

Abstract
Post-translational modifications (PTM) of proteins can control complex and dynamic cellular processes via regulating interactions between key proteins. To understand these regulatory mechanisms, it is critical that we can profile the PTM-dependent protein–protein interactions. However, identifying these interactions can be very difficult using available approaches, as PTMs can be dynamic and often mediate relatively weak protein–protein interactions. We have recently developed CLASPI (cross-linking-assisted and stable isotope labeling in cell culture-based protein identification), a chemical proteomics approach to examine protein–protein interactions mediated by methylation in human cell lysates. Here, we report three extensions of the CLASPI approach. First, we show that CLASPI can be used to analyze methylation-dependent protein–protein interactions in lysates of fission yeast, a genetically tractable model organism. For these studies, we examined trimethylated histone H3 lysine-9 (H3K9Me3)-dependent protein–protein interactions. Second, we demonstrate that CLASPI can be used to examine phosphorylation-dependent protein–protein interactions. In particular, we profile proteins recognizing phosphorylated histone H3 threonine-3 (H3T3-Phos), a mitotic histone “mark” appearing exclusively during cell division. Our approach identified survivin, the only known H3T3-Phos-binding protein, as well as other proteins, such as MCAK and KIF2A, that are likely to be involved in weak but selective interactions with this histone phosphorylation “mark”. Finally, we demonstrate that the CLASPI approach can be used to study the interplay between histone H3T3-Phos and trimethylation on the adjacent residue lysine 4 (H3K4Me3). Together, our findings indicate the CLASPI approach can be broadly applied to profile protein–protein interactions mediated by PTMs.
Keywords: post-translational modification, phosphorylation, protein–protein interaction, histone, chemical proteomics
Introduction
Protein post-translational modification (PTM)1 represents an important mechanism for regulating protein–protein interactions in cells. To develop a method for identifying PTM-dependent protein–protein interactions, we have focused on histone PTMs,2 which can recruit “effector” proteins (so-called readers) onto chromatin to regulate a variety of DNA-templated processes, such as gene transcription, DNA replication, and chromosome segregation.3–5 We recently reported a chemical probe (probe 1, Fig. 1) that captures proteins that recognize trimethylated histone H3 at lysine 4 (H3K4Me3).6 This probe carries a benzophenone moiety, which allows photo-cross-linking to convert weak interactions into covalent linkages, and a terminal alkyne group, which facilitates subsequent bio-orthogonal chemistry-mediated purifications. By combining the use of affinity chromatography and stable isotope labeling in cell culture (SILAC)-based quantitative mass spectrometry, we have used this probe to identify both known and new H3K4Me3-binding proteins in whole-cell proteomes.7 However, the applicability of this strategy to examine other histone modifications, for example, methylation at other residues or phosphorylation-dependent protein–protein interactions has not been explored. Furthermore, its use to profile PTM-dependent protein–protein interactions in genetically tractable model systems, such as fission yeast, has not been examined.
Figure 1.
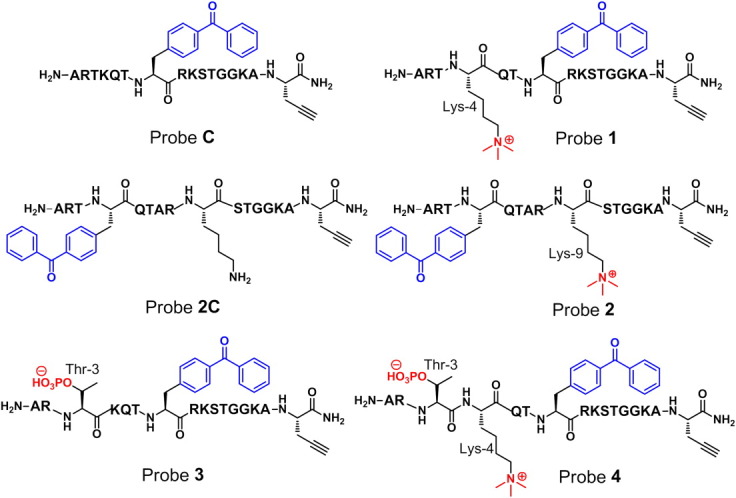
Chemical structures of probes C, 1, 2C, 2, 3, and 4. All probes are designed based on the N-terminal first 15 amino acids of histone H3. The incorporated PTMs (i.e., methylation and phosphorylation) and photo-cross-linker (i.e., benzophenone) are highlighted with red and blue colors, respectively. The key amino acids in the probes are indicated.
Protein PTMs play a central role in regulating normal and disease cell physiology.8 For example, trimethylation of histone H3 at lysine-9 (H3K9Me3) correlates with the repression of gene expression in a variety of organisms. In addition, phosphorylations of histone H3 threonine 3 (H3T3-Phos)9 and Ser10 (H3S10-Phos)10 appear exclusively during cell division,11 the stage of the cell cycle during which duplicated chromosomes are partitioned into two daughter cells. It is now clear that many of these phosphorylations play critical roles in regulating protein–protein interactions. For example, it has recently been shown that H3T3-Phos recruits chromosomal passenger complex (CPC)12 to specific sites on chromosomes to ensure the fidelity of cell division.13–15 However, we currently lack robust methods to profile phosphorylation-dependent protein–protein interactions, particularly those that occur within a narrow time-window of cell biology, such as cell division.
Here, we extend our cross-linking-assisted and SILAC-based protein identification (CLASPI) approach in three ways. First, we report a probe that can be used to identify H3K9Me3-dependent protein–protein interactions and demonstrate its use in fission yeast lysates. Second, we profile protein–protein interactions mediated by a dynamic histone phosphorylation, H3T3-Phos. Finally, we apply our method to study the interplay between histone methylation (H3K4Me3) and phosphorylation (H3T3-Phos).
Results
We first examined whether our CLASPI strategy can be used to study histone methylation-dependent protein–protein interactions other than those mediated by H3K4Me3. We focused on H3K9Me3, a “mark” associated with transcriptionally silent regions of chromatin.16 There are several proteins, such as heterochromatin protein-1 (HP1), whose interactions with histone H3 depend on this PTM. Previous structural studies have established that the chromodomain of HP1 interacts with an H3K9Me3 peptide mainly through residues between P−4 and P+1 (i.e., QTARK(Me3)S), relative to Lys-9.17, 18 We therefore synthesized probe 2 in which a benzophenone moiety replaced the side chain of Lys-4 (Fig. 1) and examined the ability of probe 2 to capture the chromodomain of HP1 in vitro. As shown in Figure 2(a), probe 2 labeled the recombinant glutathione S-transferase (GST)-tagged human HP1 chromodomain (GST-HP1CD) in a dose-dependent manner. The labeling was saturated at ∼ 5 μM of probe 2 (LC50 = 1.1 μM). Importantly, a control probe (2C) with unmodified Lys-9 failed to label the recombinant protein and the labeling of GST-HP1CD by 2 could be competed with H3K9Me3 peptide [Fig. 2(b)]. These data, together with our previous studies using probe 1, indicate that these photo-crosslinking-based chemical probes can be used to capture proteins that specifically bind histone methylation sites.
Figure 2.
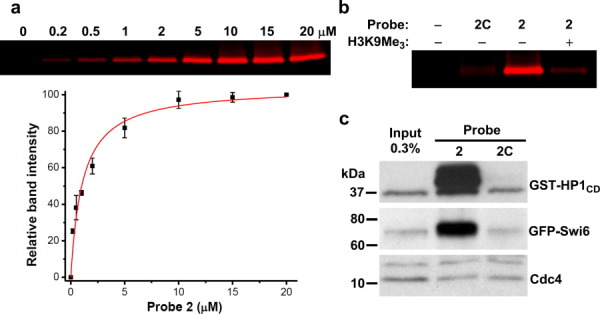
Using chemical probes to capture H3K9Me3-binding proteins. (a) The concentration-dependent labeling of purified chromodomain of HP1 (GST-HP1CD) by probe 2 (n = 2). (b) The recombinant GST-HP1CD was selectively labeled by methylated probe 2 (3 μM) but not unmodified probe 2C (3 μM) and the labeling by 2 was inhibited by H3K9Me3 peptide (30 μM). (c) Recombinant GST-HP1CD and GFP-tagged endogenous Swi6, the human HP1 homolog, were selectively captured by probe 2 from fission yeast cell lysates. Cdc4 was the loading control.
To test the potential utility of the chemical probes for identifying PTM-binding proteins in genetically tractable model organisms, we examined if probe 2 can be used to pull-down known endogenous binders of H3K9Me3 in fission yeast cell extract. We first generated a fission yeast strain expressing green fluorescent protein (GFP)-tagged Swi6, the homolog of human HP1. As a positive control, the recombinant human GST-HP1CD was also added to the yeast cell extracts. Probes 2 and 2C were then used to crosslink and pull-down associated proteins. As shown in Figure 2(c), GST-HP1CD and GFP-Swi6 were selectively captured by probe 2, whereas Cdc4, an abundant yeast protein that does not bind H3K9Me3, was not captured by the probe. We believe that these chemical probes, when combined with recently developed fission yeast SILAC technology,19 can be used to comprehensively profile H3K9Me3 “readers” in yeast lysates. However, rather than comprehensively profiling H3K9Me3-binding proteins, we shifted our focus to phosphorylation, a more dynamic PTM.
To identify H3T3-Phos-binding proteins, we designed and synthesized probe 3 (Fig. 1) based on the first 15 amino acid residues at the N-terminus of histone H3. In particular, this probe has phosphorylated Thr 3 at a stoichiometric level, a photo-crosslinking moiety (benzophenone) appended to alanine-7 (i.e., P–4) to “trap” weak interactions, and a bio-orthogonal handle (alkyne) to enable selective isolation of captured binding partners. We first asked if probe 3 could cross-link survivin, the subunit of the CPC that directly binds to H3T3-Phos. As shown in Figure 3(a), recombinant human survivin is captured by the T3-phosphorylated probe (3), whereas the unmodified control probe (probe C) shows reduced activity for labeling the protein. This result agrees well with the previous work indicating that while survivin preferentially binds H3T3-Phos, it also associates with unmodified H3.13 Importantly, the crosslinking of survivin to probe 3 was competed by H3T3-Phos peptide (20 μM), verifying the direct interaction between survivin and the phosphorylated histone “tail” peptide.
Figure 3.

Profiling H3T3-Phos-binding proteins using the CLASPI strategy. (a) Recombinant survivin, a known H3T3-Phos-binding protein, is selectively labeled in vitro by probe 3 (1 μM) and the selective labeling of the recombinant survivin by probe 3 (1 μM) can be inhibited by H3T3-Phos peptide (20 μM). (b) A 2D-plot showing the Log2 values of the SILAC ratios (L/H) of each identified protein for the forward (x axis) and the reverse (y axis) experiments. Selective H3T3-Phos binders are labeled. (c) The recombinant full-length PPM1G and N-terminal region of MCAK were selectively labeled by probe 3 (1 μM), but the labeling was not fully inhibited in the presence of excess H3T3-Phos peptide (100 μM).
We next used our CLASPI approach to examine whether probe 2 could be used to profile H3T3-Phos binders in complex proteomes. Because H3T3-Phos mark is a cell division-specific PTM,9 we arrested HeLa cells in mitosis (M-phase), during which the potential readers of this histone mark are likely accumulated. The whole-cell lysates were then prepared from the M-phase arrested HeLa cells grown in medium containing either “light” (natural isotope abundance forms) or “heavy” (13C, 15N-substitued arginine and lysine) amino acids. To identify H3T3-Phos-binding proteins, we performed two independent CLASPI experiments (Fig. 4). In a “Forward” experiment, the “light” and “heavy” lysates were photo-cross-linked with probe 3 and probe C, respectively, and pooled for subsequent steps. The cross-linked proteins were then conjugated to biotin-azide using click chemistry and enriched with streptavidin beads. After stringent washing with 6M urea to remove noncovalent interacting partners, the enriched proteins were eluted from the beads and resolved in a SDS-PAGE gel. Following in-gel trypsin digestion, the peptide mixtures were separated by HPLC and analyzed with a LTQ-Orbitrap mass spectrometer. In a “reverse” experiment, the probes were simply switched in the photo-cross-linking reaction, that is, probe 3 reacts with “heavy” lysates while probe C reacts with “light” lysates. By this method, proteins that showed a high SILAC ratio (L/H) in the “forward” experiment and a low ratio (L/H) in the “reverse” experiment were likely H3T3-Phos-binding proteins (Fig. 4).
Figure 4.
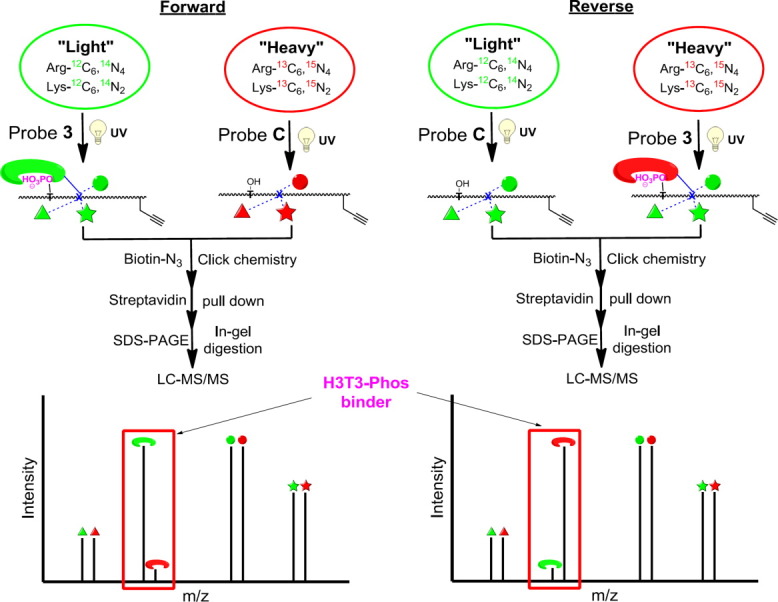
CLASPI approach to identify H3T3-Phos-binding proteins. In a “Forward” experiment, the “light” (green symbols) and “heavy” (red symbols) lysates were photo-cross-linked with probe 3 and probe C, respectively. The cross-linked proteins were then conjugated to biotin through click chemistry and isolated with streptavidin beads. After trypsin digestion, the peptide mixtures were analyzed LC-MS/MS. In a “reverse” experiment, the probes were simply switched in the photo-cross-linking reaction. Proteins that showed a high SILAC ratio (L/H) in the “forward” experiment and a low ratio (L/H) in the “reverse” experiment were likely selective H3T3-Phos-binding proteins.
A two-dimensional plot with logarithmic (Log2) SILAC ratios (L/H) of the identified proteins in the “forward” and “reverse” experiments along the x-axis and y-axis, respectively, is shown in Figure 3(b). As expected, the vast majority of identified proteins (>98%) did not show significant differences between the signal intensities of their “light” and “heavy” forms (i.e., L/H close to 1:1), indicating that they are not likely selective H3T3-Phos “readers.” In contrast, four proteins together with survivin, the only known binder of H3T3-Phos, were enriched by T3-phosphorylated probe (3) in the both experiments [Fig. 3(b)], indicating that they bind preferentially to histone H3T3-Phos “mark.”
Among the newly identified proteins, two kinesin-like motor proteins, KIF2A and MCAK, attracted our attention, as they are mitotic motor proteins that localize to centromeres, a chromosomal region where H3T3-Phos is concentrated.20 Therefore, it is important to examine whether this histone “mark” can meditate the direct interactions with these proteins in vitro. Considering the high sequence homology of KIF2A and MCAK, we focused on testing MCAK as a potential H3T3-Phos binder. We purified a recombinant MCAK N-terminal fragment that is necessary for centromere targeting.21 As shown in Figure 3(c), the recombinant MCAK was captured by probe 3 but not probe C, indicating a selective interaction between MCAK and the T3-phosphorylated H3 peptide. However, the crosslinking was not effectively competed by H3T3-Phos peptide, even at a high concentration of 100 μM. Together, these data suggest that this recombinant MCAK fragment is likely to be involved in a selective but weak interaction with T3-phosporylated H3 peptide.
This result prompted us to examine whether the interaction between endogenous MCAK and H3T3-Phos is also weak. In the previous CLASPI experiments, we compared the proteins captured by phosphorylated and unmodified probe, respectively, such that the selective interacting partners can be identified. To further distinguish the high-affinity interactions, we performed a competition CLASPI experiment (hereafter, CLASPI-C), in which both lysates were photo-crosslinked with probe 3 (2 μM), but the “heavy” sample also contains H3T3-Phos peptide as a competitor (30 μM) [Fig. 5(a)]. We anticipated that high-affinity H3T3-Phos-binding proteins should form complexes with the excessive competitor peptide while most low-affinity ones remain unbound. For example, the competitor peptide, at the concentration of 30 μM, can occupy binding sites of around 90% of protein molecules with a Kd of 3 μM and 10% of those with a Kd of 300 μM, respectively. Therefore, we expect that the addition of the competitor peptide would effectively inhibit the probe 3-induced labeling of high-affinity H3T3-Phos binders in the “heavy” extract, and should thereby, produce a high SILAC ratio of L/H for these proteins. As summarized in Figure 5(b), several proteins, including survivin, show a high ratio (L/H) in the CLASPI-C experiment, indicating that they bind tightly to H3T3-Phos peptide. Interestingly, most of these proteins, including JADE2, RBBP7, HAT1, RBBP4, UHRF1, MTA1, MTA2, and DPF2, were previously identified as unmodified histone H3 binders in our H3K4Me3 CLASPI experiments,22 suggesting that their binding to H3 is not inhibited by the T3-phosphorylation “mark.” In contrast, proteins identified as selective H3T3-Phos binders including PPM1G, KIF2A, MCAK, and ERAP1 in CLASPI-S (S, for selectivity) experiment showed SILAC ratios close to 1 in CLASPI-C experiment [Fig. 5(b)], which indicates that they are involved in weak interactions with H3T3-Phos. This result was also supported by the competition assays with recombinant MCAK and PPM1G [Fig. 3(c)]. Taken together, performing the two sets of CLASPI experiments [Fig. 5(a)], which can serve as selectivity and affinity filters, respectively, should help to identify the selective and strong interactions mediated by PTMs.
Figure 5.
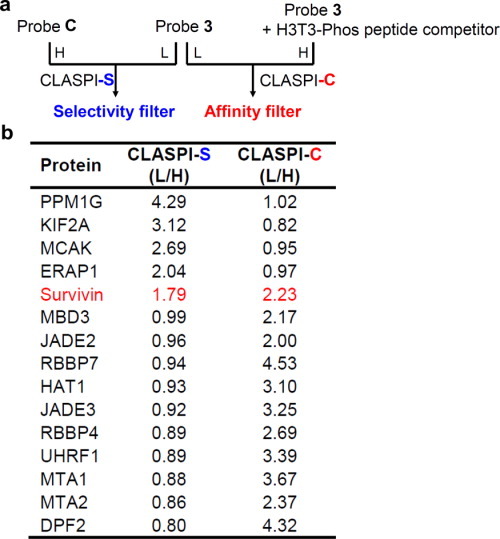
(a) Schematic for the two sets of CLASPI experiments indicates the selectivity and affinity filters used for the identification of selective and tight H3T3-Phos binders. (b) A summary of the proteins that showed significant SILAC ratios in at least one of the CLASPI experiments shown in (a).
It has been reported that the trimethylation on the adjacent lysine residue (H3K4Me3) can abolish the interaction between survivin and H3T3-Phos.14 To examine this antagonistic effect, we therefore designed and synthesized probe 4 (Fig. 1), which carries both these modifications, that is, H3T3-Phos-K4Me3. This doubly modified probe did not crosslink to recombinant survivin in vitro [Fig. 6(a)]. Moreover, the CLASPI experiment, in which the “light” lysate was crosslinked with probe 4 and the “heavy” with probe C, revealed that trimethylation on H3K4 precludes survivin binding to phosphorylated Thr 3 [Fig. 6(b)]. These data demonstrate the potential application of CLASPI for examining the interplay among PTM on the proximal protein residues.
Figure 6.
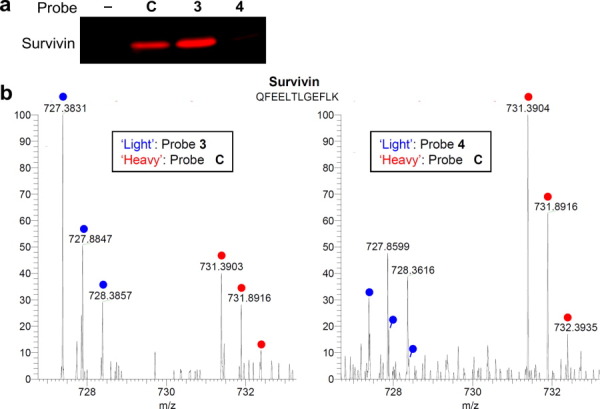
Examining the interplay between histone methylation and phosphorylation using the CLASPI strategy. (a) The double modified (i.e., T3-Phos and K4Me3) H3 probe 4 failed to label the recombinant survivin. (b) Endogenous survivin was selectively enriched by probe 3 (left panel) but not by probe 4 (right panel). The “light” and “heavy” peptide isotopes are indicated by blue and red dots, respectively.
Discussion
In summary, our CLASPI approach can be more broadly used to examine PTM-mediated protein–protein interactions. By profiling proteins that recognize H3T3-Phos, a mitotic histone “mark,” we were able to identify survivin, a known H3T3-Phos “reader,” as well as other proteins (e.g., MCAK and KIF2A) that are likely to be involved in weak but selective interactions with this histone modification in vitro. KIF2A and MCAK are substrates of the Aurora kinase and their recruitment to centromeres depends on Aurora activity.23, 24 Our findings suggest that H3T3-Phos may directly recruit not only the kinase but also its substrates (i.e., KIF2A and MCAK) to specific chromosomal sites. Testing the physiological significance of these weak but selective interactions is an important next step. In cellular contexts, inhibition of H3T3-Phos disrupts the localization of MCAK and Aurora kinase, consistent with our proposal. However, a proper test of this hypothesis will require identifying mutants that can separate the contributions of Aurora kinase-dependent phosphorylation from the H3T3-Phos-dependent recruitment.
A recent analysis on the phosphorylation sites for the cyclin-dependent kinase Cdk1, a key player in cell cycle regulation, has revealed that most Cdk1 phosphorylation sites localize in loops and disordered regions,25 which is consistent with previous analyses of phosphorylation sites in general.26 Therefore, the CLASPI approach, in which a peptide is used to mimic the endogenous phosphorylation site(s), should be effective as these regions are disordered. For cases in which a phosphorylation maps to a structured region of a protein, it should also be possible to combine the use of stabilized secondary structure mimetics27 with the CLASPI approach to profile protein interactions that depend on this PTM.
Our findings that CLASPI can be used to examine PTM-dependent protein–protein interactions in fission yeast lysates are important as it will allow the powerful genetic tools available in this model organism to be combined to examine complex cellular mechanisms, such as the cell cycle. Importantly, this model organism shares more basic mechanisms (e.g., RNAi) with humans than budding yeast, another widely used genetically tractable model organism. The role of phosphorylation in controlling the stable propagation our genomes, through replication and division, has been established in large part based on studies in this model organism. We believe that CLASPI will be useful in profiling phosphorylation-dependent protein–protein interactions required for key transitions in the cell cycle that are known to depend on the activities of conserved kinases, many of which are being evaluated as anticancer drug targets.
Given that a variety of PTMs coexist in the same protein regions, multiple biological readouts could be achieved by various combinations of these different modifications.28–30 It has been shown that modifications on proximal sites of histones can antagonize or synergize to control interactions with “reader” proteins. For example, the interaction of survivin with H3T3-Phos is abolished by trimethylation at the neighboring amino acid lysine 4.14 In support of this notion, we applied CLASPI method to show the antagonizing effect of H3K4Me3 on the survivin binding to H3T3-Phos. Together, our data indicate that CLASPI can allow profiling of single or multi-PTM-dependent protein–protein interactions. Finally, in future studies, the CLASPI method can be combined with powerful protein synthesis technologies, such as protein splicing, incorporation of unnatural amino acids, and peptidomimetic foldamers, to construct chemical probes with well-defined 3D structures to capture protein–protein interactions in vitro and in vivo.
Materials and Methods
SILAC cell culture and preparation of whole-cell lysates
Hela S3 cells were grown in suspension at 37°C in a humidified atmosphere with 5% CO2 in Dulbecco's Modified Eagle Medium (DMEM) medium (−Arg, −Lys, ThermoFisher Scientific) containing 10% dialyzed fetal bovine serum (FBS) (Atlanta Biologicals), penicillin–streptomycin, and 2 mM l-glutamine supplemented with 22 mg/L 13C615N4-l-arginine (ThermoFisher) and 50 mg/L 13C615N2-l-lysine (Sigma) or the corresponding nonlabeled amino acids (Sigma), respectively. To synchronize cells, 3 × 108 cells/L were treated with 2.2 mM thymidine (Sigma) for 22 h. Cells were then pelleted, washed with media once, and resuspended in 1 L media without antibiotics. Three hours after thymidine wash-out, nocodazole (330 nM, Sigma) was added and cells were harvested after an additional 11 h. After harvesting, cell pellets were washed twice with phosphate buffered saline (PBS) and frozen with liquid N2. The frozen cell pellets were ground into powder with a Ball Mill (Retch MM301). The cell powder was stored at −80°C until use. To prepare whole-cell lysates, the frozen cell powder was first resuspended in a hypotonic buffer (10 mM HEPES, pH 7.5, 2 mM MgCl2, 0.1% Tween-20, 20% glycerol, 2 mM phenylmethylsulfonyl fluoride (PMSF), Roche Complete EDTA-free protease inhibitors, and Pierce Halt Phosphatase Inhibitor Cocktail) and incubated for 10 min at 4°C. The suspension was centrifuged at 16,000g for 15 min at 4°C and the supernatant was kept for use later. The pellets were resuspended in a high-salt buffer (50 mM HEPES, pH 7.5, 420 mM NaCl, 2 mM MgCl2, 0.1% Tween-20, 20% glycerol, 2 mM PMSF, Roche Complete EDTA-free protease inhibitors, and Pierce Halt Phosphatase Inhibitor Cocktail) and incubated for 30 min at 4°C. The suspension was centrifuged at 16,000g for 15 min at 4°C and the supernatant was combined with the soluble fraction in hypotonic buffer to give the whole-cell lysates. The yeast cell lysates were extracted from exponentially growing swi6-GFP cells (PL52:h90ade6 leu1 swi6+-GFP≪kanr z::adh15-mcherry-atb2≪natr, gift from Watanabe lab) using glass-beads.
Photo-crosslinking
In a forward CLASPI experiment, probe 3 and probe C were incubated with 3 mL of light and heavy SILAC whole-cell lysates (2 mg/mL) (the probes were switched in a “reverse” CLASPI experiment), respectively, in the binding buffer (50 mM HEPES, pH 7.5, 168 mM NaCl, 2 mM MgCl2, 0.1% Tween-20, 20% glycerol, 2 mM PMSF, Roche Complete EDTA-free protease inhibitor cocktail, and Pierce Halt Phosphatase Inhibitor Cocktail) for 15 min at 4°C. The samples were then irradiated at 365 nm using a Spectroline ML-3500S UV lamp for 15 min on ice.
Cycloaddition reactions (click chemistry)
After UV cross-linking, the light and heavy lysates were pooled, to which 100 μM cleavable biotin-azide was added, followed by 1 mM tris(2-carboxyethyl)phosphine (TCEP) and 100 μM tris-(benzyltriazolylmethyl)amine (TBTA), and finally the reactions were initiated by the addition of 1 mM CuSO4. The reactions were incubated for 1.5 h at room temperature.
Streptavidin affinity enrichment of biotinylated proteins
The cycloaddition reactions were terminated by adding fourfold volume of ice-cold acetone, placed at −20°C overnight and centrifuged at 16,000g for 5 min at 4°C to precipitate proteins. The protein pellets were washed twice in methanol. The pellets were subsequently dissolved in PBS with 4% SDS by votexing and heating (5 min at 75°C). The protein solution was then diluted 5× with PBS to 0.8% SDS and incubated with streptavidin agarose resin (Thermo Pierce) for 1.5 h at room temperature. After washing with PBS with 0.8% SDS, 6M urea with 0.1%SDS, and 250 mM NH4HCO3 with 0.1% SDS, the enriched proteins were eluted by incubating with buffer containing 25 mM Na2S2O4, 250 mM NH4HCO3, and 0.1% SDS for 1 h. The eluted proteins were then dried down with SpeedVac.
Sample preparation for mass spectrometry
The dried proteins were resuspended in Lithium dodecyl sulfate (LDS) sample buffer (Invitrogen) with 50 mM dithiothreitol (DTT) and heated at 75°C for 8 min, then reacted with iodoacetamide in dark for 30 min to alkylate all reduced cysteines. Proteins were then separated on Bis-Tris gel (Invitrogen), followed by fixation in a 50% methanol/7% acetic acid solution. The gel was stained by GelCode Blue stain (Pierce) and sliced diced into 1-mm cubes, followed by destaining in 50 mM ammonium bicarbonate/50% acetonitrile for 1 h. The destained gel cubes were dehydrated in acetonitrile for 10 min and rehydrated in 25 mM NH4HCO3 with 125 ng trypsin (Promega) for protein digestion at 37°C overnight. The resulting peptides were enriched using the StageTips. The peptides eluted from the StageTips were dried down by SpeedVac then resuspended in 0.5% acetic acid for analysis by LC-MS/MS.
Mass spectrometry
Mass spectrometry was performed on a LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific), using a home-built micro electrospray source with a liquid junction. First, peptide samples in 0.5% acetic acid were pressure loaded onto a self-packed PicoFrit column (New Objective) with integrated emitter tip (360-μm o.d., 75-μm i.d., and 15-μm tip), packed with 6 cm of reverse-phase C18 material (Alltima C18 5-μm beads from Alltech Associates), rinsed for 10 min with 0.1M acetic acid, and subsequently gradient eluted with a linear gradient from 0 to 70% B in 150 min (A = 0.1M acetic acid, B = 70% acetonitrile in 0.1M acetic acid, flow rate 200 nL/min) into the mass spectrometer. The instrument was operated in a data-dependent mode cycling through a full scan (300–2000 m/z, single μscan) followed by 10 CID MS/MS scans on the 10 most abundant ions from the immediate preceding full scan. The cations were isolated with a 2-Da mass window and set on a dynamic exclusion list for 60 s after they were first selected for MS/MS. The raw data were processed and analyzed using MaxQuant (version 1.1.16).
Expression and purification of recombinant proteins
The human MCAK (residues 1–100) was cloned into pDONR201 and pDEST15, with a prescission protease site added to the N-terminus of the open reading frame. The protein was overexpressed in the Escherichia coli host cell BL21(Rosetta) induced overnight by 0.5 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18°C in the LB medium. After cell lysis and centrifugation, the GST-fusion proteins were purified by glutathione sepharose beads, followed by tag cleavage using PreScission™ protease. The cleaved proteins were further purified by gel-filtration chromatography on Superdex 75 column in buffer A (150 mM NaCl, 50 mM HEPES, 2 mM DTT, pH 7.5). The chromo domain (CD) of fly HP1 (residues 23–74) was cloned into pGex-6p-1 vector with an N-terminal GST tag. Proteins were overexpressed in the Escherichia coli host cell BL21 induced for 3 h by 1 mM IPTG at 37°C in the LB medium. After cell lysis and centrifugation, the GST-HP1CD proteins were purified by glutathione sepharose beads.
Acknowledgments
The authors thank Alex Kelly and Hironori Funabiki for recombinant survivin, Jamie Moseley and Paul Nurse for Cdc4 antibody, and Yoshinori Watanabe for fission yeast strains.
Glossary
Abbreviations
- CLASPI
cross-linking-assisted and SILAC-based protein identification
- CPC
chromosomal passenger complex
- H3T3-Phos
histone H3 threonine 3 phosphorylation
- H3K4Me3
histone H3 lysine 4 trimethylation
- PTM
post-translational modification
- SILAC
stable isotope labeling in cell culture
References
- 1.Walsh CT. Posttranslational modifications of proteins: expanding nature's inventory. Greenwood Village, CO: Roberts and Co. Publishers; 2006. [Google Scholar]
- 2.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 4.Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 5.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Kapoor TM. Approach to profile proteins that recognize post-translationally modified histone “tails”. J Am Chem Soc. 2010;132:2504–2505. doi: 10.1021/ja909741q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li X, Foley EA, Molloy KR, Li Y, Chait BT, Kapoor TM. Quantitative chemical proteomics approach to identify post-translational modification-mediated protein-protein interactions. J Am Chem Soc. 2012;134:1982–1985. doi: 10.1021/ja210528v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- 9.Dai J, Sultan S, Taylor SS, Higgins JM. The kinase haspin is required for mitotic histone H3 Thr 3 phosphorylation and normal metaphase chromosome alignment. Genes Dev. 2005;19:472–488. doi: 10.1101/gad.1267105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 11.Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21–32. doi: 10.1038/35048096. [DOI] [PubMed] [Google Scholar]
- 12.Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- 13.Kelly AE, Ghenoiu C, Xue JZ, Zierhut C, Kimura H, Funabiki H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science. 2010;330:235–239. doi: 10.1126/science.1189505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, Dai J, Daum JR, Niedzialkowska E, Banerjee B, Stukenberg PT, Gorbsky GJ, Higgins JM. Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science. 2010;330:231–235. doi: 10.1126/science.1189435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamagishi Y, Honda T, Tanno Y, Watanabe Y. Two histone marks establish the inner centromere and chromosome bi-orientation. Science. 2010;330:239–243. doi: 10.1126/science.1194498. [DOI] [PubMed] [Google Scholar]
- 16.Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science. 2001;292:110–113. doi: 10.1126/science.1060118. [DOI] [PubMed] [Google Scholar]
- 17.Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science. 2002;295:2080–2083. doi: 10.1126/science.1069473. [DOI] [PubMed] [Google Scholar]
- 18.Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- 19.Bicho CC, de Lima Alves F, Chen ZA, Rappsilber J, Sawin KE. A genetic engineering solution to the “arginine conversion problem” in stable isotope labeling by amino acids in cell culture (SILAC) Mol Cell Proteomics. 2010;9:1567–1577. doi: 10.1074/mcp.M110.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manning AL, Ganem NJ, Bakhoum SF, Wagenbach M, Wordeman L, Compton DA. The kinesin-13 proteins Kif2a, Kif2b, and Kif2c/MCAK have distinct roles during mitosis in human cells. Mol Biol Cell. 2007;18:2970–2979. doi: 10.1091/mbc.E07-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walczak CE, Gan EC, Desai A, Mitchison TJ, Kline-Smith SL. The microtubule-destabilizing kinesin XKCM1 is required for chromosome positioning during spindle assembly. Curr Biol. 2002;12:1885–1889. doi: 10.1016/s0960-9822(02)01227-7. [DOI] [PubMed] [Google Scholar]
- 22.Li X, Foley EA, Molloy KR, Li Y, Chait BT, Kapoor TM. Quantitative chemical proteomics approach to identify posttranslational modification-mediated protein-protein interactions. J Am Chem Soc. 2012;134:1982–1985. doi: 10.1021/ja210528v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, Luo H, Zhang Y, He W, Yang K, Zwaans BM, Tishkoff D, Ho L, Lombard D, He TC, Dai J, Verdin E, Ye Y, Zhao Y. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol Cell Proteomics. 2011;10:M111.012658. doi: 10.1074/mcp.M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holt LJ, Tuch BB, Villen J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science. 2009;325:1682–1686. doi: 10.1126/science.1172867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iakoucheva LM, Radivojac P, Brown CJ, O'Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guarracino DA, Bullock BN, Arora PS. Mini review: protein-protein interactions in transcription: a fertile ground for helix mimetics. Biopolymers. 2011;95:1–7. doi: 10.1002/bip.21546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 29.Fischle W, Wang Y, Allis CD. Histone and chromatin cross-talk. Curr Opin Cell Biol. 2003;15:172–183. doi: 10.1016/s0955-0674(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 30.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]