

Abstract
To identify candidate proteins in the nucleus accumbens (NAc) as potential pharmacotherapeutic targets for treating cocaine addition, an 8-plex iTRAQ (isobaric tag for relative and absolute quantitation) proteomic screen was performed using NAc tissue obtained from rats trained to self-administer cocaine followed by extinction training. Compared with yoked-saline controls, 42 proteins in a postsynaptic density (PSD)-enriched subfraction of the NAc from cocaine-trained animals were identified as significantly changed. Among proteins of interest whose levels were identified as increased was AKAP79/150, the rat ortholog of human AKAP5, a PSD scaffolding protein that localizes signaling molecules to the synapse. Functional downregulation of AKAP79/150 by microinjecting a cell-permeable synthetic AKAP (A-kinase anchor protein) peptide into the NAc to disrupt AKAP-dependent signaling revealed that inhibition of AKAP signaling impaired the reinstatement of cocaine seeking. Reinstatement of cocaine seeking is thought to require upregulated surface expression of AMPA glutamate receptors, and the inhibitory AKAP peptide reduced the PSD content of protein kinase A (PKA) as well as surface expression of GluR1 in NAc. However, reduced surface expression was not associated with changes in PKA phosphorylation of GluR1. This series of experiments demonstrates that proteomic analysis provides a useful tool for identifying proteins that can regulate cocaine relapse and that AKAP proteins may contribute to relapse vulnerability by promoting increased surface expression of AMPA receptors in the NAc.
Introduction
A hallmark feature of addiction to drugs of abuse is the high rate of relapse, even after prolonged abstinence (O'Brien, 2001). Thus, considerable interest lies in identifying the enduring cellular changes induced by drug use that are responsible for relapse vulnerability. Although many candidate proteins have been identified based on specific hypotheses regarding the neurobiology of addiction (Hyman et al., 2006; Kalivas, 2009), this question is ideally suited for an unbiased proteomic analysis, and to date only a limited number of studies have used this approach (Hemby, 2006, 2010; Boudreau et al., 2007; Olausson et al., 2007; Van den Oever et al., 2008). We used an eight-plex isobaric tag for relative and absolute quantification (iTRAQ) approach (Ross et al., 2004) for de novo identification of candidate proteins within a postsynaptic density (PSD) enriched subfraction of the nucleus accumbens (NAc), a region well characterized for its role in addiction (Koob and Le Moal, 2001; Kalivas and Volkow, 2005). iTRAQ uses covalent attachment of isobaric tags to individual samples, allowing for qualitative and quantitative analysis of multiple control and experimental samples simultaneously in a single solution by liquid chromatography/tandem mass spectrometry (LC-MS/MS) (Tannu and Hemby, 2006; Li and Smit, 2008).
Because synaptic plasticity has emerged as an important mechanism in the neuroadaptations believed to underlie drug seeking (Kauer and Malenka, 2007; Bowers et al., 2010), we restricted our screen to protein changes within a PSD-enriched Triton X-100-insoluble synaptic membrane fraction. Although signaling and structural proteins not restricted to the PSD contribute to synaptic plasticity, proteins enriched in the PSD, including glutamate receptors, scaffolding proteins, and kinases, have been functionally implicated in cocaine-induced plasticity (Hyman et al., 2006; Kalivas, 2009; Bowers et al., 2010). Specifically, we were interested in using the iTRAQ approach to screen for proteins that significantly changed levels in the PSD after cocaine self-administration and extinction and thereby identifying candidate proteins that might contribute to drug seeking in a reinstatement model for relapse (Shaham et al., 2003).
A total of 42 proteins were shown to be significantly changed between the cocaine and saline groups, among which 27 were upregulated and 15 were downregulated. We selected one identified protein for functional analysis, AKAP150. AKAPs (A-kinase anchor protein) provide a mechanism for localizing protein kinase A (PKA) and other signaling molecules and thereby contribute to synaptic plasticity and learning and memory (Wong and Scott, 2004; Dell'Acqua et al., 2006). Moreover, a variety of data support signaling through PKA as important for acquiring addiction-related behaviors, such as behavioral sensitization, conditioned place preference, and drug self-administration (Hyman et al., 2006). Here we report that inhibition of AKAP signaling in the NAc impaired reinstated drug seeking in animals trained to self-administer cocaine and reduced surface expression of GluR1.
Materials and Methods
Animals and behavioral analysis.
All methods were in compliance with National Institutes of Health guidelines for care of laboratory animals and approved by the Medical University of South Carolina Institutional Animal Care and Use Committee. Animals were individually housed on a 12 h reverse light/dark cycle. Male Sprague Dawley rats (∼300 g) were surgically implanted with intrajugular catheters to self-administer cocaine. For peptide microinjection into the nucleus accumbens, bilateral guide cannulas (33 gauge) were implanted 1.7 mm anterior from bregma, 1.8 mm mediolateral, and 5.5 mm ventral from skull surface (Paxinos and Watson, 2005).
Rats were trained to self-administer cocaine for 2 weeks, followed by 2 weeks of extinction training as described previously (Moussawi et al., 2009). A 15 h food training session was used before the onset of self-administration to facilitate acquisition of the task, and food was restricted during self-administration and extinction to ∼20 g of rat chow per day. Training was performed in standard operant chambers containing a house light and available light and tone cues (Med Associates). All sessions during each stage of training lasted 2 h at the same time each day, 6 d/week. Drug exposure during self-administration was performed on a fixed-ratio 1 schedule in which each active lever press resulted in a 0.05 ml infusion of 0.2 mg of cocaine in sterile saline, followed by a 20 s timeout period. Each infusion was also paired with a cue light and tone. Control animals were yoked to cocaine animals, receiving an infusion of saline with each cocaine infusion. For proteomic analysis, after 12 d of extinction training, animals were rapidly decapitated and tissue was prepared as described below. For behavioral analysis, reinstatement was performed in a 2 h session immediately after a 10 mg/kg intraperitoneal injection of cocaine and return of the light and tone cues contingent on active lever pressing. To evaluate a role for AKAP peptides in reinstated cocaine seeking, Promega InCELLect AKAP Inhibitor Peptide St-Ht31 or control peptide St-HT31P were microinjected (0.5 μl) using 26 gauge microinjectors (Plastics One). Microinjection was performed over a period of 1 min, followed by 1 min diffusion time, into the NAc (2 mm below cannula base) 30 min before reinstatement testing using a random, cross-over design separated by 3 d of additional extinction training. After the behavioral study, animals were given an overdose of pentobarbital. Brains were fixed with 4% Formalin, and coronal sections were made and stained with cresyl violet to validate cannula location in the NAc (see Fig. 4).
Figure 4.
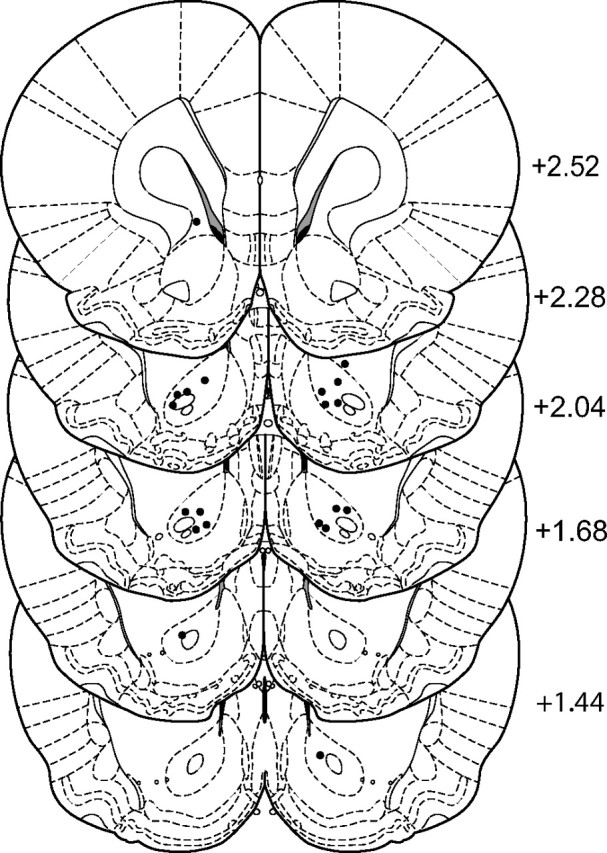
Placements of cannulas used in St-Ht31/St-Ht31P microinjections studies. Locations from bregma are indicated in millimeters at right. Cannula placement for each animal is indicated in each hemisphere (•).
Subcellular fractionation and enrichment.
Nucleus accumbens tissue samples were homogenized with 0.32 m sucrose, 10 mm HEPES, pH 7.4, and protease/phosphatase inhibitor cocktail (Thermo Fisher Scientific), and the PSD subfraction was isolated as a Triton X-100-insoluble fraction as described previously (Shen et al., 2009). For proteomic analysis, the resulting pellet containing enriched PSD and other detergent-insoluble proteins was solubilized in 1% PPS Silent surfactant in 500 mm triethylammonium bicarbonate (TEAB) buffer, and protein concentration was measured using the BCA assay (Pierce). Each sample (50 μg protein) was reduced in 5 mm Tri-(2-carboxyethyl) phosphine, alkylated with methyl methanethiosulfonate, diluted to 0.1% PPS Silent surfactant with 500 mm TEAB, and digested overnight in trypsin (1:20, enzyme/protein ratio; Applied Biosystems). Tryptic peptides were labeled with iTRAQ eight-plex reagents for 2 h at room temperature, and the contents of all digested samples were combined. The yoked saline animals (n = 3) were labeled with the 113, 115, and 118 tags, whereas the cocaine self-administration animals (n = 4) were labeled with 114, 116, 117, and 119 tags.
Strong cationic-exchange chromatography and HPLC chromatography.
The combined sample was acidified with 1% trifluoroacetic acid (TFA) and subjected to strong cationic-exchange chromatography (SCX) fractionation using HPLC-UV and a PolySULFOETHYL A column (200 × 2.1 mm inner diameter, 5 μm, 200 Å). Solvent A was 10 mm KH2PO4 and 25% acetonitrile (ACN), pH 2.7–3.0; solvent B was similar to A but with 0.5 m KCl and applied using 40 min gradient from 10 to 50% B, followed by 40 min at 50%. Five minute fractions were collected, dried, and stored at −20°C. SCX fractions were thawed, and 15 were selected based on the UV analysis for additional fractionation by HPLC. The peptides were first loaded on a C18 precolumn cartridge [5 × 300 μm inner diameter packed with PepMap 100, 5 μm, 100 Å (LC Packings)] using 2% ACN and 0.1% trifluoroacetic acid in water at 40 μl/min. After desalting, peptides were eluted from the precolumn onto a C18 (150 × 100 μm inner diameter, 3 μm, 300 Å) column (Micro-Tech Scientific) at 600 nl/min. Solvent A was 2% ACN and 0.1% TFA in water; solvent B was 85% ACN, 5% 2-propanol, and 0.1% TFA. A 50 min gradient from 12% B to 41% B at 600 nl/min was used. Peptide elution was monitored at 214 nm. The eluate from the reversed-phase HPLC separation was mixed with matrix (1:2) with 8 mg/ml α-cyano-4-hydroxy-cinnamic acid in 70% ACN, 0.1% TFA, and 0.15 mg/ml ammonium citrate, and eight mass calibration and two mass accuracy verification spots were manually spotted on stainless steel matrix assisted laser desorption ionization (MALDI) plates (Applied Biosystems).
Mass spectrometry analysis.
MS and MS/MS analyses were performed on a 4800 Proteomics Analyzer MALDI time-of-flight mass spectrometer equipped with the 4000 Series Explorer (version 3.5) data acquisition software (Applied Biosystems). After plate calibration, alignment, and default calibration update for each MALDI plate, an MS spectrum (800–3500 Da) was acquired from all 396 spots of each HPLC run using 1000 laser shots per spot. Mass spectra were subjected to an interpretation method to select the15 most intense precursors from each spot. Redundancy in the precursor selection was avoided by selecting each precursor only on the spot with maximized MS intensity. MS/MS spectra were acquired for the selected precursors (weakest to strongest intensity) on each spot using 2000 shots of the laser. Air was used as the collision induced dissociation gas during MS/MS acquisition.
Peptide and protein identification and quantification.
All MS/MS spectra were searched using Protein Pilot 2.0 software with the Paragon search algorithm (Applied Biosystems), using the RefSeq protein sequence database for Rattus norvegicus (ftp://ftp.ncbi.nih.gov/refseq). The search was done using trypsin and allowing two missed cleavages per peptide. Carbamidomethyl alkylation of cysteines and oxidation of methionine were used as fixed and variable modifications. The peptide charge was set to +1, and only the first ranking peptide with >70% confidence was used for protein identification. The identified peptides were grouped according to their parent proteins using Protein Pilot 2.0. Only proteins with a cumulative confidence >95% were reported. Reporter ion peak area measurements (Protein Pilot 2.0) were used to estimate treatment-dependent peptide and protein relative expression. In addition, all MS/MS spectra were searched with Mascot (Matrixscience) using the RefSeq protein sequence database for Rattus norvegicus (ftp://ftp.ncbi.nih.gov/refseq), and the scores for significantly (p < 0.05; i.e., credible interval ≠ 1) changed proteins are reported in Table 1. The search was done using trypsin and allowing two missed cleavages per peptide. Carbamidomethyl alkylation of cysteines was used as fixed and oxidation of methionine, iTRAQ modifications of lysine, iTRAQ–N-terminal, and iTRAQ–tyrosine were the variable modifications. Assessment of treatment-dependent changes in protein expression used a model-based approach described by Hill et al. (2008) using computational methods and software described by Schwacke et al. (2009). The linear model presented by Hill et al. was adapted for this study as follows. Reporter ion reagent cluster areas were modeled using the following expression: Ii,j(i),c,t,l = ElPiRi,cFj(i)Gj(i),cStHi,j(i),c,t(q),lK, where reporter ion cluster area (Ii,j(i),c,t,l) from channel l (e.g., 113, 114, 115, 116, 117, 118, or 119) of spectrum t, for a sample from treatment group c, for peptide j(i) associated with protein i, is written as a product of terms capturing sources of variability. The term Pi represents the protein expression for protein i in the reference group, and the factor Ri,c represents the ratio of expression for protein i in treatment group c relative to that of the reference group. Inferences on protein-level changes in expression are based on estimation of Ri,c. Term Fj(i) gives the ratio of the amount of the jth peptide to that of the ith protein, and Gj(i),c gives the ratio of the amount of the jth peptide in treatment group c to the amount of the jth peptide in the reference group after accounting for treatment-dependent differences in the associated protein. The term St gives the spectrum-to-spectrum shift in reporter ion intensity for spectra of the same peptide, El represents iTRAQ channel variation, Hi,j(i),c,t(q),l is included to represent remaining sources of variation (biological, instrument noise, etc.), and the scale factor K provides a constant capturing units and scale changes in the model. Taking logs of both sides yields the linear expression used in this analysis. Inference was accomplished with a Bayesian statistical approach using software developed in-house to overcome limitations in available tools (Schwacke et al., 2009) and inference challenges with the statistical model (Oberg et al., 2008). For each effect in the model above (p, e, r, f, g, s), parameters associated with that effect were assumed to follow a common normal prior distribution with 0 mean and unknown precision. The error term (h) was also assumed to follow a 0-mean normal distribution with unknown precision described by a gamma prior distribution, and precision parameters were estimated as part of the inference process.
Table 1.
Proteins significantly changed by cocaine self-administration and extinction
| Accession number | Protein | 95% credible intervala | No. of peptides | Protein coverage | Mascot score |
|---|---|---|---|---|---|
| Cytoskeletal | |||||
| gi|31543764 | α-Spectrin 2 | 1.01–1.09 | 81 | 0.3989 | 4868 |
| gi|13591902 | Actinin, α1 | 1.03–1.21 | 13 | 0.1839 | 549 |
| gi|9506811 | Internexin neuronal intermediate filament protein, α | 1.13–1.33 | 10 | 0.2099 | 743 |
| gi|13928704 | Myosin, heavy polypeptide 10, non-muscle | 1.01–1.20 | 10 | 0.0855 | 134 |
| gi|13929098 | Neurofilament, light polypeptide | 1.03–1.24 | 8 | 0.1439 | 350 |
| gi|11560133 | Tubulin, α1A | 0.74–1.00 | 2 | 0.0599 | 831 |
| gi|109506033; gi|109505063 | Similar to actinin α2; similar to actinin α2 | 1.03–1.34 | 4 | NaN | 451 |
| Synaptic | |||||
| gi|27465607 | Discs large (Drosophila) homolog-associated protein 3 | 1.01–1.32 | 4 | 0.0583 | 245 |
| gi|11560135 | Brain abundant, membrane attached signal protein 1 | 1.13–1.27 | 19 | 0.8227 | 1947 |
| gi|164663821 | Synaptic Ras GTPase activating protein 1 homolog isoform 1 | 1.00–1.16 | 16 | NaN | 587 |
| gi|8393415 | Growth associated protein 43 | 1.18–1.35 | 15 | 0.7168 | 1111 |
| gi|158749559 | Bassoon protein | 1.10–1.24 | 30 | 0.1353 | 1237 |
| gi|6981602 | Syntaxin binding protein 1 | 0.82–0.97 | 9 | 0.2239 | 411 |
| gi|11067399 | SH3/ankyrin domain gene 3 | 1.03–1.23 | 9 | 0.0885 | 609 |
| gi|13591936 | Drebrin 1 | 1.04–1.35 | 4 | 0.0665 | 186 |
| gi|158303339 | Synaptopodin | 1.03–1.31 | 5 | 0.1301 | 184 |
| gi|19424156 | A-kinase anchor protein 5 | 1.00–1.26 | 5 | 0.0966 | 357 |
| Cell signaling | |||||
| gi|6978593 | Calcium/calmodulin-dependent protein kinase II, α | 1.03–1.18 | 14 | 0.3054 | 2604 |
| gi|108796657 | Calcium/calmodulin-dependent protein kinase II β subunit | 1.05–1.25 | 7 | 0.2288 | 1295 |
| gi|57977323 | 2′,3′-cyclic nucleotide 3′ phosphodiesterase | 0.83–0.94 | 21 | 0.5357 | 883 |
| Channels/receptors/transporters | |||||
| gi|13786200 | Voltage-dependent anion channel 1 | 1.00–1.17 | 10 | 0.4664 | 1024 |
| gi|32189355 | Solute carrier family 25, member 4 | 0.78–0.94 | 7 | 0.2215 | 501 |
| gi|31542335 | Calcium channel, voltage-dependent, a2/d subunit 1 | 1.06–1.31 | 6 | 0.0862 | 208 |
| gi|32189350 | Solute carrier family 25, member 5 | 0.77–0.96 | 5 | 0.2081 | 332 |
| gi|9507115 | Glial high affinity glutamate transporter, member 3 (GLAST) | 0.74–0.99 | 2 | 0.0516 | 109 |
| Metabolism | |||||
| gi|16758362 | Cytochrome c oxidase subunit Vb | 1.04–1.24 | 9 | 0.6279 | 245 |
| gi|157819953; gi|109493236; gi|109493234 | ATPase, H transporting, lysosomal V1 subunit A | 1.02–1.22 | 9 | NaN | 887 |
| gi|148747253 | ATPase, Na+/K+ transporting, β1 polypeptide | 0.77–0.92 | 8 | 0.2664 | 397 |
| gi|109467443; gi|109465447; gi|109458527 | Similar to cytochrome c oxidase, subunit VIb polypeptide 1 | 0.84–1.00 | 8 | NaN | 566 |
| gi|8393180 | Cytochrome c oxidase subunit IV isoform 1 | 1.05–1.26 | 7 | 0.3728 | 346 |
| gi|170295834; gi|109487466 | NADH dehydrogenase (ubiquinone) 1α subcomplex 10 | 0.81–0.98 | 7 | NaN | 581 |
| gi|6978545 | ATPase, Na+/K+ transporting, α2 polypeptide | 0.72–0.89 | 6 | 0.0863 | 1466 |
| gi|109474252 | Similar to NADH dehydrogenase (ubiquinone) 1α subcomplex, 9 | 0.82–0.97 | 11 | 0.4138 | 475 |
| gi|6978547 | Na+/K+-ATPase a3 subunit | 0.76–0.90 | 11 | 0.1441 | 2201 |
| gi|24233541 | Cytochrome c oxidase, subunit Va | 1.07–1.23 | 11 | 0.6027 | 733 |
| gi|34866853; gi|109482351 | Similar to cytochrome c-1; similar to cytochrome c-1 | 1.00–1.25 | 5 | NaN | 324 |
| gi|114145517 | NADH dehydrogenase (ubiquinone) 1α subcomplex, 8 | 0.76–0.99 | 3 | 0.1693 | 47 |
| gi|72086149 | NADH dehydrogenase (ubiquinone) Fe-S protein 5b | 0.69–0.95 | 2 | 0.2547 | 132 |
| Cell adhesion | |||||
| gi|13591880 | Proteolipid protein (myelin) 1 | 0.70–0.89 | 4 | 0.1408 | 98 |
| gi|16923964 | Contactin 1 | 1.04–1.21 | 14 | 0.1459 | 805 |
| gi|70166255 | Myelin basic protein isoform 2 | 1.01–1.16 | 12 | 0.5799 | 769 |
| Unknown | |||||
| gi|62079059 | BM88 antigen | 1.02–1.33 | 3 | 0.2550 | 231 |
Significantly changed proteins are indicated within groups by function. Upregulated proteins are indicated in regular type, and downregulated proteins are indicated in bold type. The number of peptides identified by MS/MS for each protein are indicated, as well as average percentage change among identified proteins and fraction of the entire protein sequence covered by identified peptides. NaN indicated Not a Number, meaning that the protein was not found in the protein sequence file used to construct the report and so the sequence coverage could not be computed, in a case in which the protein database searched by Mascot is different from that used to produce the report. Assessment of treatment-dependent changes in protein expression using 95% credible intervals as described by Hill et al. (2008) using computational methods and software described by Schwacke et al. (2009). Mascot scores were calculated using the Mascot algorithm (Matrixscience).
aWhen the credible interval is 1, the protein is significantly (p < 0.05) different between the cocaine and yoked saline groups.
Tandem MS summary reports including best peptide sequence, protein name, accession number, modifications, and corrected iTRAQ reagent cluster areas were exported from the vendor-supplied software and converted into tab-delimited text files. Data from a total of 19,421 spectra were considered with 15,163 below the 70% peptide confidence threshold and thus eliminated from this analysis. Ninety-five additional spectra were eliminated as a result of disallowed modifications (iTRAQ-modified tyrosines) and 11 as a result of missing data. Using the Gibbs Sampler, 100,000 samples were collected from the Markov chain after a burn-in period of 40,000 samples. The samples were thinned by a factor of 20, leaving 5000 samples from which summary statistics were collected. For each protein and each associated peptide, mean, median, and 95% credible intervals were computed for each of the protein- and peptide-level treatment effects (ri,c and gj(i),c) and used along with previous knowledge of the pathways of interest to select proteins for additional study. A change was considered significant (p < 0.05) if the median protein ratio and 95% credible interval range did not include a ratio of 1 (a ratio of 1 = no change in relative expression between saline and cocaine groups). Although only significantly changed proteins are reported here because of space limitations, the original complete data report is available by request.
Protein interaction network model.
The network was created using the BisoGenet plugin (Martin et al., 2010) for Cytoscape (Shannon et al., 2003). The BisoGenet plugin searches a molecular interaction database (SysBiomics) that integrates data from genes and protein databases (National Center for Biotechnology Information, UniProt), pathways and ontologies (Kyoto Encyclopedia of Genes and Genomes, Gene Ontology), and molecular interactions (Database of Interacting Proteins, Biomolecular Interaction Network Database, Human Reference Protein Database). In this study, we searched for protein–protein and protein–DNA interactions using version 10.4.0 of the SysBiomics database. Differentially expressed nodes from the iTRAQ analysis were used as input, and the network was assembled by adding neighbors of input nodes up to a distance of 1. The resulting network was visualized using Cytoscape, and three additional edges, based on interactions between AKAP150 and proteins found in the literature (Colledge et al., 2000) but not present in the database, were added to connect subgraphs created in this process (see Fig. 2, shown in black on the network map).
Figure 2.

Protein network analysis using Cytoscape reveals upregulation in proteins associated with glutamatergic signaling. Upregulated proteins are indicated by red circles and downregulated proteins by dark blue circles. Additional interacting proteins are indicated by light blue font. This network analysis reveals linkage of AKAP5 with PSD scaffolding proteins and most immediately with upregulated CaMKII.
Western blotting and biotinylation.
Western blotting was performed as described previously (Shen et al., 2009). In the case of blotting for a phospho-specific antibody (GluR1 pS845), blocking was performed in 3% milk in TBS with Tween 20 containing phosphatase inhibitors (1:100; Thermo Fisher Scientific). PSD localization of PKA catalytic subunit (PKAc) was normalized against the activator of G-protein signaling 5 (AGS5/LGN/mPINS/GPSM2) (Blumer et al., 2002). Antibodies used in Western blots included the following: AGS5 (1:1000; from Drs. Joe Blumer and Stephen M. Lanier, Medical University of South Carolina), PKAc (1:1000; catalog #610981; BD Biosciences Transduction Laboratories), phospho-S845 GluR1 (1:500; Millipore), GluR1 (1:500; catalog #ab31232; Abcam), Bassoon (1:500; Assay Designs), GLAST/EAAT2 (1:200; catalog #ab416; Abcam), CaMKIIβ (1:200; catalog #ab34703; Abcam), Synaptopodin (1:1000; catalog #ab50859; Abcam), AKAP150 (1:1000; catalog #07-210; Millipore), and GAP43 (1:2000; catalog #ab50608; Abcam). In the case of Bassoon, before transfer, the gel was soaked in NuPAGE (Invitrogen) 2× transfer buffer for 20 min. For all Western blotting, band density was normalized in the yoked saline control animals and compared with cocaine-trained animals.
For the surface biotinylation assay, the NAc was dissected and sliced into prism-shaped sections (200 μm) 45 min after peptide microinjection. The tissue was incubated for 30 min in 250 μl of PBS containing 1 mg/ml Sulfo-NHS-Biotin (Pierce) at 4°C with gentle shaking. The reaction was quenched by adding ice-cold 100 mm glycine in PBS. The tissue was washed twice with ice-cold 100 mm glycine in PBS and then sonicated in 250 μl of 1% SDS in radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitor cocktail (Pierce). After incubating 30–60 min on ice, the protein-contained solution was centrifuged at 10,000 × g for 10 min at 4°C to remove insoluble material. Supernatant containing 200 μg was combined with 100 μl of NeutrAvidin agarose resin and incubated for 2 h at 4°C with gentle rotation. After washing with ice-cold PBS twice, biotinylated proteins were eluted by 35 μl of loading buffer (1% SDS and 50 mm DTT) with heating at 80°C for 5 min. Proteins in the biotinylated fraction were detected by immunoblotting.
Results
Identification of protein expression changes by iTRAQ proteomic analysis
After 2 weeks of extinction training after cocaine self-administration (Fig. 1A), NAc tissue was examined for PSD protein content. Although samples were prepared as a PSD-enriched subfraction, it is important to note that, given the limited tissue available from the NAc, only a crude subfraction containing Triton X-100-insoluble synaptosomal and glial proteins was prepared. Quantitative analysis and identification of LC-MS/MS spectra comparing cocaine-trained and yoked-saline rats revealed 442 proteins comprising 2268 unique peptides (data can be made available on request). Among the 442 proteins, 105 were identified by a single peptide and not analyzed further. Of the remaining 337 proteins, a total of 42 were significantly different between the cocaine and saline groups, including 27 upregulated and 15 downregulated (Fig. 1C; Table 1). Representative MS spectra for CaMKIIβ and AKAP5 are shown in Figure 1B. Although most of the identified proteins have not been linked previously to the cell biology of addiction, some proteins identified herein were also noted in previous studies, including CaMKII, actin, and GLT-1 (Licata et al., 2004; Toda et al., 2006; Boudreau et al., 2009; Knackstedt et al., 2009).
Figure 1.

Summary of iTRAQ proteomic results. A, Active lever presses for cocaine and yoked saline controls used for proteomic analysis (n = 4 for each group). B, Representative MS spectra profile for selected identified peptides from CaMKIIβ and AKAP5. Peaks were derived from yoked saline (S) or cocaine-trained (C) animals. Peak height indicates the relative abundance of the isobaric tag in that sample. C, Categorization of proteins whose levels were significantly upregulated or downregulated after cocaine self-administration and extinction training.
Functional categorization of protein differences revealed that the majority of cocaine-elevated proteins were cytoskeletal and synaptic (Fig. 1C). Also, the most heavily represented class of proteins were those that contribute to metabolic functions. In addition to general categorizing by function, we conducted an analysis of protein interactions in an effort to discern changes in signaling complexes that might be present. Figure 2 illustrates the largest network of functionally interacting proteins that included many of the cocaine-altered proteins. This network is primarily PSD scaffolding proteins and those related to postsynaptic glutamate signaling. Among these proteins was AKAP150, the rat ortholog of human AKAP5, which was linked to other PSD components via PSD-95. Although PSD-95 was not significantly changed in our screen, changes in expression in the NAc after cocaine self-administration and extinction have been reported (Ghasemzadeh et al., 2009; Knackstedt et al., 2010), supporting the relevance of the network implicated here. Among changed proteins identified by iTRAQ, the most heavily interacting protein in the network was CaMKII, for which mounting evidence indicates to be a common molecular node in the addiction process (Li et al., 2008).
AKAP regulates reinstated cocaine seeking
We selected AKAP for additional analysis because of characteristics conveying potential for regulating the reinstatement of cocaine seeking. AKAP150 is localized to the postsynaptic membrane via direct interactions with phospholipids, thereby providing proximal localization via protein–protein interactions with the RII regulatory subunit of PKA, PKC, and protein phosphatase 2B (calcineurin) (Wong and Scott, 2004; Dell'Acqua et al., 2006). AKAP150 also forms indirect interactions with AMPA receptor subunits via MAGUK proteins and thereby affects AMPA surface expression (Colledge et al., 2000; Wong and Scott, 2004; Snyder et al., 2005). Importantly, chronic cocaine administration has been found to alter levels and/or function of the AKAP150-associated kinase PKA and AMPA receptor surface expression (Hyman et al., 2006; Conrad et al., 2008), supporting the hypothesis that AKAP function might contribute to the cellular mechanisms of reinstatement.
To test this hypothesis, we microinjected an inhibitory peptide containing the PKA-binding sequence from human thyroid AKAP (St-Ht31) into the NAc of animals previously trained to self-administer cocaine and then extinguished (Fig. 3A). After self-administration training, animals were divided into two groups having equivalent levels of cocaine self-administration over the last 3 d of training (Fig. 3A,B). Thirty minutes before an acute injection of cocaine to reinstate extinguished lever pressing, animals were microinjected with either the active peptide or a proline mutant negative control peptide (St-Ht31P). Active lever pressing was significantly attenuated after injection of St-Ht31 compared with control peptide (Fig. 3B). No difference between groups was observed in inactive lever pressing (data not shown) or cocaine-induced locomotor behavior (Fig. 3C). These results suggest that reducing the synaptic localization of AKAP150 regulating signaling proteins, such as PKA, PKC, or calcineurin, inhibits reinstated cocaine seeking. Figure 4 shows that the microinjection cannula tips for animals used in the reinstatement study were localized to the core subcompartment of the NAc.
Figure 3.
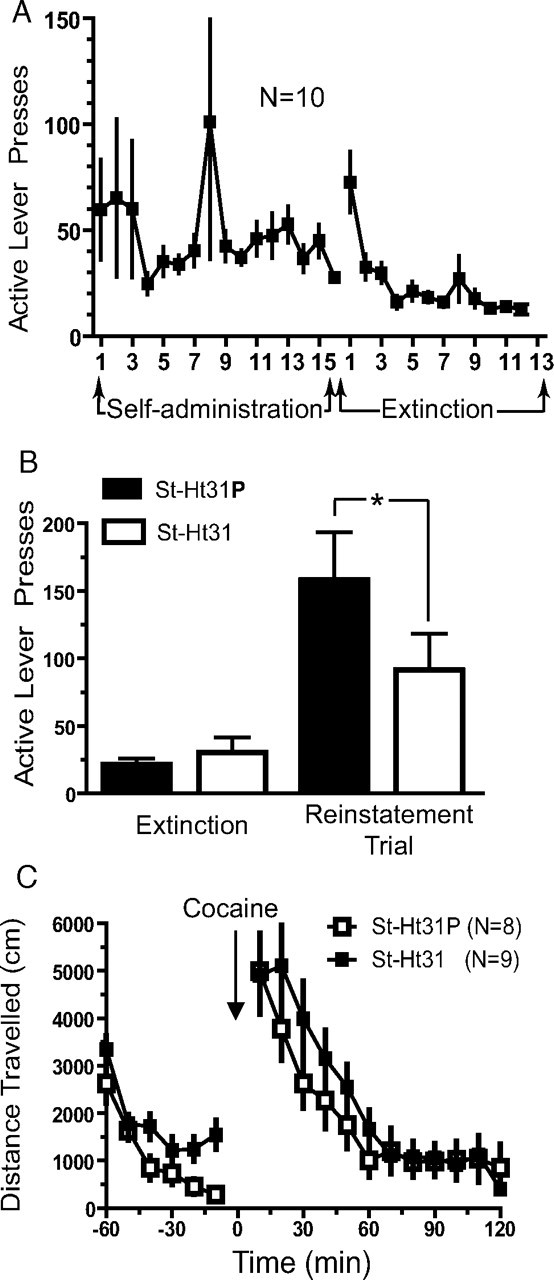
AKAP inhibitory peptide inhibits reinstated cocaine seeking but not locomotor activity. A, Active lever presses for animals used in studying the behavioral effects of St-Ht31 peptide on reinstatement of cocaine seeking. B, Microinjection of St-Ht31 peptide significantly impaired reinstatement compared with microinjection of St-Ht31P. Active lever presses are shown as total presses over a 2 h reinstatement trial. C, Lack of effect by St-Ht31 on distance traveled in response to an acute cocaine challenge (10 mg/kg, i.p.). *p ≤ 0.05, comparing control with active peptide using a two-tailed Student's paired t test.
AKAP150 regulates PKA content in the PSD and GluR1 surface expression
Regulation of surface expression of GluR1 by PKA phosphorylation is well described (Malinow and Malenka, 2002; Boehm and Malinow, 2005). Because disruption of AKAP interactions with PKA, PKC, and/or calcineurin by the St-Ht31 peptide impaired reinstatement and because increased GluR1 expression mediates drug-seeking behaviors (Conrad et al., 2008), we hypothesized that microinjection of Ht31 should result in a decrease of PKA in the PSD and a corresponding decrease in surface expression of GluR1. An injection of St-Ht31 into the NAc in drug-naive animals reduced the level of PKAc in the PSD subfraction by 30 min after peptide microinjection (Fig. 5A).
Figure 5.

Microinjection of St-Ht31 peptide reduces PSD content of PKA and surface expression of GluR1. A, Microinjections of St-Ht31 reduced PSD content for PKAc. Data were normalized to LGN content. B, Peptide microinjections were made as in A, except tissue was prepared for biotinylation and Western blotting for GluR1. Data are shown as the ratio of the density of the GluR1 band between the two treatment groups (St-Ht31/St-Ht31P). Dashed line indicates a ratio of 1.0. *p < 0.05 comparing control with active peptide using a two-tailed Student's t test.
Because the PKA-driven phosphorylation of GluR1 promotes AMPA receptor surface expression, we hypothesized that disruption of AKAP-dependent localization of PKA might reduce surface GluR1 expression. To test this, St-Ht31 and St-Ht31P were microinjected into NAc core of contralateral hemispheres within individual animals, and 45 min later surface expressed proteins were labeled using a biotinylation strategy. Indeed, microinjection of St-Ht31 reduced the surface expression of GluR1-containing AMPA receptors compared with control peptide St-Ht31P (Fig. 5B). Surprisingly however, we found that microinjection of active peptide St-Ht31 did not change the phosphorylation state of GluR1 at Ser845 versus St-Ht31P microinjection (data not shown). These data indicate that, although the inhibitory AKAP peptide moves PKAc out of the PSD, an alternative mechanism beyond PKA phosphorylation of GluR1 at residue 845 may be responsible for the internalization of GluR1. For example, in addition to PKA, AKAP localizes calcineurin and PKC to the postsynaptic membrane (Wong and Scott, 2004), and hippocampal long-term depression (LTD) requires an interaction between AKAP and calcineurin but not PKA (Jurado et al., 2010).
Western blotting
Given the potent influence of downregulating AKAP function on the capacity to reinstate cocaine seeking, we sought to further validate the cocaine-induced change in AKAP expression by Western blotting performed on accumbens PSD (Fig. 6). Surprisingly, we found that AKAP150 levels were not altered after 2 weeks of extinction training after cocaine self-administration. We then selected five addition proteins identified by the iTRAQ screen as significantly different between cocaine-trained and yoked-saline animals, including Bassoon, CaMKIIβ, GAP43, EAAT1/GLAST, and Synaptopodin for Western blot measurement. All selected proteins are involved in synaptic transmission and plasticity (Pfenninger et al., 1991; Schoch and Gundelfinger, 2006; Jedlicka et al., 2008; Robertson et al., 2009) and were chosen based on our interest in identifying candidates that might contribute to synaptic mechanisms of relapse. Western blot estimates of protein expression in the PSD subfraction of the accumbens revealed significant differences between cocaine-trained and yoked saline animals in three of the additional five proteins examined. Thus, although differences were confirmed for CaMKIIβ, GLAST/EAAT1, and Bassoon, no significant differences were observed for Synaptopodin or GAP43 (Fig. 6A). The dorsal striatum obtained from the same rats used for accumbens was also examined using Western blotting for a subset of these proteins, and, for the three proteins tested, no differences between cocaine and control animals were measured in PSD subfraction protein expression in striata for CaMKIIβ, AKAP150, or GAP43 (Fig. 6B).
Figure 6.

Western blot analysis of seven proteins identified by iTRAQ analysis as being significantly different between cocaine-trained and yoked saline subjects. Equal amounts of PSD-enriched proteins (5–10 μg) were loaded per lane. Membranes were cut horizontally for simultaneous analysis of experimental protein (top) and LGN/AGS5 (bottom). Bands from chronic cocaine (C) and saline (S) administering animals are indicated. A, PSD subfractions prepared from the nucleus accumbens. B, PSD subfractions were identically prepared from dorsal striatum. Data are shown as percentage change from yoked saline values. *p < 0.05 comparing cocaine with saline using a Student's two-tailed t test.
Discussion
Proteomic tools were used in the current study to screen for proteins that may contribute to the neural adaptations underlying cocaine addiction. Previously, Boudreau et al. (2009) performed a proteomic analysis of PKA substrates in cocaine-sensitized rats, whereas Uys et al. (2010) and Olausson et al. (2007) reported findings on changes in protein expression levels in response to cocaine-injected rats and primates. Two additional studies have used MS analysis in cocaine self-administering rhesus monkeys and human cocaine overdose victims to identify candidate proteins in the NAc (Tannu et al., 2007, 2010). However, no previous studies have extended proteomic analysis to test for a role in regulating relapse to cocaine seeking. Here we used an iTRAQ-based proteomic screen of a PSD-enriched NAc subfraction after cocaine self-administration and extinction training, followed by functional analysis of one candidate protein, AKAP150.
This particular candidate protein was selected because of its function as a regulator of PKA signaling and existing literature on the involvement of cAMP and PKA as important mediators of the effects of cocaine (Self and Nestler, 1995). Dopamine D1 receptors are coupled to adenylyl cyclase activation, resulting in cAMP activation and consequently activation of PKA. Notably, microinjection into the NAc of an inhibitor of cAMP, Rp-cAMPs, decreased cocaine self-administration but potentiated reinstated drug seeking, whereas injection of a cAMP activator, Sp-cAMPs, increased lever pressing during self-administration (Self et al., 1998). However, the cytosolic actions of cAMP and PKA include a number of target molecules compartmentalized away from the PSD (e.g., transcription factors) and therefore may mediate an entirely different series of responses than disruption of PKA localized at the synapse. Moreover, cAMP signaling is not limited to PKA but includes also other targets, most notably guanine nucleotide exchange factors (Bos, 2006). These results collectively suggest that synaptic PKA is required for reinstatement of drug seeking and that localization of PKA function by subcellular microdomains may importantly mediate opposing roles in response to drug action and in drug-seeking behavior. In addition to the studies described above, Self et al. (2004) reported that extinction training after cocaine self-administration normalized cocaine upregulated protein levels of AKAP84, indicating a possible role for AKAPs in the actions of cocaine. AKAP84, also referred to as D-AKAP1, is a widely expressed dual specificity AKAP (binds both to PKA type I and type II regulatory subunits), which has been associated with localization of PKA to organelles, including endoplasmic reticulum and mitochondria (Huang et al., 1997; Wong and Scott, 2004; Ma and Taylor, 2008).
By using the AKAP inhibitory peptide St-Ht31, a role for the AKAP family of proteins in fear learning (Moita et al., 2002; Nijholt et al., 2008) and synaptic plasticity (Génin et al., 2003; Liu et al., 2004; Snyder et al., 2005; Nie et al., 2007) has been shown. Specifically, St-Ht31 induces a rundown of EPSCs that impair the induction of hippocampal LTD and correlate with decreased synaptic localization of GluR1 and GluR2 subunits (Snyder et al., 2005). The capacity to reduce surface AMPA receptors may be critical for St-Ht31 inhibition of cocaine seeking, because AMPA receptor activation in the NAc is required for cocaine reinstatement. Blocking AMPA receptors in the NAc inhibits cocaine seeking (Cornish and Kalivas, 2000), and expression of the GluR1 subunit of the AMPA receptor is increased after chronic cocaine and withdrawal (Sutton et al., 2003; Self et al., 2004; Boudreau and Wolf, 2005; Boudreau et al., 2007; Conrad et al., 2008). Moreover, cocaine-primed reinstatement further increases GluR1 surface expression (Anderson et al., 2008). In addition to reducing GluR1 surface expression, we verified that in vivo St-Ht31 inhibition of AKAP reduces PKA sequestration in the PSD subfraction, providing a candidate mechanism for how the St-Ht31 peptide may impair reinstatement.
Despite the fact that treatment with St-Ht31 reduced the PSD PKAc content and surface levels of GluR1, we were not able to detect a significant change in GluR1 phosphorylation state at serine 845. This was a surprising result, given that stimulus-driven changes in phosphorylation of GluR1 control changes in surface expression (Lee et al., 2003; Boehm and Malinow, 2005; Man et al., 2007). However, multiple studies have indicated that unstimulated surface expression of GluR1 is not affected by conditions that should influence the basal phosphorylation state at Ser845, including genetic mutation of amino acid 845 (Lee et al., 2003; Lu et al., 2008; Sun et al., 2008), suggesting an alternative mechanism by which microinjection of St-Ht31 leads to internalization of GluR1. Along these lines, besides PKA, AKAP also localizes calcineurin and PKC to the postsynaptic membrane (Wong and Scott, 2004). Thus, although the Ht31 peptide specifically disrupts the interaction between PKA-RII and AKAP79/150 (Ruehr et al., 1999; Herberg et al., 2000), it is possible that disruption of interactions with other PSD proteins may lead to the internalization of GluR1. Furthermore, it has been shown recently that hippocampal LTD requires an interaction between AKAP150 and calcineurin but not PKA or PKC (Jurado et al., 2010), providing evidence of the functional significance of these interactions.
Including AKAP150, changes in protein expression were tested for six proteins by Western blotting and confirmed for GLAST, CaMKIIβ, and Bassoon but not for AKAP150, GAP43, or Synaptopodin. As a member of the glial high-affinity glutamate transporter family, GLAST was of interest because we previously identified another member of this family, GLT-1/EAAT2, as downregulated after exposure to chronic cocaine (Knackstedt et al., 2009). Although GLAST is predominantly expressed on glial cells (Danbolt, 2001), its identification in the iTRAQ likely results from the presence of Triton X-100-insoluble glial microdomains, akin to the neuronal PSD (Walikonis et al., 2000). Also of particular interest was the finding that both CaMKIIα and β subunits were significantly upregulated (Table 1), because numerous studies have identified a role for activation of CaMKII in drug abuse, both the holoenzyme and the CaMKIIα subunit (McClung and Nestler, 2003; Licata et al., 2004; Anderson et al., 2008; Sun et al., 2008; Boudreau et al., 2009; Liu et al., 2009; Wang et al., 2010). Thus, we chose to include the β subunit in our panel of protein for Western blotting, and the results here further underscore the developing appreciation for a role for this protein in mechanisms of addiction. Although we found no change in GAP43 by Western blotting, this protein is reported to be upregulated by self-administration and extinction from heroin in an iTRAQ proteomic screen (Van den Oever et al., 2010).
How do we consider the apparent and surprising discrepancy between iTRAQ and Western blotting results? One possible explanation is a potential differential sensitivity between the methods. iTRAQ is proposed to permit accurate quantification over at least two orders of magnitude in concentration (Casado-Vela et al., 2010). In contrast, chemiluminescent Western blotting has a dynamic linear range of just over one order of magnitude (Heinicke et al., 1992). With respect to AKAP, five individual peptides were identified by iTRAQ, with a mean increase in cocaine over saline samples of 13% and a credible interval of (1.0–1.26). Moreover, previous studies have found increased levels of AKAP150 in response to a novel environment as well as fear conditioning (Nijholt et al., 2007), and increased AKAP150 mRNA levels have been found after long-term potentiation-inducing stimuli (Génin et al., 2003), indicating that expression changes in AKAP150 may mediate other forms of experience-dependent behavioral plasticity. Alternatively, because Self et al. (2004) reported that cocaine-induced increases in expression of AKAP84 are normalized by extinction training, it is possible that both groups of rats (iTRAQ and Western blot studies) had a similar cocaine-induced change that was ameliorated to a different extent by extinction training. Ultimately, these results indicate that functional characterization is worthwhile for candidates for which proteomic expression and Western blot data are in disagreement.
Conclusions
Together, this study demonstrates the viability of using an iTRAQ proteomic screen to identify a protein regulated by cocaine self-administration, AKAP150, that promotes relapse vulnerability. Interestingly, these data also indicate that iTRAQ screening may identify proteins whose magnitude of change is at the threshold for being identified by more traditional Western blotting. Finally, although AKAP function was linked to the increase in GluR1 surface expression produced by cocaine in the NAc, it did not appear to be via a traditional PKA signaling cascade, posing a possible role for AKAP regulation of PKC or calcineurin.
Footnotes
This work was supported by National Institutes of Health Grants DA026254 (K.J.R.) and DA015369 and DA015851 (P.W.K.). We thank members of the Kalivas laboratory for critical comments on this manuscript and Charles Thomas and Megan Hensley for expert assistance with animal behavior.
References
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11:344–353. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- Blumer JB, Chandler LJ, Lanier SM. Expression analysis and subcellular distribution of the two G-protein regulators AGS3 and LGN indicate distinct functionality. Localization of LGN to the midbody during cytokinesis. J Biol Chem. 2002;277:15897–15903. doi: 10.1074/jbc.M112185200. [DOI] [PubMed] [Google Scholar]
- Boehm J, Malinow R. AMPA receptor phosphorylation during synaptic plasticity. Biochem Soc Trans. 2005;33:1354–1356. doi: 10.1042/BST0331354. [DOI] [PubMed] [Google Scholar]
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Ferrario CR, Glucksman MJ, Wolf ME. Signaling pathway adaptations and novel protein kinase A substrates related to behavioral sensitization to cocaine. J Neurochem. 2009;110:363–377. doi: 10.1111/j.1471-4159.2009.06140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers MS, Chen BT, Bonci A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron. 2010;67:11–24. doi: 10.1016/j.neuron.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado-Vela J, Martínez-Esteso MJ, Rodriguez E, Borrás E, Elortza F, Bru-Martínez R. iTRAQ-based quantitative analysis of protein mixtures with large fold change and dynamic range. Proteomics. 2010;10:343–347. doi: 10.1002/pmic.200900509. [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20(RC89):1–5. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol. 2006;85:627–633. doi: 10.1016/j.ejcb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Génin A, French P, Doyère V, Davis S, Errington ML, Maroun M, Stean T, Truchet B, Webber M, Wills T, Richter-Levin G, Sanger G, Hunt SP, Mallet J, Laroche S, Bliss TV, O'Connor V. LTP but not seizure is associated with up-regulation of AKAP-150. Eur J Neurosci. 2003;17:331–340. doi: 10.1046/j.1460-9568.2003.02462.x. [DOI] [PubMed] [Google Scholar]
- Ghasemzadeh MB, Vasudevan P, Mueller C, Seubert C, Mantsch JR. Region specific alterations in glutamate receptor expression and subcellular distribution following extinction of cocaine self-administration. Brain Res. 2009;1267:89–102. doi: 10.1016/j.brainres.2009.01.047. [DOI] [PubMed] [Google Scholar]
- Heinicke E, Kumar U, Munoz DG. Quantitative dot-blot assay for proteins using enhanced chemiluminescence. J Immunol Methods. 1992;152:227–236. doi: 10.1016/0022-1759(92)90144-i. [DOI] [PubMed] [Google Scholar]
- Hemby SE. Assessment of genome and proteome profiles in cocaine abuse. Prog Brain Res. 2006;158:173–195. doi: 10.1016/S0079-6123(06)58009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemby SE. Cocainomics: new insights into the molecular basis of cocaine addiction. J Neuroimmune Pharmacol. 2010;5:70–82. doi: 10.1007/s11481-009-9189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K. Analysis of A-kinase anchoring protein (AKAP) interaction with protein kinase A (PKA) regulatory subunits: PKA isoform specificity in AKAP binding. J Mol Biol. 2000;298:329–339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- Hill EG, Schwacke JH, Comte-Walters S, Slate EH, Oberg AL, Eckel-Passow JE, Therneau TM, Schey KL. A statistical model for iTRAQ data analysis. J Proteome Res. 2008;7:3091–3101. doi: 10.1021/pr070520u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Identification of a novel protein kinase A anchoring protein that binds both type I and type II regulatory subunits. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Jedlicka P, Vlachos A, Schwarzacher SW, Deller T. A role for the spine apparatus in LTP and spatial learning. Behav Brain Res. 2008;192:12–19. doi: 10.1016/j.bbr.2008.02.033. [DOI] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC. A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci. 2010;13:1053–1055. doi: 10.1038/nn.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, Markou A, Kalivas PW. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci. 2010;30:7984–7992. doi: 10.1523/JNEUROSCI.1244-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell. 2003;112:631–643. doi: 10.1016/s0092-8674(03)00122-3. [DOI] [PubMed] [Google Scholar]
- Li CY, Mao X, Wei L. Genes and (common) pathways underlying drug addiction. PLoS Comput Biol. 2008;4:e2. doi: 10.1371/journal.pcbi.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li KW, Smit AB. Subcellular proteomics in neuroscience. Front Biosci. 2008;13:4416–4425. doi: 10.2741/3014. [DOI] [PubMed] [Google Scholar]
- Licata SC, Schmidt HD, Pierce RC. Suppressing calcium/calmodulin-dependent protein kinase II activity in the ventral tegmental area enhances the acute behavioural response to cocaine but attenuates the initiation of cocaine-induced behavioural sensitization in rats. Eur J Neurosci. 2004;19:405–414. doi: 10.1111/j.0953-816x.2003.03110.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Hu JY, Schacher S, Schwartz JH. The two regulatory subunits of Aplysia cAMP-dependent protein kinase mediate distinct functions in producing synaptic plasticity. J Neurosci. 2004;24:2465–2474. doi: 10.1523/JNEUROSCI.4331-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Mao LM, Zhang GC, Papasian CJ, Fibuch EE, Lan HX, Zhou HF, Xu M, Wang JQ. Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron. 2009;61:425–438. doi: 10.1016/j.neuron.2008.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen M, Medvedeva Y, McKnight GS, Usachev YM, Hell JW. AKAP150-anchored PKA activity is important for LTD during its induction phase. J Physiol. 2008;586:4155–4164. doi: 10.1113/jphysiol.2008.151662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Taylor SS. A molecular switch for targeting between endoplasmic reticulum (ER) and mitochondria: conversion of a mitochondria-targeting element into an ER-targeting signal in DAKAP1. J Biol Chem. 2008;283:11743–11751. doi: 10.1074/jbc.M710494200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Man HY, Sekine-Aizawa Y, Huganir RL. Regulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc Natl Acad Sci U S A. 2007;104:3579–3584. doi: 10.1073/pnas.0611698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Ochagavia ME, Rabasa LC, Miranda J, Fernandez-de-Cossio J, Bringas R. BisoGenet: a new tool for gene network building, visualization and analysis. BMC Bioinformatics. 2010;11:91. doi: 10.1186/1471-2105-11-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Regulation of gene expression and cocaine reward by CREB and DeltaFosB. Nat Neurosci. 2003;6:1208–1215. doi: 10.1038/nn1143. [DOI] [PubMed] [Google Scholar]
- Moita MA, Lamprecht R, Nader K, LeDoux JE. A-kinase anchoring proteins in amygdala are involved in auditory fear memory. Nat Neurosci. 2002;5:837–838. doi: 10.1038/nn901. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie T, McDonough CB, Huang T, Nguyen PV, Abel T. Genetic disruption of protein kinase A anchoring reveals a role for compartmentalized kinase signaling in theta-burst long-term potentiation and spatial memory. J Neurosci. 2007;27:10278–10288. doi: 10.1523/JNEUROSCI.1602-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijholt IM, Ostroveanu A, de Bruyn M, Luiten PG, Eisel UL, Van der Zee EA. Both exposure to a novel context and associative learning induce an upregulation of AKAP150 protein in mouse hippocampus. Neurobiol Learn Mem. 2007;87:693–696. doi: 10.1016/j.nlm.2006.12.008. [DOI] [PubMed] [Google Scholar]
- Nijholt IM, Ostroveanu A, Scheper WA, Penke B, Luiten PG, Van der Zee EA, Eisel UL. Inhibition of PKA anchoring to A-kinase anchoring proteins impairs consolidation and facilitates extinction of contextual fear memories. Neurobiol Learn Mem. 2008;90:223–229. doi: 10.1016/j.nlm.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Oberg AL, Mahoney DW, Eckel-Passow JE, Malone CJ, Wolfinger RD, Hill EG, Cooper LT, Onuma OK, Spiro C, Therneau TM, Bergen HR., 3rd Statistical analysis of relative labeled mass spectrometry data from complex samples using ANOVA. J Proteome Res. 2008;7:225–233. doi: 10.1021/pr700734f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CP. Drug addiction and drug abuse. In: Hardman JG, Limbird LL, Gillman AG, editors. The pharmacological basis of therapeutics. New York: McGraw-Hill; 2001. pp. 621–642. [Google Scholar]
- Olausson P, Jentsch JD, Krueger DD, Tronson NC, Nairn AC, Taylor JR. Orbitofrontal cortex and cognitive-motivational impairments in psychostimulant addiction: evidence from experiments in the non-human primate. Ann N Y Acad Sci. 2007;1121:610–638. doi: 10.1196/annals.1401.016. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. Ed 5. Amsterdam: Elsevier Academic; 2005. The rat brain in stereotaxic coordinates. [Google Scholar]
- Pfenninger KH, de la Houssaye BA, Helmke SM, Quiroga S. Growth-regulated proteins and neuronal plasticity. A commentary. Mol Neurobiol. 1991;5:143–151. doi: 10.1007/BF02935543. [DOI] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML. Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J Neurosci. 2009;29:7929–7943. doi: 10.1523/JNEUROSCI.6093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3:1154–1169. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- Ruehr ML, Zakhary DR, Damron DS, Bond M. Cyclic AMP-dependent protein kinase binding to A-kinase anchoring proteins in living cells by fluorescence resonance energy transfer of green fluorescent protein fusion proteins. J Biol Chem. 1999;274:33092–33096. doi: 10.1074/jbc.274.46.33092. [DOI] [PubMed] [Google Scholar]
- Schoch S, Gundelfinger ED. Molecular organization of the presynaptic active zone. Cell Tissue Res. 2006;326:379–391. doi: 10.1007/s00441-006-0244-y. [DOI] [PubMed] [Google Scholar]
- Schwacke JH, Hill EG, Krug EL, Comte-Walters S, Schey KL. iQuantitator: a tool for protein expression inference using iTRAQ. BMC Bioinformatics. 2009;10:342. doi: 10.1186/1471-2105-10-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens in cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Self DW, Choi KH, Simmons D, Walker JR, Smagula CS. Extinction training regulates neuroadaptive responses to withdrawal from chronic cocaine self-administration. Learn Mem. 2004;11:648–657. doi: 10.1101/lm.81404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HW, Toda S, Moussawi K, Bouknight A, Zahm DS, Kalivas PW. Altered dendritic spine plasticity in cocaine-withdrawn rats. J Neurosci. 2009;29:2876–2884. doi: 10.1523/JNEUROSCI.5638-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Colledge M, Crozier RA, Chen WS, Scott JD, Bear MF. Role for A kinase-anchoring proteins (AKAPS) in glutamate receptor trafficking and long term synaptic depression. J Biol Chem. 2005;280:16962–16968. doi: 10.1074/jbc.M409693200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Milovanovic M, Zhao Y, Wolf ME. Acute and chronic dopamine receptor stimulation modulates AMPA receptor trafficking in nucleus accumbens neurons cocultured with prefrontal cortex neurons. J Neurosci. 2008;28:4216–4230. doi: 10.1523/JNEUROSCI.0258-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D, Karanian DA, Monteggia LM, Neve RL, Self DW. Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature. 2003;421:70–75. doi: 10.1038/nature01249. [DOI] [PubMed] [Google Scholar]
- Tannu N, Mash DC, Hemby SE. Cytosolic proteomic alterations in the nucleus accumbens of cocaine overdose victims. Mol Psychiatry. 2007;12:55–73. doi: 10.1038/sj.mp.4001914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannu NS, Hemby SE. Methods for proteomics in neuroscience. Prog Brain Res. 2006;158:41–82. doi: 10.1016/S0079-6123(06)58003-3. [DOI] [PubMed] [Google Scholar]
- Tannu NS, Howell LL, Hemby SE. Integrative proteomic analysis of the nucleus accumbens in rhesus monkeys following cocaine self-administration. Mol Psychiatry. 2010;15:185–203. doi: 10.1038/mp.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda S, Shen HW, Peters J, Cagle S, Kalivas PW. Cocaine increases actin cycling: effects in the reinstatement model of drug seeking. J Neurosci. 2006;26:1579–1587. doi: 10.1523/JNEUROSCI.4132-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uys JD, Grey AC, Wiggins A, Schwacke JH, Schey KL, Kalivas PW. Matrix-assisted laser desorption/ionization tissue profiling of secretoneurin in the nucleus accumbens shell from cocaine-sensitized rats. J Mass Spectrom. 2010;45:97–103. doi: 10.1002/jms.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, De Vries TJ. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Lubbers BR, Goriounova NA, Li KW, Van der Schors RC, Loos M, Riga D, Wiskerke J, Binnekade R, Stegeman M, Schoffelmeer AN, Mansvelder HD, Smit AB, De Vries TJ, Spijker S. Extracellular matrix plasticity and GABAergic inhibition of prefrontal cortex pyramidal cells facilitates relapse to heroin seeking. Neuropsychopharmacology. 2010;35:2120–2133. doi: 10.1038/npp.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Jensen ON, Mann M, Provance DW, Jr, Mercer JA, Kennedy MB. Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci. 2000;20:4069–4080. doi: 10.1523/JNEUROSCI.20-11-04069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J, Ma L. Chronic cocaine-induced H3 acetylation and transcriptional activation of CaMKIIalpha in the nucleus accumbens is critical for motivation for drug reinforcement. Neuropsychopharmacology. 2010;35:913–928. doi: 10.1038/npp.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]