

Abstract
Molecular recognition features (MoRFs) are intrinsically disordered protein regions that bind to partners via disorder-to-order transitions. In one-to-many binding, a single MoRF binds to two or more different partners individually. MoRF-based one-to-many protein–protein interaction (PPI) examples were collected from the Protein Data Bank, yielding 23 MoRFs bound to 2–9 partners, with all pairs of same-MoRF partners having less than 25% sequence identity. Of these, 8 MoRFs were bound to 2–9 partners having completely different folds, whereas 15 MoRFs were bound to 2–5 partners having the same folds but with low sequence identities. For both types of partner variation, backbone and side chain torsion angle rotations were used to bring about the conformational changes needed to enable close fits between a single MoRF and distinct partners. Alternative splicing events (ASEs) and posttranslational modifications (PTMs) were also found to contribute to distinct partner binding. Because ASEs and PTMs both commonly occur in disordered regions, and because both ASEs and PTMs are often tissue-specific, these data suggest that MoRFs, ASEs, and PTMs may collaborate to alter PPI networks in different cell types. These data enlarge the set of carefully studied MoRFs that use inherent flexibility and that also use ASE-based and/or PTM-based surface modifications to enable the same disordered segment to selectively associate with two or more partners. The small number of residues involved in MoRFs and in their modifications by ASEs or PTMs may simplify the evolvability of signaling network diversity.
Keywords: molecular recognition feature, MoRF, linear motif, hub protein, binding site, protein–protein interaction, intrinsically disordered protein
Introduction
Protein–protein interaction (PPI) networks underlie a wide variety of biological functions, ranging from regulating cell division to responding to external signals. High throughput methods have enabled researchers to map out sets of PPIs over entire proteomes. These studies reveal complex networks in which a few proteins, called hubs, bind to many protein partners and many other proteins bind to only a few or even just one partner. Indeed, in some cases, hubs bind to 15, 20, 50, or even more partner proteins. As expected for such network architecture, deletion of a protein with only a few partners is typically less deleterious than the deletion of a highly connected protein.1, 2
How do such networks arise from simpler precursors? Other networks of a similar architecture arise because “the rich get richer”; units with more connections have a higher probability of adding even more connections over time as compared with the units with fewer connections. This suggests that highly connected proteins have special features that facilitate their binding to multiple partners and to new partners that arise through mutation.3 What are these special features?
Theoretical arguments4, 5 and experimental data6, 7 suggest that unfolded or disordered protein can very readily change shape and thereby easily adapt to multiple, distinct partners. Thus, we proposed that the special feature of hub proteins enabling their binding to multiple partners is likely to be intrinsic disorder. We further suggested two ways that disorder could be used by hub proteins for binding to multiple partners: (1) One region of disorder could bind to many different partners (one-to-many binding), so the hub protein itself uses disorder for multiple partner binding; and (2) many different regions of disorder could bind to a single partner (many-to-one binding), so the hub protein is structured but binds to many disordered partners via interaction with disorder.8 Since this initial proposal, we9–11 and many others12–22 have provided additional evidence that hubs and/or their binding partners are especially enriched in intrinsic disorder, with both the many-to-one and one-to-many processes involving the use of intrinsic disorder.
Our initial work8–11 on disorder and PPIs focused on single binding sites that used regions of disorder. To be more complete, it is worth mentioning that, in addition to the one-to-many and many-to-one mechanisms used by single sites of disorder for multiple partner binding, hub proteins can also use multiple binding domain repeats likely connected by flexible (disordered) linkers,12 or hubs can use multiple binding sites one after another in long regions of disorder as we recently discussed.23 Of course, these additional, multisite mechanisms can be multiplexed via one-to-many and many-to-one mechanisms, thus leading to extremely complicated PPI networks.
Independent of their roles in hub protein interactions, intrinsically disordered proteins (IDPs) lack of specific structure provides the basis for important biological functions,24, 25 such as signal transduction, cell regulation, molecular recognition, and many other functions.26–33 Many of these disorder-utilizing biological functions depend ultimately on disorder-based PPIs. Thus, understanding the structural basis of PPIs involving IDPs is important for a wide variety of biological functions, not just as the mechanistic basis for hub protein function.
With regard to IDP regions involved in binding, various descriptors have been used, such as eukaryotic linear motif (ELMs),34, 35 linear motifs (LMs),36 short linear motif (SLiMs),37, 38 regions of increased structural propensity (RISPs),39 and molecular recognition features (MoRFs).40 All of these describe similar phenomena, despite differing approaches used by the various researchers for identification of binding segments. The identification of ELMs, LMs, or SLiMs starts from sequence pattern or motif-based approaches, whereas the identification of RISPs and MoRFs starts from short regions with binding indicators located within longer regions of predicted disorder.
Predicting PPI sites in proteins can be used to supplement experimental approaches.41, 42 Predicting binding sites by sequence matches to the motifs of ELMs,34, 35 LMs,36 SLiMs,37, 38 or other collections of sequence patterns43–45 provides one strategy for identifying potential binding sites located within IDPs or IDP regions. Using sequence characteristics that indicate short binding regions within longer regions of disorder offers a second strategy that does not depend on specific motifs, and several predictors have been developed that use this second strategy.46–50 Such predictors have been used by experimentalists to help with the identification of binding regions within longer regions of disorder.39, 51
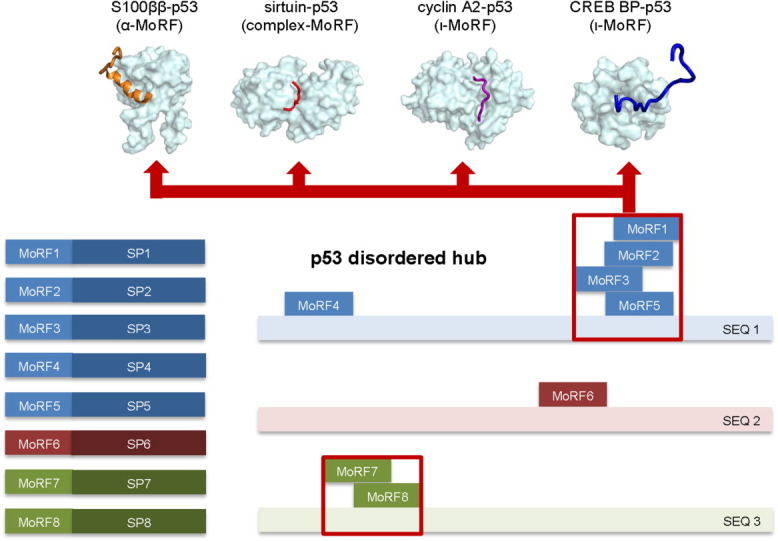
Both a hub protein's ability to bind multiple partners and the general importance of PPIs suggest that the use of flexibility for partner binding by IDPs and IDP regions is of considerable interest. However, despite the importance of understanding how one disordered region can bind to more than one partner, there have been very few structural comparisons at the atomic resolution level, either for one-to-many binding examples or for many-to-one binding examples. For the latter, we know of only two atomic resolution comparisons of more than one IDP binding to a single partner: namely, two different peptides binding to the TAZ1 domain,30 and five different peptides binding to 14-3-3ζ.48 With regard to the former, we likewise know of just three published examples: namely, a short segment from HIF1α bound to two partners, the TAZ1 domain and the asparagine hydroxylase FIH protein,30 a short segment from the C-terminus of p53 bound to four partners, S100ββ, sirtuin, CREB binding protein, and cyclin A2,48 and a larger collection of various short segments bound to multiple partners.52
We have carried out data mining on the Protein Data Bank (PDB) to find additional examples of both one-to-many and many-to-one complexes at atomic resolution. While both datasets are assembled, our focus herein is on the collected examples of one-to-many interactions. Our work on the many-to-one examples is in progress and will be published at a later date.
We have found well over 300 sets that contain segments having the same sequence bound to two or more partners, but here we are focusing on unambiguously the same protein bound to highly divergent partners (e.g., partner pairs with less than 25% sequence identity), thus reducing the numbers down to 23 sets of segments that bind to 2–9 partners. The goal is to provide detailed analyses of the conformational changes enabling the same disordered segment to bind to more than one protein partner. Overall these data support the view that the flexibility of disordered regions is a significant factor in the ability of IDPs to bind to two or more partners. As we assembled this dataset, we also found that ASEs and PTMs were also involved in the process of enabling one disordered region to bind to more than one protein partner. These latter findings suggest that interplay of multiple factors has participated in the evolution of complex PPI networks and might be important in the development of tissue- specific signaling networks.
Results
Summary of our MoRF dataset involving one-to-many binding
We identified 4289 MoRFs from PDB based on their sequence length (5–25 residues). Of these, 452 complexes with small surface areas of interaction were eliminated due to uncertainty regarding the biological significance of the interactions. An additional 689 complexes were excluded because their partners were nonglobular.
To identify overlapping MoRFs, MoRF sequences were mapped back to their parent sequences. A short segment will give exact matches to many unrelated sequences. Because many of the MoRFs are short, only 1805 of the remaining 3148 MoRFs could be unambiguously mapped in an automated fashion to their parent sequences in the Universal Protein Resource (UniProt) database. Based on the overlapping regions in parent sequence mapping (at least one residue), 298 MoRF sets with multiple partnerships were obtained. Structurally redundant partners were discarded from our final dataset based on imposing an upper bound of 25% pairwise sequence identity for every pair of partners.
Finally, 23 MoRF clusters with 61 partners were confirmed by manual inspection to further insure that short peptides were bound to globular partners. Thus, for the dataset investigated herein, each MoRF associates with 2–3 distinct partners on average. A summary of the development of the dataset is given in Table I. The 23 MoRF examples are listed in Table II. The previous two partnerships involving HIF1α were not found in this study because the length of the peptide, 51 amino acids, exceeded the upper bound of 25 residues used in this study. On the other hand, note that the previously described four partnerships involving the carboxy terminal tail of p53 were all found in our dataset,53 showing that our overall strategy found a previously known example the length of which was between the upper and lower thresholds used herein.
Table I.
Description of MoRF Dataset
| Data set | MoRFs | Clusters | MoRFs per cluster |
|---|---|---|---|
| Initial MoRF dataset (5–25)a | 4289 | ||
| MoRF dataset with biological interaction (>400 Å2)b | 3837 | ||
| MoRF dataset with globular partner (>70)c | 3148 | ||
| MoRFs mapped to UniProt sequence databased | 1805 | ||
| MoRFs with overlapped region in mappinge | 1493 | 298 | 5.01 |
| MoRFs without 100% sequence identity in partners | 248 | 87 | 2.85 |
| MoRFs without 25% sequence identity in partners | 214 | 77 | 2.78 |
| MoRFs without atypical casesf | 61 | 23 | 2.65 |
MoRFs with 5–25 residues are the focus of this study.
400 Å2 cutoff was set to filter out the spurious interactions caused by crystal contacts.
Binding partners of MoRF are supposed to be globular proteins having more than 70 residues to fold into a certain conformation. The excluded ones includes interactions between short domain like SH3, chromo domain, A/B chain of insulin, Gramicidin-form ion channels, peptides forming amyloid-like fibril, alpha-helical coiled coil, and de novo proteins.
Most MoRFs cannot be mapped to UniProt are 5–9 residues in length.
MoRFs having one or more overlapping residues with each other.
Atypical cases include, for example, one MoRF bound to more than one partner in the same PDB entry and partners with subsequences that exactly match the entire sequence of another partner.
Table II.
Twenty-Three Examples of MoRFs and Their Secondary Structures
| Bound conformation | Partners | MoRFs | |||||||
|---|---|---|---|---|---|---|---|---|---|
| MoRF examples | N | Helix | Sheet | Coil | Complex | RMSD | Coverage | PTM | AS |
| 8 MoRFs with differently folded partners | 26 | 11 | 1 | ||||||
| 1. Histone H3—N-terminal I | 9 | 0 | 0 | 9 | 0 | 7.07 | 0.21 | 5 | — |
| 2. p53—near C-terminal | 4 | 1 | 0 | 2 | 1 | 6.80 | 0.39 | 1 | Na |
| 3. CTD of RNA polymerase II | 3 | 0 | 0 | 3 | 0 | 8.35 | 0.26 | 3 | — |
| 4. Angiotensin | 2 | 0 | 0 | 2 | 0 | 7.74 | 0.27 | 0 | — |
| 5. HIV envelope glycoprotein | 2 | 0 | 0 | 2 | 0 | 4.16 | 0.41 | 0 | — |
| 6. Histone H3—N-terminal II | 2 | 0 | 0 | 2 | 0 | 8.25 | 0.22 | 2 | — |
| 7. Vasopressin | 2 | 0 | 0 | 2 | 0 | 8.69 | 0.37 | 0 | — |
| 8. p53—near N-terminal | 2 | 2 | 0 | 0 | 0 | 6.18 | 0.62b | 0 | Ya |
| 15 MoRFs with similarly folded partners | 35 | 2 | 4 | ||||||
| 9. Nuclear receptor coactivator 1 and 2 | 5 | 2 | 0 | 2 | 1 | 3.94 | 0.92 | 0 | — |
| 10. Nuclear receptor corepressor 2 | 3 | 2 | 0 | 1 | 0 | 3.43 | 0.85 | 0 | TSa |
| 11. Thyroid receptor associated protein 220 | 3 | 3 | 0 | 0 | 0 | 3.05 | 0.91 | 0 | Yc |
| 12. Nuclear receptor coactivator 1 | 2 | 2 | 0 | 0 | 0 | 5.49 | 0.85 | 0 | — |
| 13. BAK peptide | 2 | 2 | 0 | 0 | 0 | 5.50 | 0.73 | 0 | Na |
| 14. Nuclear receptor 0B2—near N-terminal | 2 | 2 | 0 | 0 | 0 | 3.74 | 0.86 | 0 | Na |
| 15. Troponin I, cardiac muscles | 2 | 0 | 0 | 1 | 1 | 3.01 | 0.79 | 0 | — |
| 16. Nuclear receptor 0B2—near C-terminal | 2 | 1 | 0 | 1 | 0 | 3.88 | 0.80 | 0 | Na |
| 17. Cell death protein GRIM | 2 | 0 | 2 | 0 | 0 | 2.33 | 0.79 | 0 | — |
| 18. Beclin-1 | 2 | 2 | 0 | 0 | 0 | 4.10 | 0.84 | 0 | — |
| 19. Histone H4 | 2 | 0 | 0 | 2 | 0 | 3.93 | 0.50d | 0 | — |
| 20. Bcl-2-like protein 11 (Bim) | 2 | 2 | 0 | 0 | 0 | 2.72 | 0.90 | 0 | Ya |
| 21. Amyloid beta A4 protein | 2 | 0 | 0 | 2 | 0 | 2.93 | 0.84 | 0 | Ya |
| 22. Rhodopsin | 2 | 2 | 0 | 0 | 0 | 4.25 | 0.86 | 0 | — |
| 23. DNA repair protein RAD9 | 2 | 0 | 0 | 2 | 0 | 3.53 | 0.36d | 2 | — |
N, numbers of MoRFs in the set; PTM, post-translation modification; AS, alternative splicing; TS, tissue-specific alternative splicing; —, MoRFs from other species (not from human or mouse).
MoRFs from human.
Although most residues within the two partners can be roughly aligned together, their individual structure varies a lot.
MoRFs from mouse.
Within these two sets, the coverage of alignments is low because one partner is a sub-domain of the other partner but with low sequence identity.
Most sets contain one MoRF interacting individually with two partners, but six of the sets have more than two partners. These are the N-terminus of histone H3, nuclear receptor coactivator 1 and 2, the C-terminus of p53, the NR corepressor 2, the thyroid receptor associated protein 220, and the carboxyl-terminal domain (CTD) of RNA polymerase II (RNAP II). Because MoRFs in the NR coactivator 1 and 2 share similar sequences and can be mapped to the same parent sequence, our method clustered them together as a single set. Most clusters have MoRFs with similar secondary structures in different complexes. Only five of them exhibit a mixture of different secondary structures (Table III).
Table III.
The Combination of Secondary Structure Types in the 23 MoRFs
| Secondary structure | Clusters | Similarly folded partners | Differently folded partners |
|---|---|---|---|
| α + β + ι + Complex | 0 | 0 | 0 |
| α + β + ι | 0 | 0 | 0 |
| α + β + Complex | 0 | 0 | 0 |
| α + ι + Complex | 2 | 1 | 1 |
| β + ι + Complex | 0 | 0 | 0 |
| α + β | 0 | 0 | 0 |
| α + ι | 2 | 2 | 0 |
| α + Complex | 0 | 0 | 0 |
| β + ι | 0 | 0 | 0 |
| β + Complex | 0 | 0 | 0 |
| ι + Complex | 1 | 1 | 0 |
| α | 8 | 7 | 1 |
| β | 1 | 1 | 0 |
| ι | 9 | 3 | 6 |
| Complex | 0 | 0 | 0 |
The goal here was to find the same MoRF sequence bound to structurally distinct partners, so partners having low sequence identity were selected. A sequence identity of 25% was chosen as the upper bound because proteins with sequence identities higher than this value are almost always similar in structure.54 Nevertheless, even though the partners of each MoRF set were selected to have low sequence identity, several partner conformations turned out to exhibit structural similarity. Based on the structure alignment of their partners, the 23 MoRF sets can roughly be grouped into 15 MoRFs with similarly folded partners (with ∼19% sequence identity on average) and 8 MoRFs with differently folded partners (with ∼10% sequence identity on average). Notice that MoRFs with differently folded partners apparently prefer to form irregular secondary structure upon binding, whereas MoRFs with similarly folded but sequence diverse partners tend to prefer to form helix or sheet (Table III).
Two predictors, ANCHOR49 and MoRFpred,55 have been developed to predict partner binding sites within longer regions of disorder. Application of these predictors to the MoRF-containing sequences herein shows that, while both predictors typically indicate binding sites corresponding to the observed MoRFs, neither predictor is particularly accurate with respect to the locations of the binding sites (data not shown). Interestingly, the locations of the MoRFs with similarly folded partners are predicted with slightly greater accuracy by both predictors as compared with the locations of MoRFs that bind to differently folded partners.
15 MoRF sets with partner pairs exhibiting similar folds
Among the 15 MoRFs with partners having similar folds, similar binding profiles and common interacting residues were observed. Partner pairs within 11 of these MoRFs have both a relatively low RMSD and a relatively good structural alignment. The mean sequence identity for structurally aligned binding and nonbinding residues are 42 ± 6% and 20 ± 3%, respectively, within these 11 sets. Binding residues, which are usually on the surface, have about 2.5-fold higher sequence identity than nonbinding surface residues, indicating that these interactions are likely to be biologically significant. For the same MoRF bound to structurally similar partners, only slight conformational changes of MoRF side chains were observed, whereas the backbone conformations of the same MoRF between various complexes are relatively uniform.
Figures 1 and 2 show two examples for which the flexibility needed to accommodate different partner surface features is manifested as side chain rotations. Lysine in nuclear receptor corepressor 2 has different conformations to stretch into the opposite cleft in three complexes to form the associations between the three receptors (Fig. 1). Histidine and arginine in nuclear receptor coactivator 1 (NCOA1) and 2 (NCOA2) also act in a similar way in Figure 2. Here, the two different proteins NCOA 1 and 2 are grouped into one cluster in our dataset because both of them have similar conserved binding sequences containing LxxLL motifs (“HKILHRLLQD” and “HKILHRLLQE”) like other NR-boxes.56 The side chain conformations of the three leucine residues stay nearly the same except for the ones that interact with the androgen receptor.
Figure 1.
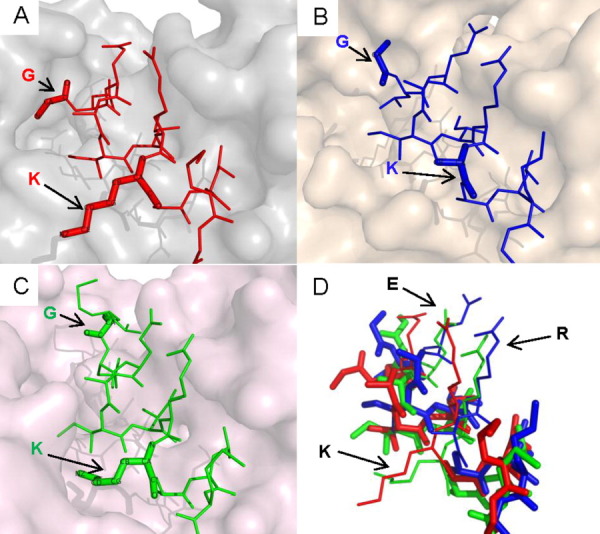
MoRFs in nuclear receptor corepressor 2 bind to three different but structurally similar nuclear receptors. They are (A) estrogen-related receptor gamma (with α-MoRF in 2GPV), (B) progesterone receptor (with α-MoRF in 2OVH), and (C) peroxisome proliferator activated receptor (with ι-MoRF in 1KKQ). (D) In the superimposition of the three complexes, the uncharged residues (in bold) in the core MoRF region maintain relatively stable conformation. An interactive view is available in the electronic version of the article.
Figure 2.
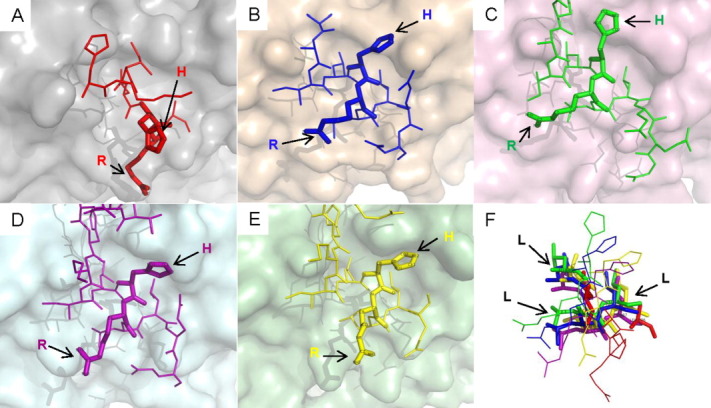
The diagram shows a variety of interactions of MoRFs with highly similar sequences in nuclear receptor coactivator 1 and nuclear receptor coactivator 2. (A) ι-MoRF in nuclear receptor coactivator 2 interacts with androgen receptor (1T65). (B) α-MoRF in glucocorticoid receptor-interacting protein 1 (alternative name of NCOA2) interacts with estrogen receptor (1L2I). (C) complex-MoRF in Nuclear receptor coactivator 1 isoform 1 interacts with orphan nuclear receptor NR1I3 (1XV9). (D) α-MoRF in nuclear receptor coactivator 1 interacts with bile acid receptor (2O9I). (E) ι-MoRF in nuclear receptor coactivator 1 isoform 3 interacts with orphan nuclear receptor pregnane X receptor (3BEJ). (F) The three leucine residues (in bold) of the LxxLL motif are superimposed well in the five complexes. An interactive view is available in the electronic version of the article. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
This example demonstrates that the same protein can be involved in both one-to-many and also many-to-one binding, thus raising the level of network complexity and leading to multiprotein regulatory complexes that can respond to environmental signals. Comparing our one-to-many dataset described herein with our many-to-one dataset (manuscript in preparation) reveals that, of the 23 examples in Table II, there are 12 cases of proteins involved in both one-to-many and many-to-one binding. That is, 12 of the MoRFs in Table II bind to a structured partner that also binds to additional MoRFs having different sequences. Because our identification of one-to-many and many-to-one examples did not involve any steps for identifying MoRFs involved in both mechanisms, we find this number of 12 of 23 involved in both mechanisms to be quite high and to suggest that such dual use of both mechanisms is likely to be a very common feature of PPI networks.
In summary, NCOA binding molecules include many kinds of nuclear receptors, including androgen receptor, estrogen receptor, nuclear receptor subfamily 1, group I member 3 (NR1I3/CAR), bile acid receptor, and pregnane X receptor.57–61 Other detailed investigation into the MoRFs with similar-fold partners was performed and discussed in our previous work.52
Conformational changes of MoRFs with differently folded partners in various interaction complexes
Eight MoRFs in our dataset converted into significantly different conformations to fit onto the surfaces of structurally different molecular partners. For these examples, only a small portion of their partners' residues can be structurally aligned. We selected the three examples with the largest number of partnerships (p53, RNAP II, and histone H3) to illustrate the variable buried surface area of each MoRF residue upon diverse binding (Fig. 3).
Figure 3.
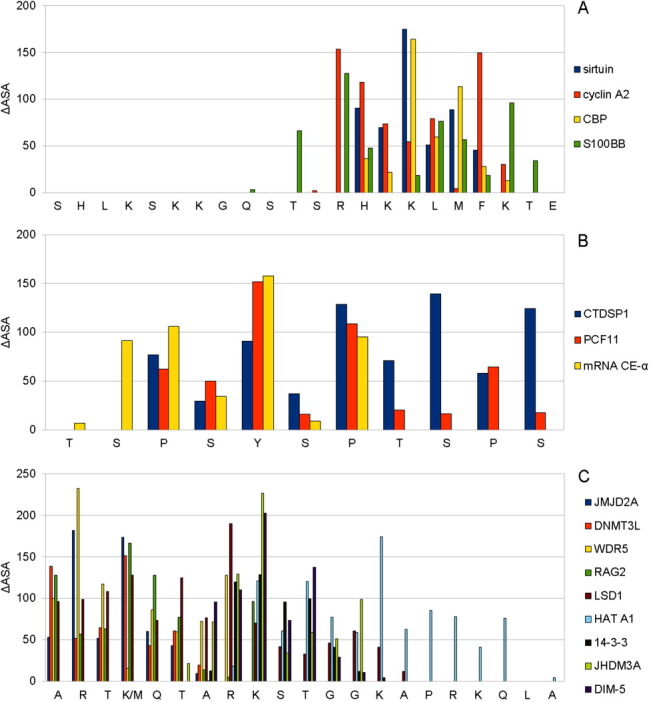
The profiles of solvent surface area changes within three selected MoRF clusters with structurally different partners: (A) p53, (B) RNAP II, and (C) H3. The Y axis gives the change in surface area of each entire residue upon binding, whereas the X axis gives the residues. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Charged residues (R, H, and K), aromatic residues (F and Y), and phosphorylation-related residues (S, T, Y, H, R, and K) in MoRF regions vary substantially in their contributions to binding different partners. In contrast, proline contributions to the different interfaces involving RNAP II remain relatively stable. Unlike MoRFs with similarly folded partners, which generally use their various residues in quite similar ways to associate with relatively conserved interacting residues, each partnership within this set uses conformationally distinct MoRFs and different residues or the same residues with different degrees of burial in their associations with their very distinct partners. That is, the same MoRFs show large variability in their side chain burial and exposure and even shifts in the binding region when binding to structurally divergent partners.
In addition to differential side chain burial and rotations, PTMs are also observed to be associated with the conformational alterations that are observed when the same MoRF binds to different partners, especially for those MoRFs that bind to structurally distinct partners. That is, of the 26 complexes involving differently folded partners, 11 have posttranslationally modified residues. On the other hand, for the MoRFs with similarly folded partners, just 2 of the 35 complexes contain PTMs.
The C-terminus of p53 illustrates the conformational changes of a single MoRF within different partnerships. It was observed to transform either into a complex MoRF, an ι-MoRF (irregular MoRF), or an α-MoRF (helix), in four different structures in our dataset (Fig. 4). The complex MoRF is composed of three residues of β-strand and three residues of coil and was classified as a β-MoRF in our previous work.53 This change from the previous work arose because here we use automated secondary structure assignment (DSSP), whereas the previous work used the crystallographer's assignment of secondary structure.
Figure 4.
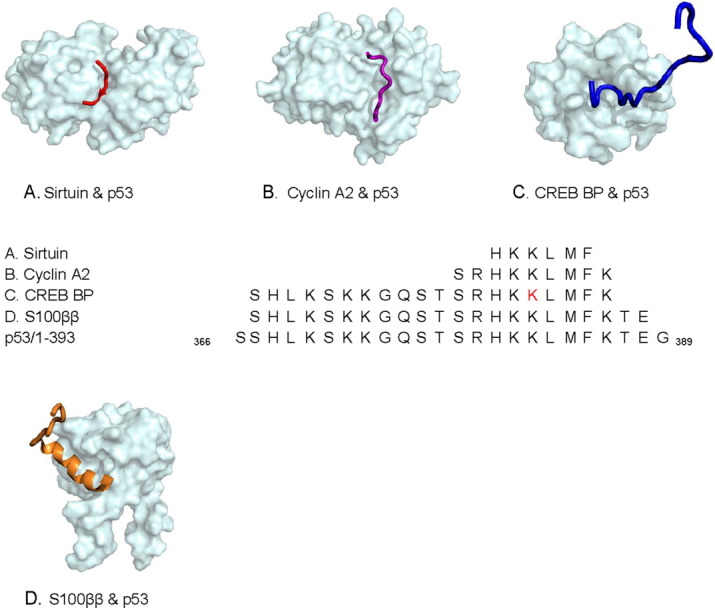
Four different biological molecules interact with C-terminus of p53. (A) Sirtuin: an NAD-dependent deacetylase (with complex-MoRF), 2H59, (B) cyclin A2 (with ι-MoRF), 1H26, (C) CREB binding protein (with ι-MoRF), 1JSP, and (D) S100 calcium-binding protein (α-MoRF), 1DT7. In the sequence alignments, a residue having a posttranslational modification in PDB is indicated in red. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Examination of the MoRFs in RNAP II and Histone H3
Although the two other MoRFs, RNAP II, and H3, with distinctly folded partners have coiled structures for all of their three and nine complexes, respectively, the backbone conformations differ markedly between any two pairs of structure.
Phosphorylation of specific serines (red letters in Fig. 5 in online version) in the carboxyl-terminal domain (CTD) of RNAP II affects not only partner binding to the MoRF but also provides important regulation of transcriptional activity. The CTD in RNAP II is composed of up to 52 heptapeptide repeats (YSPTSPS), which are important for polymerase activity.62 Efficient capping, splicing, and polyadenylation of mRNAs all require the CTD portion of RNAP II. For example, the CTD small phosphatase 1 (CTDSP1) catalyzes the dephosphorylation of Ser5 within the tandem seven residues repeats, causing the initiation of RNAP II transcription [Fig. 5(A)].63 The Ser2-phosphorylated CTD binds to a CTD-interacting domain (CID) in protein1 of cleavage and polyadenylation factor I (PCF11), which is essential for transcription elongation 3′ and RNA processing [Fig. 5(B)].64 The mRNA capping enzyme (mRNA CE) is recruited to the transcription complex, catalyzing its reaction through the binding of the phosphorylated Ser5 in CTD of RNAP II [Fig. 5(C)].65 The capping modification is helpful in the recognition and attachment of mRNA to the ribosome as well as protection from exonucleases.
Figure 5.
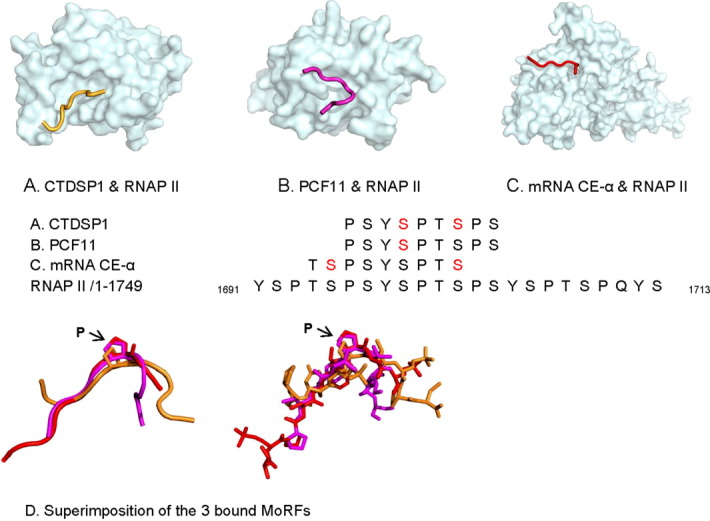
The MoRF mechanism plays a role in mediating interactions involving the CTD of RNA polymerase II. (A) CTD small phosphatase 1 (with ι-MoRF), 2GHQ, (B) protein 1 of cleavage and polyadenylation factor I (with ι-MoRF), 1SZA, (C) mRNA capping enzyme alpha subunit (with ι-MoRF), 1P16, and (D) similar bends near Pro 1700 occurs in all three bound MoRFs. In the sequence alignments, residues in red indicate residues with PTMs in PDB. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The three bound MoRFs in RNAP II all seem to exhibit a bend at a similar location. To gain greater insight, these three MoRFs were structurally aligned [Fig. 5(D)]. Two of the MoRFs (bound to PCF11 and mRNA CE-α) show very similar backbone traces with bends at Pro 1700. The third MoRF (bound to CTDSP1) also shows a bend near Pro 1700, but the backbone trace and location of the bend relative to Pro 1700 are different from the other two examples [Fig. 5(D)].
Because these sequences typically contain just one MoRF binding site for multiple partners, this raises the possibility that partner competition for the single site could be an important regulatory feature these binding interactions. In contrast, for the CTD of RNAP II, the MoRF sequence is repeated more than 50 times. These MoRFs may adapt different structures as they bind to alternative partners. The interplay between partner competition and repeated binding sites may provide a mechanism for subtle and tunable regulation of MoRF/partner interactions.
The MoRF in Histone H3, which contains the maximal number of partners in our dataset, interacts with nine structurally different partners using residues from 2 to 22 in the sequence (Fig. 6). Even though all nine MoRFs are classified as coiled structures, some residues within the MoRF region form helical or strand-like structures upon binding to the different partner proteins. Among the nine binding partners of the N-terminal tail of histone H3, there are several enzymes that are implicated in PTMs. This N-terminal tail that protrudes from the globular nucleosome core can undergo several different types of epigenetic modifications that influence cellular processes. These modifications include the covalent attachment of methyl or acetyl groups to lysine and arginine amino acids and the phosphorylation of serine or threonine. Some of these modifications are included in our data set and characterized in Figure 6 (with the modified residues marked in red).
Figure 6.

Nine different binding partners of ι-MoRFs in the N-terminus of histone H3. Its partners include (A) Jumonji domain-containing protein 2A, 2GFA, (B) DNA-methyltransferase 3-like, 2PVC, (C) WD-repeat protein 5, 2H6K, (D) VDJ recombination-activating protein 2, 2V83, (E) lysine-specific demethylase 1, 2V1D, (F) histone acetyltransferase (HAT A1), 1PU9, (G) 14-3-3 protein zeta/delta, 2C1J, (H) Jmjc domain-containing histone demethylation protein 3A, 2Q8C, and (I) histone H3 methyltransferase DIM-5, 1PEG. (J) Schematic diagram of histone H3 protein shows its predicted and validated disordered tails and a central folded domain. Structural data and various disordered binding site predictors reveal the potential binding regions of H3 are highly associated with posttranslationally modified sites. The residues in red in (A–I) are PTM sites in PDB, and the methionine in gray is a residue that was mutated for the structural study. The annotated PTM sites on the entire H3 in J is from UniProt. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The double Tudor domain of JMJD2A, a Jmjc domain-containing histone demethylase, binds methylated Lys 5 on Histone H3. This complex functions as a transcription repressor [Fig. 6(A)].66 The DNA-methyltransferase 3-like (DNMT3L) protein recognizes the histone H3 tails with unmethylated Lys5 and stimulates de novo DNA methylation by engaging the DNMT3A2 molecule [Fig. 6(B)].67 The WD-repeat protein 5 (WDR5) is a core component of SET1-family complexes that achieve transcriptional activation via methylation of histone H3 on Lys 5 [Fig. 6(C)].68 The recombination activating gene 2 contains a plant homeodomain that recognizes histone H3 methylated at Lys 5 and influences V(D)J recombination [Fig. 6(D)].69 Histone demethylase LSD1 regulates transcription by demethylating Lys5 of histone H3 [Fig. 6(E)].70 A substrate-like peptide was generated by a K5M mutation (marked in gray in Fig. 6) because this mutation led to 30-fold increase in binding affinity thereby helping to stabilize the complex. Phosphorylation at Ser 11 of histone H3 enhances GCN5 histone acetyltransferase mediated Lys 15 acetylation, promoting transcription [Fig. 6(F)].71 The 14-3-3 isoforms present a class of proteins that mediate the effect of Ser 11 phosphorylated histone H3 [Fig. 6(G)].72 The jumonji domain of JHDM3A (JMJD2A) catalyzes the demethylation of di- and tri-methylated Lys10 and Lys 37 in histone H3 [Fig. 6(H)].73 DIM-5 is a histone H3 Lys 9 methyltransferase, that is, essential for DNA methylation [Fig. 6(I)].74
Figure 6(J) summarizes the results of disorder/order predictions, potential interacting regions, and annotated PTM sites in UniProt in human histone H3. In general, H3 has a central structural region (residue 58–132) that matches to a Pfam family (histone: core histone H2A/H2B/H3/H4) and a long N-term disordered tail (around 38–48 residues in length). A similar disorder/order estimate was given by PONDR VSL2B. Within current 294 PDB entries related to human histone H3 (27-Mar-12), 40 complexes were found to include H3 fragments (MoRFs) between residue 2 and 34. This N-terminal binding region was not recognized by both MoRF1 and MoRF2 predictors,47, 75 but we claim the reasons may be because these two predictors were built specifically for helix MoRFs, not coil MoRFs like the ones in H3. Figure 6(A–I) shows the nine MoRFs found in the same region are all coil MoRFs. Part of the binding region can be predicted by ANCHOR,49 whereas the entire region can be found by MoRFpred50 method. Based on the sequence annotations of UniProt database, most PTM sites of H3 are located in the N-terminus of H3, implying the functionally regulation sites may highly tie with MoRFs within disordered regions.
Discussion
Our 23 MoRF examples of one-to-many binding comprise a special set, containing partners with little sequence similarity that bind to MoRFs with identical sequences. This approach is distinct from the concept of structural compensation or coadaptation, for which mutations on one partner are linked to compensating mutations on the partner.76 It would certainly be possible to lift the requirement of MoRF sequence identity to thereby study coadaptation in complexes involving disordered proteins. Indeed, we have work in progress along these lines for a few specific examples to determine whether coadaptation between two structured proteins is different from coadaptation between structured proteins and MoRFs.
There have been several previous bioinformatics investigations of large numbers of IDP-involving PPIs at a high level, without paying attention to the structural details.47, 75, 77, 78 Instead, our approach here is to investigate fewer MoRF examples, but in greater detail in order to develop a deeper understanding of how IDPs can alter their conformations so as to be able to bind to structurally distinct partners. Our observations demonstrated that, in general, conformation flexibility allows for both subtle and complex structural variation, thereby enabling the same sequence to transform onto the diverse and distinctively shaped binding sites provided by their partners.
The MoRFs collected and grouped into one cluster herein are typically gathered from different organisms. As suggested by others, through parallel or convergent evolution, such MoRFs can exist as conserved functional motifs or regions among various species, such as human, mouse, yeast, E. coli, or even viruses.79
As pointed out previously,77 such short linear motifs are amenable to convergent evolution due to the limited number of mutations that are necessary for the generation of a useful motif. In fact, motifs are commonly used as adding new functional modules within a proteome, especially in higher eukaryotes.80 These short functional linear motifs are hypothesized to have higher levels of conservation, to frequently evolve convergently, to preferentially occur in disordered regions and to often form a specific secondary structure when bound to interaction partners.79 This observation fits in with the conception that alternative inclusion of exons in different tissues provides functional diversity of proteins. In fact, embedded conserved binding motifs and PTM sites are both rich in tissue-dependent protein segments.81 The tissue-dependent spliced regions have higher percentage of protein disorder that likely form conserved interaction surface and participate significantly more protein interactions.82
Among the 23 MoRFs in our dataset, three MoRFs (TRAP220, Bim, and amyloid A4 protein) were annotated in UniProt to be located in alternatively spliced regions. Alternative splicing has the potential to add or delete an entire MoRF region. In addition, MoRF-related functions could be modulated by alternative splicing by changing the expression patterns, localization and regulation. These complex mechanisms could lead to broad functional and regulatory diversity. For example, pro-apoptosis protein Bim has 17 isoforms. Its predominant three isoforms, BimEL, BimL and BimS, all have the MoRF region (BH3 ligand) “DMRPEIWIAQELRRIGDEFNAYYAR,” which is responsible for binding selectivity for their pro-survival protein binding targets and starting Bcl-2 regulated apoptosis. Those Bim isoforms lacking the BH3 ligand, for example, Bimβ1-7, also lack pro-apoptotic activities.
Two additional MoRFs were reported to have ASEs based on studies of the tissue-specific splicing exon data set.81 A MoRF region from nuclear receptor corepressor 2 is specifically expressed in only 1 of 14 tissue types. As was pointed out,81 the tissue-specific alternative splicing that leads to presence and absence of binding sites in disordered protein regions leads to the “rewiring” of PPI networks and may, therefore, contribute fundamentally to tissue development. It would be very interesting to develop models for the alterations in PPI networks in different tissues that arise from alternative splicing, but unfortunately the partners for the tissue-specific MoRFs are simply not known.
In a previous study, we found that alternatively spliced regions of RNA code for protein disorder much more often than for regions of structure, and we showed that such alternative splicing could lead to inclusion or exclusion of binding sites within the disordered regions.83 Interestingly, of the human MoRFs studied here, 50% (4 of 8) are in exon regions that have been identified as included or excluded by alternative splicing. The discussion in the previous paragraph suggests that a concerted effort should be made to identify additional MoRFs that map to tissue-specific alternatively spliced regions and to identify their partners as well.
In our previous study of the carboxy terminal tail of p53 bound to four different partners, we noticed that two of the complexes were distinguished by having PTMs, namely lysine acetylations for both examples. Furthermore, the acetate groups both became buried in the interfaces between the two MoRFs and their respective partners.48 In this study, we discovered that differences in PTMs occur commonly when MoRFs bind to alternative partners. Furthermore, this use of modified side chains to bind to one of two partners is most common when the two partners are structurally distinct. Indeed in this study, of 13 MoRFs containing PTMs, 11 involve MoRFs that bind to differently folded partners, thus providing additional observations in support of this concept. Finally, the chemical group added via the modification is typically found buried or partially buried in the interface between the MoRF and its partner, which strongly suggests that PTM provides an important part of the signal for the MoRF to bind to an alternative partner.
Phosphorylation occurs much more often in intrinsically disordered as compared with structured regions of proteins.84, 85 Recently, several other types of PTM have been shown to prefer disorder over structure.86 The results presented herein suggest that such a modification can be used to change the partner preference of a given MoRF, thus leading to switching the connections of a PPI network.
Our results contribute to a better understanding of the role of disorder binding regions (MoRFs) that may serve as protein interaction hubs. Exploring the diverse binding partners of our collected MoRF sets and the corresponding complex conformations definitely give us a general Rosetta stone to interpret the underlying biological mechanisms and evolutional aptness. The importance and indispensability of hub proteins is apparent as they appear to evolve more slowly and are more likely to be vital for survival. Given their importance, many human disease-associated proteins related to cancer, diabetes, autoimmune disease, neurodegenerative disease, and cardiovascular disease are found to have predicted disordered binding regions (MoRFs) as we expect.87 These MoRFs associate with other structured partners and considered as promising druggable interactions because of their high specificity and low affinity for binding. Binding with relatively low affinity is an advantageous attribute for transient, conditional, and tunable interactions, which is needed for many regulatory events. Therefore, this study will help to pave the way for the development of novel pathways by designing intervening disordered peptides having binding sites for particular partners but with tighter interactions.
Materials and Methods
MoRF data sets
Our disordered hub dataset was extracted from PDB by analyzing the complex structures that have short nonglobular protein fragments bound to large globular structured partners. In this article, we concentrated on those MoRFs which are short nonglobular protein fragments whose visible residues in crystallographic electron density maps included between 5 and 25 residues and binding partners are globular proteins greater than 70 amino acids in length. The PDB entries we used were released on March 28, 2008.
An interface size (ΔASA) of 400 Å2 was used to discriminate biologically relevant interactions and nonbiological interactions caused by crystal packing contacts in this study.88 The same cutoff was previously chosen by the authors of the protein quaternary structure file server, because the minimal ΔASA of homodimers and heterodimer are about 370 Å2 and 640 Å2, respectively.89
Characterization of MoRF clusters that perform one-to-many binding like p53
To discover specific disordered regions binding to multiple structured partners like p53, we used a FASTA program to align each MoRF sequence to the UniProt sequence database. This database encompasses the UniProtKB/Swiss-Prot and UniProtKB/TrEMBL databases. The e-value was set at 1000 while carrying out the similarity search. Following that, we only kept those MoRFs which had overlapping regions (circled ones in Fig. 7) in their parent sequence mapping and used a cluster algorithm (wherein at least one residue overlapped with the rest of the MoRFs in the same cluster).
Figure 7.
A schematic diagram to show how we constructed our disordered hub dataset by aligning and clustering MoRF sequences from complex structures. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Removal of redundant MoRFs in MoRF clusters based on sequence identity
As our research is focused upon those MoRFs from the same disordered region which bind to structurally different partners, we used the BLASTCLUST program to remove any redundant structured partners in our dataset based on 100% and 25% sequence identity. That means that those specific MoRFs are in one disordered region, but they use distinct residues to form bonding with different structured partners.
Removal of atypical MoRFs in MoRF clusters
After examination of the entire MoRF dataset manually, we found there were several unanticipated cases that were not consistent and needed to be removed from our dataset. They include the cases involving one MoRF interacting with more than one partner in a single PDB entry or a partner molecule which may be a subset of another partner in the same cluster.
Secondary structure assignment of MoRF
We classified MoRFs into four different types (α, β, ι, and complex) based on their secondary structure type, which has the largest percentage value of the four types mentioned above. If there is no clear preponderance of any one secondary type (which is at least 1% greater than the other two types), we classified it as a complex-MoRF. Only the residues on the interface were counted. DSSP was used as the secondary structure assignment program here.
Acknowledgments
The authors are very grateful to Dr. Thomas D. Hurley and Dr. Yaoqi Zhou for providing helpful suggestions and discussions and Dr. M. Madan Babu and Ms. Marija Buljan for helping with the use of their tissue-specific alternative splicing dataset.
References
- 1.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 2.Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 3.Hasty J, Collins JJ. Protein interactions. Unspinning the web. Nature. 2001;411:30–31. doi: 10.1038/35075182. [DOI] [PubMed] [Google Scholar]
- 4.Pauling L. A theory of the structure and process of formation of antibodies. J Am Chem Soc. 1940;62:2643–2657. [Google Scholar]
- 5.Dunker AK, Garner E, Guilliot S, Romero P, Albrecht K, Hart J, Obradovic Z, Kissinger C, Villafranca JE. Protein disorder and the evolution of molecular recognition: theory, predictions and observations. Pac Symp Biocomput. 1998:473–484. [PubMed] [Google Scholar]
- 6.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci USA. 1996;93:11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James LC, Roversi P, Tawfik DS. Antibody multispecificity mediated by conformational diversity. Science. 2003;299:1362–1367. doi: 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- 8.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 9.Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dosztanyi Z, Chen J, Dunker AK, Simon I, Tompa P. Disorder and sequence repeats in hub proteins and their implications for network evolution. J Proteome Res. 2006;5:2985–2995. doi: 10.1021/pr060171o. [DOI] [PubMed] [Google Scholar]
- 11.Boxem M, Maliga Z, Klitgord N, Li N, Lemmens I, Mana M, de Lichtervelde L, Mul JD, van de Peut D, Devos M, Simonis N, Yildirim MA, Cokol M, Kao HL, de Smet AS, Wang HD, Schlaitz AL, Hao T, Milstein S, Fan CY, Tipsword M, Drew K, Galli M, Rhrissorrakrai K, Drechsel D, Koller D, Roth FP, Iakoucheva LM, Dunker AK, Bonneau R, Gunsalus KC, Hill DE, Piano F, Tavernier J, van den Heuvel S, Hyman AA, Vidal M. A protein domain-based interactome network for C-elegans early embryogenesis. Cell. 2008;134:534–545. doi: 10.1016/j.cell.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ekman D, Light S, Bjorklund AK, Elofsson A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae. Genome Biol. 2006;7:R45. doi: 10.1186/gb-2006-7-6-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patil A, Nakamura H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006;580:2041–2045. doi: 10.1016/j.febslet.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 14.Singh GP, Dash D. Intrinsic disorder in yeast transcriptional regulatory network. Proteins. 2007;68:602–605. doi: 10.1002/prot.21497. [DOI] [PubMed] [Google Scholar]
- 15.Singh GP, Ganapathi M, Dash D. Role of intrinsic disorder in transient interactions of hub proteins. Proteins. 2007;66:761–765. doi: 10.1002/prot.21281. [DOI] [PubMed] [Google Scholar]
- 16.Kim PM, Sboner A, Xia Y, Gerstein M. The role of disorder in interaction networks: a structural analysis. Mol Syst Biol. 2008;4:179. doi: 10.1038/msb.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjorklund AK, Light S, Hedin L, Elofsson A. Quantitative assessment of the structural bias in protein-protein interaction assays. Proteomics. 2008;8:4657–4667. doi: 10.1002/pmic.200800150. [DOI] [PubMed] [Google Scholar]
- 18.Higurashi M, Ishida T, Kinoshita K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 2008;17:72–78. doi: 10.1110/ps.073196308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kahali B, Ahmad S, Ghosh TC. Exploring the evolutionary rate differences of party hub and date hub proteins in Saccharomyces cerevisiae protein-protein interaction network. Gene. 2009;429:18–22. doi: 10.1016/j.gene.2008.09.032. [DOI] [PubMed] [Google Scholar]
- 20.Manna B, Bhattacharya T, Kahali B, Ghosh TC. Evolutionary constraints on hub and non-hub proteins in human protein interaction network: insight from protein connectivity and intrinsic disorder. Gene. 2009;434:50–55. doi: 10.1016/j.gene.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Patil A, Kinoshita K, Nakamura H. Domain distribution and intrinsic disorder in hubs in the human protein-protein interaction network. Protein Sci. 2010;19:1461–1468. doi: 10.1002/pro.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patil A, Kinoshita K, Nakamura H. Hub promiscuity in protein-protein interaction networks. Intl J Mol Sci. 2010;11:1930–1943. doi: 10.3390/ijms11041930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uversky VN, Dunker AK. Understanding protein non-folding. Biochim Biophys Acta. 2010;1804:1231–1264. doi: 10.1016/j.bbapap.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 25.Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 26.Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 27.Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Prot Chem. 2002;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- 28.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 29.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 31.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Uversky VN, Obradovic Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J Proteome Res. 2007;6:1882–1898. doi: 10.1021/pr060392u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vucetic S, Xie H, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 2. Cellular components, domains, technical terms, developmental processes, and coding sequence diversities correlated with long disordered regions. J Proteome Res. 2007;6:1899–1916. doi: 10.1021/pr060393m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie H, Vucetic S, Iakoucheva LM, Oldfield CJ, Dunker AK, Obradovic Z, Uversky VN. Functional anthology of intrinsic disorder. 3. Ligands, post-translational modifications, and diseases associated with intrinsically disordered proteins. J Proteome Res. 2007;6:1917–1932. doi: 10.1021/pr060394e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puntervoll P, Linding R, Gemund C, Chabanis-Davidson S, Mattingsdal M, Cameron S, Martin DM, Ausiello G, Brannetti B, Costantini A, Ferre F, Maselli V, Via A, Cesareni G, Diella F, Superti-Furga G, Wyrwicz L, Ramu C, McGuigan C, Gudavalli R, Letunic I, Bork P, Rychlewski L, Kuster B, Helmer-Citterich M, Hunter WN, Aasland R, Gibson TJ. ELM server: a new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 2003;31:3625–3630. doi: 10.1093/nar/gkg545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gould CM, Diella F, Via A, Puntervoll P, Gemund C, Chabanis-Davidson S, Michael S, Sayadi A, Bryne JC, Chica C, Seiler M, Davey NE, Haslam N, Weatheritt RJ, Budd A, Hughes T, Pas J, Rychlewski L, Trave G, Aasland R, Helmer-Citterich M, Linding R, Gibson TJ. ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res. 2010;38:D167–D180. doi: 10.1093/nar/gkp1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuxreiter M, Tompa P, Simon I. Local structural disorder imparts plasticity on linear motifs. Bioinformatics. 2007;23:950–956. doi: 10.1093/bioinformatics/btm035. [DOI] [PubMed] [Google Scholar]
- 37.Davey NE, Shields DC, Edwards RJ. SLiMDisc: short, linear motif discovery, correcting for common evolutionary descent. Nucleic Acids Res. 2006;34:3546–3554. doi: 10.1093/nar/gkl486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edwards RJ, Davey NE, Shields DC. SLiMFinder: a probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins. PLoS One. 2007;2:e967. doi: 10.1371/journal.pone.0000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callaghan AJ, Aurikko JP, Ilag LL, Gunter Grossmann J, Chandran V, Kuhnel K, Poljak L, Carpousis AJ, Robinson CV, Symmons MF, Luisi BF. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J Mol Biol. 2004;340:965–979. doi: 10.1016/j.jmb.2004.05.046. [DOI] [PubMed] [Google Scholar]
- 40.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 41.Obenauer JC, Yaffe MB. Computational prediction of protein-protein interactions. Methods Mol Biol. 2004;261:445–468. doi: 10.1385/1-59259-762-9:445. [DOI] [PubMed] [Google Scholar]
- 42.Valencia A, Pazos F. Computational methods to predict protein interaction partners. In: Panchenko A, Przytycka TM, editors. Protein-protein interactions and networks. London: Springer-Verlag; 2008. pp. 67–81. [Google Scholar]
- 43.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kadaveru K, Vyas J, Schiller MR. Viral infection and human disease—insights from minimotifs. Frontiers Biosci. 2008;13:6455–6471. doi: 10.2741/3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mi T, Merlin JC, Deverasetty S, Gryk MR, Bill TJ, Brooks AW, Lee LY, Rathnayake V, Ross CA, Sargeant DP, Strong CL, Watts P, Rajasekaran S, Schiller MR. Minimotif Miner 3.0: database expansion and significantly improved reduction of false-positive predictions from consensus sequences. Nucleic Acids Res. 2012;40:D252–D260. doi: 10.1093/nar/gkr1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garner E, Romero P, Dunker AK, Brown C, Obradovic Z. Predicting binding regions within disordered proteins. Genome Inform Ser Workshop Genome Inform. 1999;10:41–50. [PubMed] [Google Scholar]
- 47.Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 48.Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK. Flexible nets: disorder and induced fit in the associations of p53 and 14-3–3 with their partners. BMC Genomics. 2008;9:S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics. 2009;25:2745–2746. doi: 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Disfani FM, Hsu WL, Mizianty MJ, Oldfield CJ, Xue B, Dunker A, Uversky V, Kurgan L. MoRFpred, a computational tool for sequence-based prediction and characterization of disorder-to-order transitioning binding sites in proteins. Bioinformatics. 2012;28:i75–i83. doi: 10.1093/bioinformatics/bts209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourhis J-M, Johansson K, Receveur-Brechot V, Oldfield CJ, Dunker KA, Canard B, Longhi S. The C-terminal domain of measles virus nucleoprotein belongs to the class of intrinsically disordered proteins that fold upon binding to their physiological partner. Virus Res. 2004;99:157–167. doi: 10.1016/j.virusres.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 52.Hsu WL, Oldfield C, Meng J, Huang F, Xue B, Uversky VN, Romero P, Dunker AK. Intrinsic protein disorder and protein-protein interactions. Pac Symp Biocomput. 2012:116–127. [PubMed] [Google Scholar]
- 53.Oldfield CJ, Meng J, Yang JY, Uversky VN, Dunker AK. Intrinsic disorder in protein-protein interaction networks: case studies of complexes involving p53 and 14-3-3. BIOCOMP. 2007;07:553–566. [Google Scholar]
- 54.Doolittle RF. Of Urfs and Orfs: a primer on how to analyze derived amino acid sequences. Mill Valley, California: University Science Books; 1886. [Google Scholar]
- 55.Disfani FM, Hsu WL, Mizianty MJ, Oldfield CJ, Xue B, Dunker AK, Uversky VN, Kurgan L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics. 2012;28:i75–i83. doi: 10.1093/bioinformatics/bts209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, Katzenellenbogen JA, Agard DA, Greene GL. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–364. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 58.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol Cell. 2004;16:919–928. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 59.Estebanez-Perpina E, Moore JM, Mar E, Delgado-Rodrigues E, Nguyen P, Baxter JD, Buehrer BM, Webb P, Fletterick RJ, Guy RK. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J Biol Chem. 2005;280:8060–8068. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]
- 60.Soisson SM, Parthasarathy G, Adams AD, Sahoo S, Sitlani A, Sparrow C, Cui J, Becker JW. Identification of a potent synthetic FXR agonist with an unexpected mode of binding and activation. Proc Natl Acad Sci USA. 2008;105:5337–5342. doi: 10.1073/pnas.0710981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xue Y, Chao E, Zuercher WJ, Willson TM, Collins JL, Redinbo MR. Crystal structure of the PXR-T1317 complex provides a scaffold to examine the potential for receptor antagonism. Bioorg Med Chem. 2007;15:2156–2166. doi: 10.1016/j.bmc.2006.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosonina E, Blencowe BJ. Analysis of the requirement for RNA polymerase II CTD heptapeptide repeats in pre-mRNA splicing and 3′-end cleavage. RNA. 2004;10:581–589. doi: 10.1261/rna.5207204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Kim Y, Genoud N, Gao J, Kelly JW, Pfaff SL, Gill GN, Dixon JE, Noel JP. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol Cell. 2006;24:759–770. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meinhart A, Cramer P. Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature. 2004;430:223–226. doi: 10.1038/nature02679. [DOI] [PubMed] [Google Scholar]
- 65.Fabrega C, Shen V, Shuman S, Lima CD. Structure of an mRNA capping enzyme bound to the phosphorylated carboxy-terminal domain of RNA polymerase II. Mol Cell. 2003;11:1549–1561. doi: 10.1016/s1097-2765(03)00187-4. [DOI] [PubMed] [Google Scholar]
- 66.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 67.Ooi SK, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, Cheng X, Bestor TH. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruthenburg AJ, Wang W, Graybosch DM, Li H, Allis CD, Patel DJ, Verdine GL. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol. 2006;13:704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramon-Maiques S, Kuo AJ, Carney D, Matthews AG, Oettinger MA, Gozani O, Yang W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci USA. 2007;104:18993–18998. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Forneris F, Binda C, Adamo A, Battaglioli E, Mattevi A. Structural basis of LSD1-CoREST selectivity in histone H3 recognition. J Biol Chem. 2007;282:20070–20074. doi: 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- 71.Clements A, Poux AN, Lo WS, Pillus L, Berger SL, Marmorstein R. Structural basis for histone and phosphohistone binding by the GCN5 histone acetyltransferase. Mol Cell. 2003;12:461–473. doi: 10.1016/s1097-2765(03)00288-0. [DOI] [PubMed] [Google Scholar]
- 72.Macdonald N, Welburn JP, Noble ME, Nguyen A, Yaffe MB, Clynes D, Moggs JG, Orphanides G, Thomson S, Edmunds JW, Clayton AL, Endicott JA, Mahadevan LC. Molecular basis for the recognition of phosphorylated and phosphoacetylated histone h3 by 14-3-3. Mol Cell. 2005;20:199–211. doi: 10.1016/j.molcel.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 73.Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC. Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol. 2007;14:689–695. doi: 10.1038/nsmb1273. [DOI] [PubMed] [Google Scholar]
- 74.Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell. 2003;12:177–185. doi: 10.1016/s1097-2765(03)00224-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cheng Y, Oldfield CJ, Meng J, Romero P, Uversky VN, Dunker AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry. 2007;46:13468–13477. doi: 10.1021/bi7012273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fares MA, Ruiz-Gonzalez MX, Labrador JP. Protein coadaptation and the design of novel approaches to identify protein-protein interactions. IUBMB Life. 2011;63:264–271. doi: 10.1002/iub.455. [DOI] [PubMed] [Google Scholar]
- 77.Diella F, Haslam N, Chica C, Budd A, Michael S, Brown NP, Trave G, Gibson TJ. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- 78.Gfeller D, Butty F, Wierzbicka M, Verschueren E, Vanhee P, Huang HM, Ernst A, Dar N, Stagljar I, Serrano L, Sidhu SS, Bader GD, Kim PM. The multiple-specificity landscape of modular peptide recognition domains. Mol Syst Biol. 2011;7:484. doi: 10.1038/msb.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Davey NE, Van Roey K, Weatheritt RJ, Toedt G, Uyar B, Altenberg B, Budd A, Diella F, Dinkel H, Gibson TJ. Attributes of short linear motifs. Mol Biosyst. 2012;8:268–281. doi: 10.1039/c1mb05231d. [DOI] [PubMed] [Google Scholar]
- 80.Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, Toedt G, Uyar B, Seiler M, Budd A, Jodicke L, Dammert MA, Schroeter C, Hammer M, Schmidt T, Jehl P, McGuigan C, Dymecka M, Chica C, Luck K, Via A, Chatr-Aryamontri A, Haslam N, Grebnev G, Edwards RJ, Steinmetz MO, Meiselbach H, Diella F, Gibson TJ. ELM—the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40:D242–D251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buljan M, Chalancon G, Eustermann S, Wagner GP, Fuxreiter M, Bateman A, Babu MM. Tissue-specific splicing of disordered segments that embed binding motifs rewires protein interaction networks. Mol Cell. 2012;46:871–883. doi: 10.1016/j.molcel.2012.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ellis JD, Barrios-Rodiles M, Colak R, Irimia M, Kim T, Calarco JA, Wang X, Pan Q, O'Hanlon D, Kim PM, Wrana JL, Blencowe BJ. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell. 2012;46:884–892. doi: 10.1016/j.molcel.2012.05.037. [DOI] [PubMed] [Google Scholar]
- 83.Romero PR, Zaidi S, Fang YY, Uversky VN, Radivojac P, Oldfield CJ, Cortese MS, Sickmeier M, LeGall T, Obradovic Z, Dunker AK. Alternative splicing in concert with protein intrinsic disorder enables increased functional diversity in multicellular organisms. Proc Natl Acad Sci USA. 2006;103:8390–8395. doi: 10.1073/pnas.0507916103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gao J, Thelen JJ, Dunker AK, Xu D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol Cell Proteomics. 2010;9:2586–2600. doi: 10.1074/mcp.M110.001388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao J, Xu D. Correlation between posttranslational modification and intrinsic disorder in protein. Pac Symp Biocomput. 2012:94–103. [PMC free article] [PubMed] [Google Scholar]
- 87.Cheng Y, LeGall T, Oldfield CJ, Mueller JP, Van YY, Romero P, Cortese MS, Uversky VN, Dunker AK. Rational drug design via intrinsically disordered protein. Trends Biotechnol. 2006;24:435–442. doi: 10.1016/j.tibtech.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 88.Carugo O, Argos P. Protein-protein crystal-packing contacts. Protein Sci. 1997;6:2261–2263. doi: 10.1002/pro.5560061021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]