropyl β-d-1-thiogalctopyranoside (IPTG) at 25°C for 3 h. The 15N-labeled proteins were purified as described above. The proteins were buffer exchanged using Millipore Ultra-free 10K NMWL membrane concentrators into a buffer of 10 mM NaCl, 2.7 mM KCl, and 6 mM NaH2PO4 pH 7.2 at a concentration of 60 μM. 1H–15N Transverse relaxation optimized spectroscopy (TROSY) spectra in this study were obtained at 37°C on a 700-MHz Varian NMR system equipped with a cryogenically cooled triple-resonance probe. Spectra were processed using NMRpipe33 and analyzed using Cara.34
Size exclusion chromatography
The curves for CA-GFP, GFP, and cleaved CAGFP were previously reported in Nicholls et al.1 The size of each protein was determined by size exclusion chromatography using a Superdex 200 10/100 GL column in a buffer of 100 mM NaCl, 20 mM Tris pH 8. The molecular weight of each was determined by comparison to molecular weight standards albumin (66 kDa), carbonic anhydrase (29 kDa), ovalbumin (45 kDa), and ribonuclease A (14.7 kDa) with blue dextran 2000 used to determine the void volume of the column (Sigma).
Lysate-based CA-GFP fluorescence assays
Expression constructs for CA-GFP with the peptide on either the C- or N-terminus, CA-GFP and nCA-GFP, respectively, in pET21b (Amp) was co-transformed in the Bl21(DE3) strain of E. coli with either a constitutive two chain version of caspase-7 (C7 CT) or full-length inactive caspase-7 where the active site cysteine has been substituted with an alanine (C186A). Both versions of caspase-7 were contained in the vector pBB75 (Kan). 50 mL cultures were inoculated from dense overnight cultures and grown at 37°C to an OD600 of 0.6. Cultures were then induced with 1 mM IPTG at 25°C for 18 h. Two milliliters of each culture was centrifuged and resuspended in 800 μL a buffer of 0.5 mg/ml lysozyme (sigma) and 2 units DNAase (NEB). Cells were lysed using four cycles of freeze-thaw and the supernatant (100 μL) was analyzed for fluorescence (Ex. 475 nm/Em. 512 nm) in a co-star 96-well black plate on a Molecular Devices Spectramax M5 spectrophotometer. Fluorescence values were normalized based on the relative OD of each culture.
Western blotting
Samples of supernatant prepared as described for the lysate fluorescence assays of CA-GFP or nCA-GFP each co-transformed with either the active constitutively two-chain caspase-7 or the inactive C186A caspase-7 were taken. Three identical SDS-PAGE gels were run of the four samples. One was stained with coomassie, two gels were transferred onto Hybond-ECL™ nitrocellulose membrane (GE Healthcare) and blocked overnight in a solution of 30 mg/ml BSA in TBS. One was then blotted with an anti-GFP (Millipore) monoclonal mouse primary antibody, and the other was blotted with a mouse primary antibody recognizing the large subunit of caspase-7 (sigma). The western blots were then treated with anti-mouse IgG alkaline phosphatase produced in goat (Sigma) and visualized using 1-Step™ NBT/BCIP (Thermo Scientific).
Acknowledgments
The authors thank Anastasia Zhuravleva for advice in the preparation of NMR samples, for collecting NMR spectra and instruction in the use of NMRpipe and CARA as well as helpful discussions.
Supplementary material
Additional Supporting Information may be found in the online version of this article.
References
- 1.Nicholls SB, Chu J, Abbruzzese G, Tremblay KD, Hardy JA. Mechanism of a genetically encoded dark-to-bright reporter of caspase activity. J Biol Chem. 2011;286:24977–24986. doi: 10.1074/jbc.M111.221648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.I G, AD H, Regan L. Antiparallel leucine zipper-directed protein reassembly: application to the green fluorescent protein. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 3.Hu C, Chinenov Y, Kerppola T. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 4.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nature Biotech. 2004;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 5.Wood TI, Barondeau Dp, Hitomi C, Kassmann CJ, Trainer JA, Getzoff ED. Defining the role of arginine 96 in green fluorescent protein fluorophore biosynthesis. Biochemistry. 2005;44:16211–16220. doi: 10.1021/bi051388j. [DOI] [PubMed] [Google Scholar]
- 6.Enoki S, Saeki K, Maki K, Kuwajima K. Acid denaturation and refolding of green fluorescent protein. Biochemistry. 2004;43:14238–14248. doi: 10.1021/bi048733+. [DOI] [PubMed] [Google Scholar]
- 7.Cannone F, Bologna S, Campanini B, Diaspro A, Bettati S, Mozzarelli A, Chirico G. Tracking unfolding and refolding of single GFPmut2 molecules. Biophys J. 2005;89:2033–2045. doi: 10.1529/biophysj.105.064584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldini G, Canonne F, Chirico G, Collini M, Campanini B, Battati S, Mozzarelli A. Evidence of discrete substates and unfolding pathways in green fluorescent protein. Biophys J. 2007;92:1724–1731. doi: 10.1529/biophysj.106.093567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mickler M, Dima RI, Dietz H, Hyeon C, Thirumalai D, Rief M. Revealing the bifurcation in the unfolding pathways of GFP by using single-molecule experiments and simulations. Proc Natl Acad Sci USA. 2007;104:20268–20273. doi: 10.1073/pnas.0705458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang J-r, Hsu S-T, Chistodoulou J, Jackson SE. The extremely slow-exchanging core and acid-denatured state of green fluorescent protein. HFSP J. 2008;2:378–387. doi: 10.2976/1.2976660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orte A, Craggs TD, White SS, Jackson SE, Klenerman D. Evidence of an Intermediate and parallel pathways protein unfolding from single-molecule fluorescence. J Am Chem Soc. 2008;130:7898–7907. doi: 10.1021/ja709973m. [DOI] [PubMed] [Google Scholar]
- 12.Benjamin TAndrews, Roy M, Jennings PA. Chromophore packing leads to hysteresis to GFP. J Mol Biol. 2009;392:218–227. doi: 10.1016/j.jmb.2009.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ugrinov KG, Clark PL. Cotranslational folding increases GFP folding yield. Biophys J. 2010;98:1312–1320. doi: 10.1016/j.bpj.2009.12.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelkar DA, Khushoo A, Yang Z, Skach WR. Kinetic analysis of ribosome-bound fluorescent proteins reveals an early, stable, cotranslational folding intermediate. J Biol Chem. 2012;287:2568–2578. doi: 10.1074/jbc.M111.318766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ward WW, Prentice H, Roth A, Cody C, Reeves S. Spectral perturbations of the aequorea green-fluorescent protein. Photochem Photobiol. 1982;35:803–808. [Google Scholar]
- 16.Tsein RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 17.Bokman SH, Ward WW. Renaturation of Aequorea green-fluorescent protein. Biochem Biophys Res Commun. 1981;101:1372–1380. doi: 10.1016/0006-291x(81)91599-0. [DOI] [PubMed] [Google Scholar]
- 18.Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA. Global sequencing of proteolytic cleavage sites in apoptosis by specific labeling of protein N termini. Cell. 2008;134:866–876. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan F, Stott K, Jackson S. Letter to the Editor: 1H, 15N and 13C backbone assignment of the green fluorescent protein (GFP) J Biomol NMR. 2003;26:281–282. doi: 10.1023/a:1023817001154. [DOI] [PubMed] [Google Scholar]
- 20.Jian Yao, Dyson HJ, Wright PF. Chemical shift dispersion and secondary structure prediction in unfolded and partly folded proteins. FEBS Lett. 1997;419:285–289. doi: 10.1016/s0014-5793(97)01474-9. [DOI] [PubMed] [Google Scholar]
- 21.Jean-Denis Pedelacq SC, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nature Biotech. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 22.Melnik BS, Povarnitsyna TV, Melnik TN. Can the fluorescence of green fluorescent protein chromophore be related directly to the nativity of protein structure? Biochem Biophys Res Commun. 2009;390:1167–1170. doi: 10.1016/j.bbrc.2009.10.109. [DOI] [PubMed] [Google Scholar]
- 23.Kent KP, Oltrogge LM, Boxer SG. Synthetic control of green fluorescent protein. J Am Chem Soc. 2009;131:15988–15989. doi: 10.1021/ja906303f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clark PL. Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 25.Fulle S, Gohlke H. Statics of the ribosomal exit tunnel: Implications for cotranslational peptide folding, elongation regulation, and antibiotics binding. J Mol Biol. 2009;387:502–517. doi: 10.1016/j.jmb.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 26.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nature Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz R, Instrail S, King J. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci. 2001;10:1023–1031. doi: 10.1110/ps.33201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nature Biotech. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 29.Vasiljevic S, Ren J, Yao Y, Dalton K, Adamson CS, Jones IM. Green fluorescent protein as a reporter of prion protein folding. Virology J. 2006;3:59–68. doi: 10.1186/1743-422X-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson VL, Webb WW. A desolvation model for trifluoroethanol-induced aggregation of enhanced green fluorescent protein. Biophys J. 2012;102:897–906. doi: 10.1016/j.bpj.2012.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tugarinov V, Kanelis V, Kay L. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nature Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- 32.Paliy O, Gunasekera TS. Growth of E. coli BL21 in minimal media with different gluconeogenic carbon sources and salt contents. Appl Microb Cell Physiol. 2007;73:1169–1172. doi: 10.1007/s00253-006-0554-8. [DOI] [PubMed] [Google Scholar]
- 33.Delaglio F, Grzesiek S, Vulster GW, Zhu G, Pfeifer J, Bax A. NMRpipe – a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 34.Keller R. Optimizing the process of nuclear magnetic resonance spectrum analysis and computer aided resonance assignment. Doctoral Thesis. Swiss Federal Institute of Technology Zurich (ETH), Zurich. 2004 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.