

Abstract
Promyelocytic leukemia (PML) is a major component of macromolecular multiprotein complexes called PML nuclear-bodies (PML-NBs). These PML-NBs recruit numerous proteins including CBP, p53 and HIPK2 in response to DNA damage, senescence and apoptosis. In this study, we investigated the effect of presenilin (PS), the main component of the γ-secretase complex, in PML/p53 expression and downstream consequences during DNA damage-induced cell death using camptothecin (CPT). We found that the loss of PS in PS knockout (KO) MEFs (mouse embryonic fibroblasts) results in severely blunted PML expression and attenuated cell death upon CPT exposure, a phenotype that is fully reversed by re-expression of PS1 in PS KO cells and recapitulated by γ-secretase inhibitors in hPS1 MEFs. Interestingly, the γ-secretase cleavage product, APP intracellular domain (AICD), together with Fe65-induced PML expression at the protein and transcriptional levels in PS KO cells. PML and p53 reciprocally positively regulated each other during CPT-induced DNA damage, both of which were dependent on PS. Finally, elevated levels of PML-NB, PML protein and PML mRNA were detected in the brain tissues from Alzheimer's disease (AD) patients, where γ-secretase activity is essential for pathogenesis. Our data provide for the first time, a critical role of the PS/AICD-PML/p53 pathway in DNA damage-induced apoptosis, and implicate this pathway in AD pathogenesis.
Keywords: presenilin, promyelocytic leukemia, cell death, DNA damage, Alzheimer's disease
Promyelocytic leukemia (PML) protein is a well-known tumor suppressor, which functions by its association with PML-nuclear bodies (PML-NBs).1, 2 PML regulates a variety of cellular processes such as induction of apoptosis, cellular senescence, inhibition of proliferation, transcription, response to DNA-damage and antiviral response.1, 2 PML-NBs, which are spherical shape with a diameter of 0.2–1.0 μm, possess dynamic and heterogeneous structures.1 PML-NBs are present in most mammalian cells' nuclei, numbers that typically range from 5–15 bodies per nucleus depending on the cell type, cell cycle and differentiation stage of the cell.1 As PML KO mice are resistant to proapoptotic stimuli3 and overexpression of PML prohibits tumor formation in nude mice,4 the tumor suppressive and pro-apoptotic roles of these structures are well established. Indeed, PML KO cells are protected against both p53-dependent and -independent apoptotic stimuli, such as infrared, interferon, TNF-α and ceramide.5 Moreover, PML has been found to physically associate with critical apoptosis regulators including p53, pRb, c-Jun and Daxx.6
Our previous studies have shown that genotoxic stressors such as camptothecin (CPT) and etoposide enhance γ-secretase activity and Aβ generation,7, 8 suggesting that modulation of γ-secretase activity is a component of the genotoxic cell death pathway. As presenilin (PS) contains the core catalytic activity within the γ-secretase complex, we hypothesized that PS has a key role in mediating DNA damage-induced cell death. Importantly, a recent study by Konitzko et al.9 revealed a close physical apposition between AFT (AICD, Fe65, Tip60) complexes and PML-NBs in HEK293 cells, suggestive a mechanistic link between APP processing and PML-NBs. Indeed in the present study, we present evidence that PS-dependent γ-secretase activity is a critical mediator of genotoxic stress-induced PML induction and apoptosis. Specifically, we found that the loss of PS diminishes DNA damage-induced cell death and attenuates PML expression, effects that were restored by exogenous expression of PS in PS-deficient cells. The diminished expression of PML upon DNA damage in PS-deficient cells was due to reduced PML mRNA, an effect that could be mimicked by γ-secretase inhibitors and reversed by APP intracellular domain (AICD). Moreover, CPT-induced p53 and PML expression depended on each other as positive reciprocal regulators. Finally, brain samples from AD patients showed increased level of PML protein. These results demonstrate for the first time the critical role of PS in the modulation of PML expression via the γ-secretase and AICD-dependent pathway, mechanisms that link PS to PML/p53 induction and DNA damage-induced cell death.
Results
The absence of PS-diminished DNA damage-induced cell death and PML expression
Western blot analysis revealed that PS expression was not detected in PS dKO MEFs (Supplementary Figure S1) as we had reported previously.10, 11 Exogenous expression of human PS1 in PS dKO MEFs (hPS1 MEFs) restored PS1 expression (Supplementary Figure S1). As the loss of PS resulted in the activation of Cyclin D1, hyperproliferation and skin tumorigenesis,12 we tested whether the absence of PS is less sensitive to CPT-induced cell death in PS dKO MEFs. CPT is a cytotoxic quinoline alkaloid that inhibits the DNA enzyme topoisomerase. CPT consequently causes DNA damage that leads to apoptosis. Indeed, cell viability assay confirmed that PS dKO MEFs were less sensitive to CPT (24 h) in various doses compared with PS WT MEFs (Figure 1a). We further tested our hypothesis that this attenuated sensitivity to CPT in PS dKO MEFs might be achieved by regulating the expression level of PML. Western blot analysis showed that PML protein level in CPT-induced DNA damage condition was not dramatically increased in PS dKO MEFs up to 200 nM, whereas PML expression by CPT was increased in a dose-dependent manner at PS WT MEFs (Figures 1b and d). Less-increased PML level was detected in PS dKO MEFs compared with PS WT MEFs in response to 150 nM of CPT with 36 h exposure (Figures 1c and e). Cisplatin (CDDP, 24 h), another DNA damage-inducing agent, showed similar findings to that of CPT in PS dKO MEFs, whereas PS WT and hPS1 MEFs showed increased PML expression by CDDP in a dose-dependent manner (Supplementary Figure S2). Therefore, these results clearly indicate the absence of PS results in reduced vulnerability to DNA damage, and that DNA damage-induced PML expression is also dependent on PS expression.
Figure 1.

The absence of PS-diminished CPT-induced cell death and PML expression. (a) Cell viability assay revealed that PS dKO MEFs was less sensitive to CPT-induced cell death compared with PS WT MEFs. Representative MEFs exposed to 150 nM of CPT for 24 h. Cell viability induced by CPT is normalized as % of vehicle control. Bar graphs were presented as the mean±S.E.M. (n=3 for each dose). *P<0.05, ***P<0.001. (b) Western blot analysis revealed that PML protein level in CPT-induced DNA damage condition remained constant throughout entire dose ranges at PS dKO MEFs, whereas PML expression by CPT increased in a dose-dependent manner at PS WT MEFs. (c) PS dKO MEFs treated with 150 nM of CPT showed less increased level of PML than WT MEFs during 36 h. (d and e) Densitometric analysis of PML protein expression from three separate experiments. PML was normalized to β-actin. *P<0.05, **P<0.01, ***P<0.001
PS1 restores DNA damage-induced cell death, PML expression and PML-NB formation in PS dKO cells
We further examined whether restoring the PS function would influence the DNA damage-induced cell death and PML expression using hPS1 MEF cells. Cell viability assay revealed that hPS1 MEFs were vulnerable to DNA damage-induced cell death. Specifically, the degree of cell death by 150 nM of CPT for 24 h in hPS1 MEFs was similar to that in PS WT MEFs, demonstrating that PS is required for regulating DNA damage-induced cell viability (Figure 2a). In addition, PML protein levels were fully restored in hPS1 MEFs, indistinguishable from that in WT cells (Figures 2b and c). Several studies have demonstrated markedly increased PML levels and formation of PML-NB under stress conditions, such as DNA damage, senescence and viral infection.13, 14, 15 Therefore, immunofluorescence assay was performed to detect PML-NB in the three different MEFs. Upon CPT treatment, PS WT and hPS1 MEFs clearly showed punctuated PML-NB structures (red positive staining) in nuclei (blue positive staining), whereas PS dKO MEFs had aberrantly decreased the numbers of PML-NB (Figure 2d). These data suggest that PS could contribute to CPT-induced cell death by tight regulation of PML protein expression and PML-NB formation.
Figure 2.

Rescued PS1 restored CPT-induced cell death, PML expression and PML-NB. (a) Cell viability assay showed that rescued hPS1 in PS dKO MEFs (hPS1) was more sensitive to CPT-induced cell death than PS dKO. Three different MEFs were incubated with 150 nM of CPT for 24 h and compared. The viability is normalized as % of vehicle control. Data were represented as mean±S.E.M. values of three independent experiments. ***P<0.001, ###P<0.001. (b) Western blot analysis of PML revealed that hPS1-rescued PS dKO MEFs (hPS1) restored PML protein levels by CPT-induced DNA damage. PS expression is not altered by CPT treatment. (c) Densitometric analysis of PML protein expression from three separate experiments. PML was normalized to α-tubulin. ***P<0.001, ###P<0.001. (d) Micrograph of immunostaining with PML in MEFs clearly showed that PML-NB was formed within nucleus by 150 nM of CPT for 24 h in PS WT MEFs or hPS1 MEFs, whereas the formation of these condensed PML-NBs were significantly decreased in PS dKO MEFs by CPT-induced DNA damage. PML: red, DAPI: blue. Scale bar=50 μm. (e) Real-time PCR analysis showed that 150 nM of CPT treatment for 24 h did not increase the level of PML mRNA in PS dKO MEFs, whereas CPT enhanced (2.2- and 2.1-fold) PML mRNA level in PS WT and hPS1 MEFs. Bar graph represented as mean±S.E.M. from three independent experiments and normalized as fold of vehicle control. ***P<0.001, ###P<0.001
Depletion of PS decreases PML mRNA levels
We further investigated how PS can influence the expression level of PML and PML-NB formation with real-time PCR analysis. 150 nM CPT treatment for 24 h significantly increased PML mRNA in PS WT MEFs and hPS1 MEFs compared with vehicle treatment (2.2-fold and 2.1-fold, ***P<0.001), whereas CPT did not induce any change of PML mRNA in PS dKO MEFs relative to vehicle control (Figure 2e). These results indicate that PS regulates PML protein levels by the modulation of PML transcription under CPT-induced DNA damage condition.
CPT-induced PML expression is reduced by γ-secretase dependent pathway
As PS is a key component of the γ-secretase complexes, which hydrolyzes multiple substrates including the amyloid precursor protein (APP), we, therefore, tested whether γ-secretase dependent pathway is involved in the modulation of PML expression level. We measured PML expression in CPT-treated hPS1 MEFs in the presence of two different γ-secretase inhibitors; DAPT (functions as a suppressor for Aβ production by blocking γ-secretase), L-685,458 (functions as a transition state analog mimic at the catalytic site of aspartyl protease). hPS1 MEFs cells treated with DAPT or L-685,458 for 24 h have shown the accumulation of APP-CTF (APP C-terminal fragment), which is the substrate of γ-secretase, compared with Veh or CPT-treated group (Figure 3a). Given that γ-secretase inhibitors effectively blocked the activity of γ-secretase without changing PS1 expression level (Supplementary Figure S3a), γ-secretase dependent signaling pathway is closely associated with the modulation of PML expression. When PML protein expression level was measured by western blot analysis under DAPT or L-685458-treated condition in the presence of CPT, both γ-secretase inhibitors significantly reduced CPT-induced PML protein expression levels compared with only CPT-treated groups (Figure 3b). Whether PML regulation is affected by another γ-secretase component, nicastrin (NCT),16 we measured CPT-induced PML expression level in NCT KO MEFs compared with NCT WT MEFs. Western blot analysis revealed that PML expression was not upregulated by CPT in NCT KO MEFs similar to PS dKO MEFs (Supplementary Figure S4). In addition, CPT-induced increase in PML mRNA expression was also significantly decreased by both γ-secretase inhibitors in hPS1 MEFs (Figure 3c), indicating that γ-secretase cleavage-dependent transcriptional regulation is involved in the regulation of PML expression. Collectively, these data demonstrated that γ-secretase activity has an important role in CPT-induced PML expression.
Figure 3.

PML expression was regulated by γ-secretase inhibitors. (a) Western blot analysis showed that two different γ-secretase inhibitors (DAPT, L-685,458) decreased CPT-induced PML expression in hPS1 MEFs. (b) Densitometric analysis of PML protein expression from three independent experiments. PML was normalized to α-tubulin. ***P<0.001, #P<0.05, ##P<0.01. (c) Enhanced PML mRNA levels in hPS1 MEFs with CPT treatment were also decreased by γ-secretase inhibitors. Bar graph showed mean±S.E.M. from the value of three independent experiments and normalized as fold increase of vehicle control. ***P<0.001, #P<0.05, ##P<0.01
Overexpression of AICD (APP intracellular domains) restores the CPT-induced PML expression in PS dKO cells
As an important end product of activated γ-secretase is APP intracellular domains (AICD), a transcription factor to regulate gene expression,17 we hypothesized that AICD might be involved in the modulation of PML expression. In order to test this possibility, we transfected AICD and examined whether CPT-induced PML expression is influenced by AICD transfection. Indeed, AICD overexpression markedly restored the CPT-induced PML expression in PS dKO cells (Figures 4a and b). The importance of Fe65 as an adaptor protein in AICD expression and AICD translocation and stabilization is well established.18 Therefore, we transfected AICD together with Fe65 into HEK293 cells and measured the activity of PML promoter in these cells. Although AICD alone significantly increased the PML promoter activity, such an increase was dramatically potentiated by co-expression of AICD with Fe65; CPT-induced PML expression is largely dependent on γ-secretase activity and that the γ-secretase cleavage product, AICD together with Fe65 can mediate such activity at the transcriptional level (Figure 4c). When we measured PML protein levels in HEK293 cells overexpressing AICD (lane 2, Figure 4d) or AICD+Fe65 (lane 3, Figure 4d), PML protein levels were also significantly increased in cells overexpressing AICD. In addition, cells overxpressing AICD+Fe65 showed even higher PML expression, which strongly suggests that increased AICD stability by Fe65 further enhances PML expression. These results indicate that AICD is closely associated with PML expression.
Figure 4.

Overexpression of AICD restores the CPT-induced PML expression. (a) Western blot analysis revealed that the transient transfection of AICD cDNA into PS dKO MEFs upregulated CPT-induced PML expression compared with mock transfection. (b) Densitometric analysis of PML protein expression from four independent experiments. PML was normalized to α-tubulin. ***P<0.001, ###P<0.001. (c) Cotransfection with AICD and Fe65, which is known as AICD adaptor protein, into HEK293 cells significantly increased the transactivation of PML promoter compared with control or AICD-transfected groups. The values of three independent experiments were represented as mean±SEM. **P<0.01, ***P<0.001, ###P<0.001. (d) Transfection efficiency of PML, AICD and Fe65 was confirmed by western blot analysis. (e) APP siRNA into PS WT MEFs downregulated CPT-induced PML, p53 expression compared with control siRNA. (f) Densitometric analysis of PML protein from three independent experiments. PML was normalized to α-tubulin. **P<0.01, #P<0.05
We then hypothesized that the absence of APP/AICD/γ-secretase pathway might reduce the CPT-induced PML expression in hPS1 MEFs. We silenced APP function using APP siRNA and measured CPT-induced PML expression. In APP siRNA-transfected hPS1 MEFs, PML expression was significantly reduced compared with control siRNA-transfected hPS1 MEFs (Figures 4e and f). These data reinforced the close association between APP/AICD/γ-secretase pathway and PML induction.
DNA damage-induced PML expression via p53 dependent signaling pathway
p53, a well-known tumor suppressor protein, has been associated with the transcriptional activation of PML.6 To test whether p53 could be involved in PS-mediated regulation of PML transcription, we measured mRNA levels of p53 in CPT (150 nM, 24 h)-treated hPS1 and PS dKO MEFs using real-time PCR. CPT treatment significantly increased p53 mRNA in hPS1 MEFs compared with vehicle control (3.18 fold, ***P<0.001, Figure 5a). However, in PS dKO MEFs, CPT significantly reduced p53 mRNA compared with vehicle control (0.8 fold, ***P<0.001, Figure 5a). In addition, we measured PML expression in the presence of p53 inhibitor, Pifithrin-α (PFT-α) with/without treatment of CPT. Western blot analysis clearly revealed that the CPT-induced increase in PML protein was drastically decreased by PFT-α (20 μM) treatment in hPS1 cells (Figures 5b and c), whereas PML expression was not significantly changed regardless of PFT-α treatment in PS dKO MEFs (Figure 5b). Similar pattern of p21, a downstream protein of p53, was also observed (Figure 5b). Under PFT-α-treated condition, hPS1 level was not changed (Supplementary Figure S3b). We further observed significant decreases in CPT (150 nM for 24 h)-induced PML and p53 mRNAs by PFT-α(20 μM) in hPS1 MEFs using real-time PCR analysis (Figures 5d and e). However, CPT-induced changes in PML or p53 mRNAs after PFT-α treatment were not detected in PS dKO MEFs (data not shown). As PML not only acts as a p53 functional effector, but also is an upstream regulator of p53,19 we tested whether PML is capable of modulating p53 expression using shRNA technique. In hPS1 MEFs, PML shRNA downregulated p53 protein and p53 mRNA levels under CPT-treated condition compared with control shRNA-treated condition (Figures 5f and g), suggesting that PML has an important role in regulating p53 expression. Importantly, p53 expression level in APP siRNA-transfected hPS1 MEFs was also attenuated than in control siRNA-transfected hPS1 MEFs (Figure 4e), suggesting that the absence of APP/AICD results in a failure of CPT-induced PML and p53 expression. Collectively, these data clearly indicate that reciprocal interactions between p53 and PML are critical for PS/γ-secretase-mediated regulation of CPT-induced cell death.
Figure 5.

PS controlled the expressions of PML and p53. (a) Real-time PCR analysis clearly indicated that the level of p53 mRNA in hPS1 MEFs was significantly increased (3.18-fold) by CPT 150 nM for 24 h compared with vehicle treatment. On the contrary, p53 mRNA level of PS dKO MEFs induced by CPT treatment was significantly attenuated (0.8-fold) than by vehicle treatment. Bar graph represents as mean±S.E.M.. ***P<0.001, ###P<0.001. (b) Western blot analysis showed that p53 inhibitor, PFT-α(20 μM) decreased the induction of p21 (downstream protein of p53) by CPT in hPS1 MEFs. Any amount of p21 was not detected by western blot analysis with PS dKO MEFs treated with/without CPT and PFT-α. (c) Densitometric analysis of PML protein expression from six independent experiments. PML was normalized to β-actin. **P<0.01, #P<0.05, ##P<0.01. (d and e) Real-time PCR analysis showed that treatment of 20 μM PFT-α significantly decreased both PML and p53 mRNA levels in CPT (150 nM for 24 h) pre-treated hPS1 MEFs. The bar graph is represented as normalized as fold of vehicle control. Data were collected from three to six independent experiments. **P<0.01, #P<0.05, ##P<0.01. (f) Transfection with shRNA of PML significantly decreased p53 immunoreactivity compared with transfection with control shRNA. The bar graph was represented as mean±S.E.M. and normalized % of control shRNA. *P<0.05. (g) Enhanced p53 mRNA level by CPT was significantly attenuated by PML shRNA transfection at hPS1 MEFs. Each experimental data were collected from 3–6 independent experiments. **P<0.01, ##P<0.01
PML KO MEFs are less sensitive to CPT-induced cell death
To test whether PML is involved in CPT-induced cell death, we established a stable PML KO MEFs cell line. As expected and observed in Figures 5f and g, decreased p53 protein levels were detected by western blot analysis in PML KO cells (Figure 6a). WST-1 cell viability assay revealed that PML KO MEFs were also less sensitive to CPT-induced cell death compared with PML WT MEFs in a dose-dependent manner (Figure 6b). In addition, we examined whether exogenously expressed PML in PML KO cells is able to restore p53 activity and CPT-induced cell death. As it turned out to be extremely difficult to establish PML insertion in PML KO MEF cells, as an alternative, we tested our hypothesis using control (shCon) and PML-knockdown (shPML) human foreskin fibroblast (HFF) cells that were transduced with either empty retroviral vector or a vector expressing PML-IV. These cells were treated with vehicle or CPT (400 and 600 nM) for 24 h. Cell death assay revealed that both shPML HFF cells and shPML HFF cells with empty vector were less sensitive to CPT-induced cell death compared with shCon HFF cells. However, when PML protein level is restored by reinserting PML-IV into the shPML HFF cells (shPML-PML IV HFF), it showed similar level of cell death in shCon HFF cells (Figure 6c) and recovered p53 protein expression levels (Figure 6d). Therefore, these data indicate that PML has a critical role in p53 induction and consequently in DNA damage-induced cell death.
Figure 6.

PML KO MEFs are less sensitive to CPT-induced cell death. (a) Western blot analysis clearly indicated that PML was not expressed in PML KO MEFs. Absence of PML also decreased p53 protein expression in these cells. (b) PML KO MEFs were less sensitive to CPT treatment (24 h) compared with PML WT MEFs. Data are represented as mean±S.E.M. from the value of three independent experiments. ***P<0.001. (c) Control (shCon) and PML-knockdown (shPML) HFF cells and shPML HFF cells transduced with empty retroviral vector or a vector expressing PML-IV were vehicle-treated or treated with CPT for 24 h. *P<0.05, ###P<0.001. (d) The bands expected to large PML isoforms (PML-I and –II), other smaller PML isoforms, and SUMO-modified PML proteins are indicated. The exogenously expressed PML-IV was indicated as an arrow. p53 protein level was rescued in shPML HFF cells transduced with PML-IV expressing vector
PML protein and PML mRNA level are upregulated in human AD brains
We previously reported that Aβ treatment upregulates γ-secretase activity in cell culture systems7 and recent study reported that γ-secretase activity might be upregulated in human AD brains.20 As PML expression was regulated by γ-secretase activity, we further hypothesized that PML expression might be elevated in human AD patients. In order to establish our hypothesis, we measured PML immunoreactivity in human AD brains. Increased level of PML and PML-NBs were detected in neurons of the temporal cortex of familial AD (FAD) and sporadic AD (SAD) brains by immunohistochemistry (Figure 7a). We observed that PML and PML-NBs were increased in neurons of the temporal cortex of FAD and SAD patients' brains. Summarized bar graph of western blot analysis indicated that PML expression was significantly upregulated in SAD human brains compared with control human brains (*P<0.05, Figure 7b, n=5 for each case). Moreover PML mRNA levels were also significantly elevated in SAD human brains (***P<0.001, Figure 7c, n=6). In addition, Upregulated PML level was also observed in FAD animal mouse model (5 × FAD, Tg6799 mice) compared with non-transgenic littermates (Supplementary Figures S5a and b ). Moreover, we confirmed that γ-secretase activity was enhanced in the brains of Tg6799 mice (Supplementary Figure S5c).
Figure 7.

PML protein and PML mRNA level are increased in human AD brains. (a) Micrograph of immunostaining with PML in the brains of human AD clearly showed that PML was increased in neurons of the temporal cortex in familial AD (FAD) and sporadic AD (SAD) brains. Scale bars=upper 1 cm; lower 20 μm. (b) Western blot analysis reveals that PML protein level was significantly elevated in the superior temporal cortex of SAD brains (n=5) in comparison to control (n=5). (c) Real-time PCR analysis shows that PML mRNA level is upregulated in the temporal cortex of sporadic AD brains (n=6) in comparison to controls (n=6). Data are represented as mean±S.E.M. *P<0.05, ***P<0.001
Discussion
PS is a multipass membrane protein that is involved in a variety of cellular events under physiological or pathological conditions.21 Mutations in two PS genes (PSEN1 and PSEN2) cause the majority of FAD, implicating a critical role of PS in AD pathogenesis. Elevated level of Aβ liberated from APP by sequential cleavage of β- and γ-secretase was found in these mutations.22 PS provides the catalytic core of the γ-secretase complex that cleaves short transmembrane proteins, in particular APP and Notch.16, 23 The cleavage of APP by PS-dependent γ-secretase activity also releases AICD from the membrane.24 AICD has been proposed as a transcriptional factor that moves to the nucleus with the adaptor protein Fe6525 and regulates the expression of various proteins such as p53, cellular prion protein (PrPc) and Pen2.26, 27, 28 Although the function of PS is well documented as a γ-secretase component, PS also regulates various cellular functions including apoptosis, Ca2+ homeostasis, MEK/ERK and AKT/phosphatidylinositol 3-kinase (PI3 kinase) signaling.26, 29, 30 Importantly, several studies indicated that PS can modulate apoptotic cell death. Overexpression of PS1 into cortical neurons has a protective effect against apoptosis-inducing agent such as etoposide and staurosporine.31 Unlike PS1, PS2 shows a proapoptotic feature in a p53-depenedent manner in various cell types.32, 33 Collectively, p53 could be an important mediator of PS function in apoptotic cell death induced by DNA damage, as p53 is involved in apoptotic signaling of both PS1 and PS2.
The tumor suppressor protein p53 is a key regulator of apoptotic signaling pathway and is involved in both extrinsic and intrinsic apoptosis.34 In a healthy state, the level of p53 remains low via proteasomal degradation. However, once p53 is activated and stabilized under stressed conditions, such as those induced by UV, infrared, hypoxia and DNA-damaging chemotherapeutic drugs.35, 36 Consequently, activated p53 induces the transcription of target gene, which leads to cell cycle arrest, senescence and apoptosis. The association with p53 and PML or PML-NB was well documented.37 Especially under conditions of DNA damage, activated and stabilized form of p53 is recruited into PML-NB through interaction with PML. p53 can be phosphorylated on serine 46 in PML-NB and further can be acetylated by acetyl transferases including Tip60 (Tat-interactive protein 60), CBP (CREB-binding protein) or p300, which facilitates the activity of p53 towards its proapoptotic target genes. Interestingly, PML is itself a p53 target gene that also acts downstream of p53 to potentiate its antiproliferative effects,19 representing the positive-feedback mechanism. Indeed, our data (Figure 5) clearly demonstrated the presence of this reciprocal and functional interaction between PML and p53.
Our previous study indicated that genotoxic stressors such as CPT and etoposide enhance γ-secretase activity and Aβ generation mediated by oxidative stress.7, 8 As PS is a major catalytic core in the γ-secretase complex, it might act as a possible mediator between γ-secretase activity and DNA-damaged apoptotic signaling via p53 tumor suppressor proteins. Recently, it was reported that AICD together with Fe65 and Tip60 (AFT complex) showed a close physical apposition in PML-NB bodies.9 Based on these studies, we hypothesized PML could be a possible mediator between genotoxic stress-induced apoptosis and PS. As PS1 knock-out mice die in late embryogenesis,38 we employed PS dKO MEFs to explore the relationship between PML and PS. PS dKO MEFs and PS WT MEFs were exposed to CPT, a DNA damaging agent, which unmasked a clear mechanistic relationship between PS and PML. Modulation of PML expression by CPT was also confirmed in neuronal cell line, HT22 cells (data not shown), suggesting that the neuronal system might have similar pathway to modulate PML expression under DNA damage condition in PS WT MEFs. Also, as γ-secretase cleaves multiple substrates and releases their intracellular domain, such as AICD and NICD (notch intracellular domain), the expression of NICD for PML expression by CPT was also measured by western blot analysis. Even though NICD expression was increased in both PS WT MEFs and hPS1 MEFs by CPT treatment (Figure 2b), it is unclear whether upregulated NICD expression can modulate PML expression as NICD expression itself is also modulated by DNA damaging agent.39 Involvement of NICD in PML expression under CPT-treated condition needs to be clarified by further studies. Figure 8 shows a schematic diagram, describing the role of PS-dependent γ-secretase activity and subsequent AICD generation in transcriptional activation of PML. AICD together with Fe65 functions as downstream signaling molecule to regulate either p53 or PML expression leading to DNA damage-induced cell death.
Figure 8.
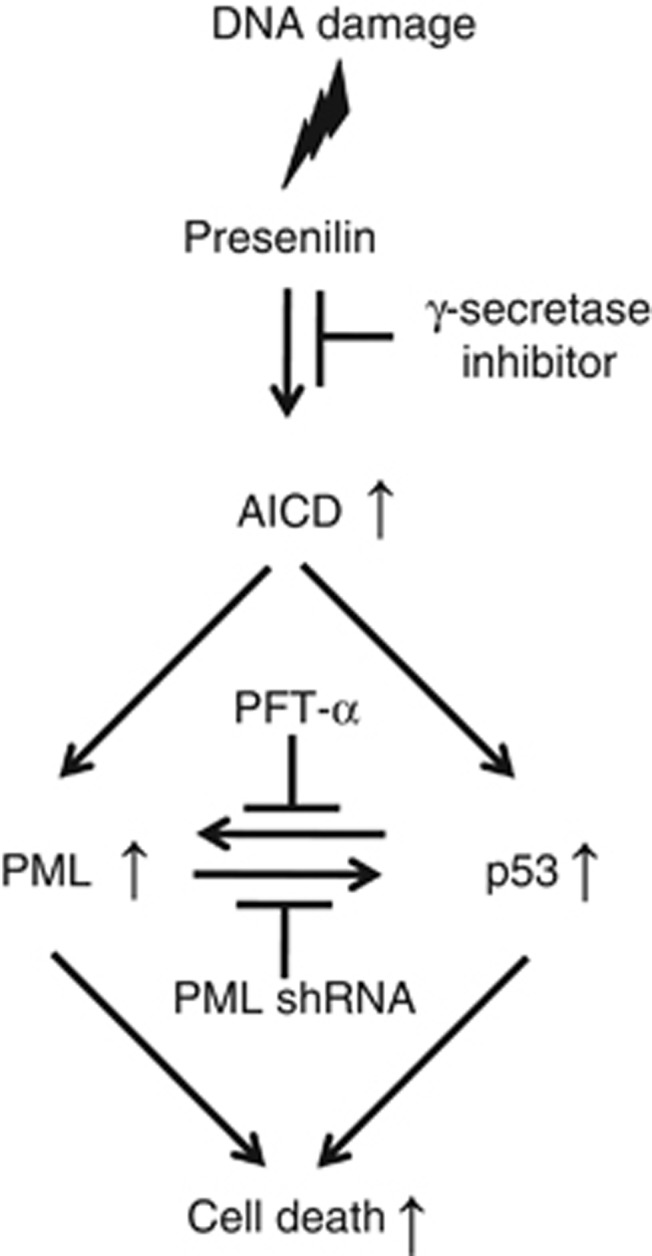
Schematic diagram for role of PS in PML expression under DNA-damaged condition. Diagram describes signaling cascade between PS and PML unmasked by DNA damage. Based on our experimental data, PS with γ-secretase activity and AICD can regulate PML and p53 at the transcriptional level under DNA damaged condition. Reciprocal regulation of PML and p53 is also observed
As PS mutations promote the generation of reactive oxygen species (ROS) and induction of neuronal apoptosis,40, 41 it is conceivable that apoptotic stressors similar to those inducing DNA damage (i.e. CPT) promote signaling via the PS-PML pathway in AD pathogenesis. A recent study reported that neurons harboring the PS1 M146V FAD mutation show enhanced neuronal apoptosis associated with abnormal induction of neuronal cell cycle proteins.42 Consistent with the role of PS-dependent regulation of PML, significantly increased levels of PML protein and mRNA were observed in samples from AD patients (FAD and SAD) compared with controls (Figures 7a and b). Interestingly, polarized PML expression was also observed in SAD patients, suggesting that different mechanisms of PML-NB signaling could exist between SAD and FAD, the latter potentially due to a direct effect of PS mutations. This present study provides for the first time, a critical functional relationship between PS-dependent γ-secretase activity/AICD and PML/p53 expression in DNA damage-induced cell death, and suggests that the PS/AICD-PML/p53 pathway may have an important role in AD pathogenesis.
Materials and Methods
Human brain samples
Biochemical analysis of PML immunoreactivity and immunostain was performed on superior temporal cortex from normal and AD human brain samples. Human brain samples were obtained from Boston University Alzheimer's Disease Center and followed guidelines by Boston University School of Medicine. The detail information of brain tissues is described in the Supplementary information (Supplementary Table S1). Five control brain (female, 101, 87 and 78 years old/ male, 88 and 68 years old) and five sporadic AD brain (female, 90, 100 and 79 years old/ male, 75 and 83 years old), Braak stage V and VI were used.
Cell culture and transient transfection
PS wild-type (PS+/+) MEFs (PS WT MEFs), PS double knockout (PS−/− PS1 and PS2 doubly deficient) MEFs (PS dKO MEFs), PS−/− MEFs expressing human wild-type PS1 (hPS1 MEFs), PML−/− MEFs (PML KO MEFs), NCT−/− MEFs (NCT KO MEFs), human foreskin fibroblast (HFF) and HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; HyClone, Salt Lake City, Utah, USA) supplemented with 10% fetal bovine serum (FBS; HyClone) and 100 units penicillin and 100 μg/ml streptomycin (Sigma-Aldrich, St. Louis, MO, USA) at 37 °C in a 5% CO2 incubator. Transient transfection with AICD, Fe65 and mouse anti-PML shRNA (5′-TAGTGAAGCCACAGATGTA-3′) in MEFs was performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) under guidance of manufacturer's protocol.
Plasmids and luciferase reporter gene assay
AICD, Fe65 and hPML-Luc were transfected into HEK293 cells. For luciferase reporter assays, HEK293 cells were transfected using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) under guidance of manufacturer's protocol. At 24 h after transfection, cells were lysed in passive lysis buffer (PLB), and luciferase activities in the cell extracts were measured by a dual-luciferase reporter assay system (Promega, Madison, WI, USA).
Reagents and antibodies
Camptothecin (CPT), DAPT, Cisplatin (CDDP) and L-685,458 were purchased from Sigma-Aldrich and Calbiochem (La Jolla, CA, USA). The following antibodies were used for immunodetection; mouse monoclonal anti-PML (MBL international corporation, Woburn, MA, USA), mouse monoclonal anti-p53 (Cell signaling, Beverly, MA, USA), rat monoclonal anti-PS1 NTF, mouse monoclonal anti-PS1 loop (Chemicon, Temecula, CA, USA), rabbit polyclonal anti-Nicastrin (ABR, Golden, CO, USA), rabbit polyclonal anti-p21 (Santa Cruz Biotechnologies Inc., Santa Cruz, CA, USA), mouse monoclonal anti-β-Actin and anti-α-Tubulin (Sigma-Aldrich)
PML-depleted human foreskin fibroblast (HFF) cells
PML-depleted HFF cells were produced using a retroviral vector (pSIREN-RetroQ with a puromycine-resistance marker) (BD Bioscience, Franklin Lakes, NJ, USA) expressing anti-PML shRNA as described previously.43 The coding strand sequence of anti-PML shRNA was 5′-AGATGCAGCTGTAT CCAAG-3′, which lies in exon 4. The pMIN retroviral vector with a G418-resistance marker44 was used to express human PML-IV that has been shown to regulate p53 activity.45, 46 The cDNA encoding PML-IV isoform was made resistant to the anti-PML shRNA by introducing two silent point mutations. The altered sequence was 5′-AGATGCGGCCGTATAACCG-3′. Retroviral stocks were prepared by cotransfecting 293T cells with pMIN-PML-IV plasmid along with the packaging plasmids pHIT60 (expressing murine leukemia virus gal/pol) and pMD-G (expressing the envelope G protein of vesicular stomatitis virus)43 using Metafectene reagents (Biotex, Munich, Germany). Cell supernatants were harvested at 48 h after transfection and used to transduce HFF cells in the presence of polybrene (7.5 mg/ml). Cells were selected with G418 (0.4 mg/ml) plus puromycine (0.5 g/m1) (Calbiochem), and the selected cells were maintained in medium containing G418 (0.1 mg/ml) and puromycine (0.5 g/m1).
Cell viability assay
PS MEFs cells were seeded at 4 × 103 cells/well in 96-well flat-bottomed plates (BD falcon, BD Bioscience). After 24 h at 37 °C, the medium was replaced with media containing CPT diluted to the appropriate concentrations. Control cells were treated with DMSO (vehicle) at a final concentration of 1%. After 24 h treatment with CPT (0–200 nM), 10 μl WST-1 solution (EZ-Cytox, Daeil Lab Service, Seoul, Korea) was added to the 96-well cell culture, which was incubated at 37 °C in 5% CO2 for 2 h. The colormetric determination of WST-1 was measured at 460 nm with plate reader (PowerWave XS; BIO-TEK, Winooski, VT, USA).
Western blot analysis
CPT-treated cells were scraped and resuspended in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.5% Deoxicholic acid sodium salt, pH 7.4)-containing protease inhibitor cocktail (Sigma-Aldrich) and incubated on ice for 15 min. After centrifugation at 17 000 g for 15 min, the supernatant was collected and protein concentration was determined by a BCA assay kit (Pierce, Rockford, IL, USA). For western blot analysis, equal amounts of protein (30 μg per lane) were loaded on each Tris-glycine polyacrylamide gel. The target protein bands were visualized by enhanced chemiluminescence (ECL, Amersham Pharmacia Biotech, Buckinghamshire, England) and quantified by a imaging analyzer (LAS-3000; Fuji, Tokyo, Japan).
Immunocytochemistry
Immunofluorescence detection using primary antibody of PML-NB was followed as previously described.10 Briefly, CPT-treated cells were washed twice with ice-cold PBS. Following fixation (4% paraformaldehyde, 10 min), the cells were thoroughly washed with PBS, permeabilized (0.5% Triton X-100), blocked (PBS containing 2% horse serum and 2% goat serum and 2% fetal bovine serum, 1 h) and incubated with primary antibody for overnight at 4°C, followed by incubation with Cy3-conjugated secondary antibody (1:500, Jackson Immuno Research, West Grove, PA, USA) for 1 h. Additional DAPI (4′-6-Diamidino-2-phenylindole, 1:1,000, Sigma-Aldrich, 15 min) was incubated for counter staining. Fluorescent signals were visualized with fluorescent microscopy (Olympus, Tokyo, Japan).
Real-time PCR
RNA was isolated by NucleoSpin Total RNA isolation kit (Macherey-Nagel, Duren, Germany) and cDNA was generated using Maxime RT PreMix_Oligo dT primer kit (Intron biotechnology, Seoul, Korea). Real-time PCR was performed on the cDNA samples using an ABI stepone 2.1 (Applied Biosystems, Foster City, CA, USA). Specific primers (Forward: AACCCTGCGC TGACTGACA/RV: CGCGCATGTGCAACACA) for PML, and p53 (FW: CCCAGCCA AAGAAGAAACCA/RV: TTCCAAGGCCTCATTCAGCT) were used. GAPDH (FW: ACAGCCGCATCTTCTTGTGCAGTG/ RV: GGCCTTGACTGTGCCGTT GAATTT) gene was used as endogenous control to standardize the amount of RNA in each reaction. PML and p53 primers were tested on serial dilutions and primer efficiency was calculated (Supplementary Figure S6).
Statistical analysis
For statistical analysis, Student's t-test or one-way ANOVA test were executed and followed by the Turkey post-test. (*P<0.05, **P<0.01, ***P<0.001 or #P<0.05, ##P<0.01, ###P<0.001) using GraphPad Prism Version 4.0 (GraphPad Software, San Diego, CA, USA).
Acknowledgments
PS wild-type (PS+/+) MEFs (PS WT MEFs), PS double knockout (PS−/− PS1 and PS2 doubly deficient) MEFs (PS dKO MEFs) were generous gifts from Dr. Bart De Strooper (Katholieke, University of Leuven, Leuven, Belgium), NCT−/− MEFs were generous gift from Dr. Phillip Wong of Johns Hopkins University. AICD-GFP cDNA was a generous gift from Dr. YH Suh (Seoul National University, Korea) and hPML-Luc cDNA was a generous gift from Dr. Giovanni Blandino (Regina Elena Cancer Institute, Italy). This work was supported by grants from NRF (2012R1A2A1A01002881, 2008-05943, MRC (2011-0030738), WCU (R32-10084)); KNIH ROAD R&D Program Project (A092058) for I.M-J. Also, it was partly supported by Basic Science Research Program (2012-002551) from NRF for J-H.A and by Woo Jang Choon Project (PJ009103) from RDA for K.H.K.
Glossary
- PML
Promyelocytic leukemia
- NB
Nuclear-body
- PS
Presenilin
- CPT
Camptothecin
- KO
Knock out
- APP
Amyloid precursor protein
- AICD
APP intracellular domain
- AD
Alzheimer's disease
- Topo
Topoisomerase
- CDDP
Cisplatin
- PFT-α
Pifithrin-α
- FAD
Familial Alzheimer's disease
- SAD
Sporadic Alzheimer's disease
- PrPc
Cellular prion protein
- PI3 kinase
phosphatidylinositol 3-kinase
- Tip60
Tat-interactive protein 60
- CBP
Creb-binding protein
- ROS
Reactive oxygen species
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by N Bazan
Supplementary Material
References
- Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007;8:1006–1016. doi: 10.1038/nrm2277. [DOI] [PubMed] [Google Scholar]
- Hofmann TG, Will H. Body language: the function of PML nuclear bodies in apoptosis regulation. Cell Death Differ. 2003;10:1290–1299. doi: 10.1038/sj.cdd.4401313. [DOI] [PubMed] [Google Scholar]
- Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- Mu ZM, Chin KV, Liu JH, Lozano G, Chang KS. PML a growth suppressor disrupted in acute promyelocytic leukemia. Mol Cell Biol. 1994;14:6858–6867. doi: 10.1128/mcb.14.10.6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, et al. PML is essential for multiple apoptotic pathways. Nat Genet. 1998;20:266–272. doi: 10.1038/3073. [DOI] [PubMed] [Google Scholar]
- Schreck KC, Gaiano N. PML: a tumor suppressor essential for neocortical development. Nat Neurosci. 2009;12:108–110. doi: 10.1038/nn0209-108. [DOI] [PubMed] [Google Scholar]
- Jin SM, Cho HJ, Jung ES, Shim MY, Mook-Jung I. DNA damage-inducing agents elicit gamma-secretase activation mediated by oxidative stress. Cell Death Differ. 2008;15:1375–1384. doi: 10.1038/cdd.2008.49. [DOI] [PubMed] [Google Scholar]
- Jin SM, Cho HJ, Jung MW, Mook-Jung I. DNA damage-inducing agent-elicited gamma-secretase activity is dependent on Bax/Bcl-2 pathway but not on caspase cascades. Cell Death Differ. 2007;14:189–192. doi: 10.1038/sj.cdd.4402003. [DOI] [PubMed] [Google Scholar]
- Konietzko U, Goodger ZV, Meyer M, Kohli BM, Bosset J, Lahiri DK, et al. Co-localization of the amyloid precursor protein and Notch intracellular domains in nuclear transcription factories. Neurobiol Aging. 2010;31:58–73. doi: 10.1016/j.neurobiolaging.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boo JH, Song H, Kim JE, Kang DE, Mook-Jung I. Accumulation of phosphorylated beta-catenin enhances ROS-induced cell death in presenilin-deficient cells. PloS one. 2009;4:e4172. doi: 10.1371/journal.pone.0004172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DE, Soriano S, Xia X, Eberhart CG, De Strooper B, Zheng H, et al. Presenilin couples the paired phosphorylation of beta-catenin independent of axin: implications for beta-catenin activation in tumorigenesis. Cell. 2002;110:751–762. doi: 10.1016/s0092-8674(02)00970-4. [DOI] [PubMed] [Google Scholar]
- Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, et al. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci USA. 2001;98:10863–10868. doi: 10.1073/pnas.191284198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Jeong JH, Brown AL, Lee CH, Pandolfi PP, Chung JH, et al. Promyelocytic leukemia activates Chk2 by mediating Chk2 autophosphorylation. J Biol Chem. 2006;281:26645–26654. doi: 10.1074/jbc.M604391200. [DOI] [PubMed] [Google Scholar]
- Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. Deconstructing PML-induced premature senescence. EMBO J. 2002;21:3358–3369. doi: 10.1093/emboj/cdf341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005;3:799–808. doi: 10.1038/nrmicro1248. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, et al. PML is a direct p53 target that modulates p53 effector functions. Mol Cell. 2004;13:523–535. doi: 10.1016/s1097-2765(04)00062-0. [DOI] [PubMed] [Google Scholar]
- Rothhaar TL, Grosgen S, Haupenthal VJ, Burg VK, Hundsdorfer B, Mett J, et al. Plasmalogens inhibit APP processing by directly affecting gamma-secretase activity in Alzheimer's disease. ScientificWorldJournal. 2012;2012:141240. doi: 10.1100/2012/141240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, et al. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer's amyloid beta protein precursor. JAlzheimers Dis. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- Slomnicki LP, Lesniak W. A putative role of the amyloid precursor protein intracellular domain (AICD) in transcription. Acta Neurobiol Exp. 2008;68:219–228. doi: 10.55782/ane-2008-1691. [DOI] [PubMed] [Google Scholar]
- Alves da Costa C, Sunyach C, Pardossi-Piquard R, Sevalle J, Vincent B, Boyer N, et al. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer's disease. J Neurosci. 2006;26:6377–6385. doi: 10.1523/JNEUROSCI.0651-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent B, Sunyach C, Orzechowski HD, George-Hyslop P, Checler F. p53-Dependent transcriptional control of cellular prion by presenilins. J Neurosci. 2009;29:6752–6760. doi: 10.1523/JNEUROSCI.0789-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunys J, Sevalle J, Giaime E, Pardossi-Piquard R, Vitek MP, Renbaum P, et al. p53-dependent control of transactivation of the Pen2 promoter by presenilins. J Cell Sci. 2009;122:4003–4008. doi: 10.1242/jcs.051169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DE, Yoon IS, Repetto E, Busse T, Yermian N, Ie L, et al. Presenilins mediate phosphatidylinositol 3-kinase/AKT and ERK activation via select signaling receptors. Selectivity of PS2 in platelet-derived growth factor signaling. J Biol Chem. 2005;280:31537–31547. doi: 10.1074/jbc.M500833200. [DOI] [PubMed] [Google Scholar]
- Pack-Chung E, Meyers MB, Pettingell WP, Moir RD, Brownawell AM, Cheng I, et al. Presenilin 2 interacts with sorcin, a modulator of the ryanodine receptor. J Biol Chem. 2000;275:14440–14445. doi: 10.1074/jbc.m909882199. [DOI] [PubMed] [Google Scholar]
- Bursztajn S, DeSouza R, McPhie DL, Berman SA, Shioi J, Robakis NK, et al. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J Neurosci. 1998;18:9790–9799. doi: 10.1523/JNEUROSCI.18-23-09790.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves da Costa C, Mattson MP, Ancolio K, Checler F. The C-terminal fragment of presenilin 2 triggers p53-mediated staurosporine-induced apoptosis, a function independent of the presenilinase-derived N-terminal counterpart. J Biol Chem. 2003;278:12064–12069. doi: 10.1074/jbc.M212379200. [DOI] [PubMed] [Google Scholar]
- Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, et al. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W. New insights into p53 activation. Cell Res. 2010;20:614–621. doi: 10.1038/cr.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Mol Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieghoff-Henning E, Hofmann TG. Role of nuclear bodies in apoptosis signalling. Biochim Biophys Acta. 2008;1783:2185–2194. doi: 10.1016/j.bbamcr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, et al. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009;69:573–582. doi: 10.1158/0008-5472.CAN-08-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessel K, Frey C, Jourdan C, Keil U, Weber CC, Muller-Spahn F, et al. Aging sensitizes toward ROS formation and lipid peroxidation in PS1M146L transgenic mice. Free Radic Biol Med. 2006;40:850–862. doi: 10.1016/j.freeradbiomed.2005.10.041. [DOI] [PubMed] [Google Scholar]
- Popescu BO, Ankarcrona M. Mechanisms of cell death in Alzheimer's disease: role of presenilins. Journal of Alzheimer's disease: JAD. 2004;6:123–128. doi: 10.3233/jad-2004-6203. [DOI] [PubMed] [Google Scholar]
- Malik B, Currais A, Andres A, Towlson C, Pitsi D, Nunes A, et al. Loss of neuronal cell cycle control as a mechanism of neurodegeneration in the presenilin-1 Alzheimer's disease brain. Cell Cycle. 2008;7:637–646. doi: 10.4161/cc.7.5.5427. [DOI] [PubMed] [Google Scholar]
- Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, et al. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol. 2011;85:11928–11937. doi: 10.1128/JVI.00758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SS, Kim JM, Kim S. High efficiency retroviral vectors that contain no viral coding sequences. Gene Ther. 2000;7:797–804. doi: 10.1038/sj.gt.3301164. [DOI] [PubMed] [Google Scholar]
- Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, et al. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, et al. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.