

Abstract
We introduce a mass spectrometry-based method that provides residue-resolved quantitative information about protein phosphorylation. In this FLEXIQinase assay we combined our Full-Length Expressed Stable Isotope-labeled Protein for Quantification strategy (FLEXIQuant) with a traditional kinase assay to determine the mechanisms of multi-kinase substrate phosphorylation such as priming-dependent kinase activities. The assay monitors the decrease in signal intensity of the substrate peptides and the concomitant increase in the (n×80 Da)-shifted phosphorylated peptide. We analyzed the c-Jun N-terminal Kinase (JNK)-dependent glycogen synthase kinase 3β (GSK3β) activity on doublecortin (DCX) revealing mechanistic details about the role of phosphorylation cross-talk in GSK3β activity and permitting an advanced model for GSK3β-mediated signaling.
Keywords: in vitro, phosphorylation, quantitative proteomics, mass spectrometry, full length stable isotope labeled protein standards
INTRODUCTION
Protein phosphorylation is one of the most studied molecular mechanisms of the cell, yet the exact details of protein phosphorylation-based signaling mechanisms can be difficult to elucidate for a number of reasons. For example, ,substrates are often phosphorylated by a combination of kinases, however, kinase assays have yet to be coupled to a read-out that can localize a phosphorylation event to a specific amino acid residue of a substrate and determine the extent of the phosphorylation. Traditionally, once candidate kinase-substrate partners are inferred, biologists turn to an in vitro kinase assay for validation1, 2. However, information regarding the location and abundance of phosphorylation sites, and the dependence on cross-talk between sites is not readily determined. One could investigate specific sites by immuno-based detection methods, however, the use of antibodies to detect posttranslational modifications may be suboptimal due to epitope occlusion, non-quantitative binding behavior, and/or suboptimal sensitivity and specificity3, 4. To fully understand the mechanisms of kinase activity that regulate cells and their signaling cascades, we need to extract quantitative information on phosphorylation sites. Mass spectrometry (MS)-based phosphorylation analysis is currently the most efficient method to readily identify, localize and quantify numerous phosphorylation sites on a substrate in a single experiment5, 6, however it is challenging to extract mechanistic details about kinase activities from standard mass spectrometry workflows. We designed FLEXIQinase, a strategy that combines the power of soluble protein standards7 and mass spectrometry, and is easily implemented into the well-established in vitro kinase assay workflow1, 8. To test its versatility we analyzed serial kinase activities of the two kinases JNK and GSK3β on DCX and uncovered a surprisingly complex interaction of the kinases with their substrate.
RESULTS
FLEXIQinase for 1-kinase assays
Traditionally, kinase activity is evidenced by the addition of 32P-labeled phosphate onto the candidate substrate and one uses an inactive form (or dead kinase) as a control in a parallel experiment (Supplementary Fig. 1a). FLEXIQinase adopts a similar, dead versus active kinase strategy however the assay does not rely on the incorporation of 32P-labeled phosphate. Instead parallel reactions with the dead and active form of the kinase are performed on two distinct stable isotope labeled protein substrates, “light” and “heavy”, which are synthesized using light or heavy (for example, 12C6,14N2-Lys and 12C6,14N4-Arg vs. 13C6,15N2-Lys and 13C6,15N4-Arg) labeled amino acids, respectively (Supplementary Fig. 1a). Differential labeling of the control and phosphorylated substrate allows for simultaneous sample processing prior to analysis by mass spectrometry. This mass spectrometry-based quantification result in three basic FLEXIQinase readouts (Supplementary Fig. 1b): 1) no change in peak ratio of unphosphorylated peptides between samples, if the kinase does not phosphorylate a particular peptide 2) A decreased or absent heavy ion signal of the unphosphorylated peptide with respect to the light control indicates a phosphorylation event within the observed peptide. This decrease in a heavy signal intensity is accompanied by 3) a concomitant increase in the ion signal of the heavy (+n×80 Da)-shifted phosphorylated peptide (n = number of phosphorylation sites).
FLEXIQinase for serial kinase assays
Some kinases require their substrates be primed by another kinase; in this case we use a 2-kinase assay with which additional peak pair patterns emerge (Fig. 1a). First, unchanged peak ratios are no longer unique to unmodified peptides, but can also represent phospho-peptides resulting from priming kinase activity only (Fig. 1a). Second, phosphorylation by the second kinase can manifest itself by a decrease of the heavy peptide signal, an increase of a heavy phospho-peptide signal, and/or the appearance of a heavy phospho-peptide peak without a light complement (Fig. 1b).
Figure 1.

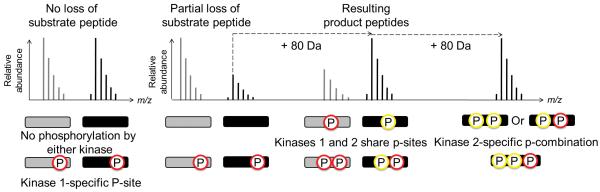
The 2-kinase assay. (a) Comparison between the traditional radioactivity and the FLEXIQinase workflow. Grey and black circles indicate light and heavy labeled proteins, respectively. Grey and black bands at the bottom indicate bands after gel electrophoresis. (b) Peak chase profiling. Grey and black bars (and their corresponding spectra above), indicate light and heavy peptides, respectively. Phospho-sites are colored according to their respective kinases, represented by the FLEXIQinase workflow in (a). y-axis shows relative peak abundance.
When broken down further these three scenarios reveal mechanistic details: a (phospho-)peptide peak pair with no change in signal intensity indicates that the peptide is not phosphorylated by the second kinase (Fig. 1b). A decrease in the heavy phospho-peptide peak signal intensity indicates an additional kinase 2-dependent phosphorylation on another site within the same peptide (Fig. 1b) which is associated with a concomitant increase or appearance of an n×80 Da heavy phospho-peptide: the increased intensity of the heavy phospho-peptide signal indicates Kinase 2-dependent additional phosphorylation on the same phosphorylation site that is shared with kinase 1 (Fig. 1b). The appearance of a heavy-only peak is the signature for a phospho-peptide with at least one kinase 2-specific phosphorylation site (Fig. 1b). These simple changes in the peptide peak ratios can therefore provide relevant details about kinase activities and specificities.
MS1 peak chasing and insights into phosphorylation cross-talk
The peak ratio scenarios outlined above show that phospho-peptides can in turn serve as substrates for further phosphorylation events. This results in the observation of various phosphorylated -species for the same peptide, differing in retention time and in mass (+n×80 Da) depending on the possible phosphorylation sites. We call the process of identifying substrate and product (phospho)-peak pairs (MS1) ‘peak chasing’. The peak chase profile, that is the extent of signal loss or gain, can vary among the peptides, indicating that a kinase can have different phosphorylation profiles on a single protein substrate.
To investigate the effects of prior priming phosphorylation events on subsequent kinase activities, FLEXIQinase experiments can be combined with site-directed mutagenesis (Fig. 2). Assume three phosphorylation sites (P-1, P-2 and P-3) across two peptides: P-1, P-2 on Peptide A and P-3 on Peptide B. The second phosphorylation site (P-2) is observed only post kinase 2 treatment (Fig. 2a). The novel phospho-site occurs on peptide A, while nothing changes on peptide B (control peptide ratio). Two kinase activity models can explain this observation (Fig. 2b): 1) kinase 2 phosphorylated P-2 without influence by primed P-1 or P-3; or 2) kinase 2 activity was dependent on priming on P-1, or P-3. Site-directed mutagenesis of P-1 or P-3 in combination with FLEXIQinase analysis will enable us to differentiate between the models (Fig. 2b). If a mutation does not perturb the substrate peptide ratios when compared to the wildtype substrate (Fig. 2b), it can be concluded that this site has no influence on kinase 2 activity. If however kinase 2-dependent phosphorylation on P-2 is not observed post mutation, that is, the substrate peptide ratio is that of the control ratio (Fig. 2b), it can be concluded that this is the bona fide priming site (Fig. 2b).
Figure 2.


A theoretical approach to interpreting a serial kinase mechanism. (a) Upper panels – the resulting phospho-profile imprinted by the serial activity of Kinases 1 and 2. The three possible phosphorylation sites are labeled, P-site 1, 2 and 3. Lower panel – the substrate and product peak profiles that result from the scenario above. (b) Site-directed mutagenesis confirms the serial kinase mechanism. Upper panel – two theoretical phospho-profiles that could result due to the mutation in the candidate priming site, P-site 1. Lower panel – the substrate and product profiles that would arise from each possible phospho-profile. Red and yellow phospho-sites are phosphorylated by Kinase 1 or 2, respectively. The x indicates that the light peptide peak is not observed.
JNK-dependent phosphorylation of DCX by GSK3β is localized to the C-terminal end
We quantified peptide peak pairs corresponding to 80-90% of the DCX protein sequence and identified over 20 phosphorylation sites (Supplementary Table 1). Most of these sites were localized to the C-terminal domain spanning amino acids 299-340 (Fig. 3a). The N-terminal half of DCX has unperturbed mixing ratios indicating the absence of GSK3β activity. The phosphorylated peptides covering the amino acid range 299-340 display various MS1 peak pair ratios indicating that JNK-induced GSK3β activity is specific to the C terminus (Fig. 3a).
Figure 3.



FLEXIQinase analysis of JNK/GSK3β activity on DCX. (a) A plot of the light/heavy ratios of WT DCX tryptic peptides 1-22 (18-22 are labeled and amino acids within 19 to 21 are indicated) – how do the three tryptic peptides correlated to Peptide A and B? what is the significance of peptides 18 and 22; why are they labeled?. Unmodified peptides plots are black, singly phosphorylated peptides are blue, doubly phosphorylated peptides are red, and triply phosphorylated peptides are green. (b, c) Peak chase profile for peptides A and B for WT (b) and mutant (c) DCX. The phosphorylation status is profiled on top where a dash indicates an unmodified peptide, and the number of (P’s) indicate the number of phosphorylation sites on the peptides below. Representative light-to-heavy peak profiles [label the axis, indicating real numbers for m/z since this is real data not schemata] are shown for the phosphorylation sites indicated below them. Green and blue boxes include all possible phospho-acceptor sites for peptides B and A, respectively, a simplified view of the tryptic peptides in (a) which peptides in (a) do these correspond to, please give the number. Dashed lines connect heavy substrate peaks with their expected heavy product peaks, until an end product is observed. A product peak that should be present but not observed with MS is indicated by, (not observed). JNK-dependent sites are red, GSK3β-sites are yellow, and black indicates either JNK or GSK3β-dependent phosphorylation.
JNK and GSK3β share a number of phosphorylation sites
The DCX sequence corresponding to amino acid range 299-340 produced two tryptic peptides each with unique peak chase profiles (Fig. 3b). In most cases, MS2 and/or MS3 spectra provided precise localization of the phosphorylation sites (Supplementary Table 1). The peak chase profile for Peptide B shows GSK3β phosphorylates this region and that activity is dependent on JNK priming as indicated by the unchanged peak pair ratio for the unmodified peptide (Supplementary Fig. 2a). The highest order phosphorylation product observed was a triply phosphorylated peptide, and it has a higher heavy peak signal with respect to the light counterpart. This ratio indicates that for DCX, amino acids S327, T331 and S334 are target sites for both JNK and GSK3β. Thus, GSK3β further phosphorylates these sites beyond the initial priming by JNK. However, phosphorylation by GSK3β is abrogated once particular doubly or triply phosphorylated species are generated (Fig. 3b).
Identification of novel GSK3β target sites
The peak chase profile for the peptide cohort corresponding to amino acids 299-323 (Peptide A) of DCX is distinct from that of peptide B (Fig. 3c), as GSK3β activity is observed irrespective of the priming status of peptide A (Fig. 3b). The intensity of the heavy unmodified peptide signal is low suggesting that priming within this amino acid range is not a requirement for GSK3β activity. We do not observe the heavy peak until a triply phosphorylated peptide is obtained; the relative peak height of the corresponding light peptide is less than 1 % indicating that GSK3β activity heavily favors the formation of a triply phosphorylated species. We determined the triply phosphorylated peptide to be (pS316)(pS317)QLS(pT321) corresponding to unexpected GSK3β-specific sites. The ability of GSK3β to phosphorylate the non-primed peptide A sequence is most likely due to the priming of amino acids downstream within peptide B, however alternative mechanisms may also be active such as direct binding of GSK3β within peptide A (Supplementary Fig. 2a), irrespective of priming status. Site-directed mutagenesis experiments will assist in differentiating among the models.
GSK3β specific activity is dependent on downstream priming cross-talk
To test whether GSK3β-specific activity on the SSxxxT motif is dependent on remote effects of the downstream (Peptide B) priming status, we mutated the proposed priming region by substituting one (T331, Supplementary Fig.3) or two (S327 and T331, Fig. 3c) assumed priming sites with alanine. If the GSK3β-specific activity on the Peptide A motif, SSxxxT, is dependent on the overall extent of phosphorylation of the proposed priming region on Peptide B, we would expect mutation-dependent attenuated activity by GSK3β on this region. The results of the double mutant show two major effects: the highest order phospho-peptide that we could readily capture for Peptide B was a doubly phosphorylated form rather than the triply phosphorylated form observed in wildtype DCX (Fig. 3b). In contrast, GSK3β activity on the sequence, SSxxxT of Peptide A, was attenuated as a function of these mutations (Fig. 3c and Supplementary Fig. 3), confirming that GSK3β-activity on Peptide A is dependent on these remote priming events (Supplementary Fig. 2b). The effect observed for the single mutant was less than that of the double mutant, indicating that GSK3β-activity can be modulated by the degree of remote, downstream priming (Supplementary Fig. 3).
pT321 phosphorylation status can drive the priming response
Based on the data above, we hypothesized that GSK3β-specific activity is influenced by more than one mechanism. This notion is underscored by the observation that phospho-peptides of peptide A harboring a JNK-primed pT321, a previously described in vivo JNK phospho-site11, were further phosphorylated by GSK3β, irrespective of the mutation status downstream (Fig. 3c, Supplementary Fig. 3). These data reveal that T321 is part of the priming hot spot which promotes GSK3β activity to the upstream SSxxxT motif. Interestingly, the relative abundance of pT321 relative to the other singly phosphorylated peptide forms provided by JNK priming is rather low (⪡ 10 % of the total light population, Supplementary Fig. 4). Upon removal of downstream priming sites, GSK3β-dependent activity relies more on the low abundant pT321, which might explain why we were not able to observe the final (pS316)(pS317)QLS(pT321) form in the DCX mutants.
The mutant analyses suggest that even for wildtype DCX, a primed T321 is sufficient to trigger GSK3β-specific activity, giving rise to an “immediate priming” response (Fig. 4). However, the relatively low abundance of the pT321 observed in our quantitative analysis of wildtype DCX (Supplementary Fig. 4), also indicates that GSK3β–specific activity is most likely driven by binding to and subsequent phosphorylation of the downstream amino acids, performing a type of “secondary priming” in response to initial JNK priming (Fig. 4). If these Ser/Thr sites are removed, we are less likely to observe the GSK3β-specific phospho-peptide, indicating that secondary priming by GSK3β accounts for most of its (pS316)(pS317)QLS(pT321) in the in vitro system.
Figure 4.

Model for JNK-dependent GSK3β activity on DCX. Position of phosphorylation sites (P) on the two peptide (blue and green) are indicated. Red circles represent JNK sites, yellow circles GSK3β sites. The yellow arrow depicts the net movement of GSKβ.
DISCUSSION
Elucidating the mechanistic details of priming-dependent kinases is crucial to understanding the basic biology of these kinases and to design inhibitors. FLEXIQinase allows the qualitative and quantitative analysis of a single kinase target with multiple acceptor sites, as are often observed in high throughput mass spectrometry-based kinase screens5, 6. FLEXIQinase can also serve as a follow up study on kinase-substrate pairs identified in kinase screens. Once one has determined the phosphorylation dynamics in the controlled environment of the in vitro system, specific phospho-peptides of interest can be monitored and absolute quantification can be determined in vivo17. Moreover, FLEXIQinase can be readily adapted to more complex in vivo experiments. Finally, we anticipate that FLEXIQinase will serve as a basis for even more sophisticated applications of the peak chasing strategy while remaining a simple and practical approach. By combining multiplexed labeling and FLEXIQinase, for example, detailed kinetic information about the phosphorylation site dynamics can be obtained in temporal experiments. The general approach is not limited to a single modification such as phosphorylation, but can also be extended to several other posttranslational modifications and combinations thereof.
ONLINE METHODS
In vitro kinase assay
GST-tagged DCX (GST-DCX)10_ENREF_10 was sub-cloned into the CellFree Systems (CFS) pEU-E0-MCS vector, in vitro synthesized, as either light or heavy labeled (13C6,15N2-Lys and 13C6,15N4-Arg), using the CFS wheat germ system (WEPRO2240), and purified and retained on glutathione sepharose beads (GE Healthcare). FLAG-JNK1 was expressed in 293T cells which were UV irradiated for 30 minutes (to activate JNK1) prior to its purification: agarose beads were conjugated with the FLAG antibody (Sigma) and mixed with 293T lysate. The beads were washed in high salt (50 mM Tris pH 7.5, 2 mM EDTA, 1 % Triton X-100, 500 mM NaCl) and JNK1 was eluted with the 3×FLAG peptide (Sigma). The JNK priming reaction was carried out under the following conditions: 20 mM HEPES pH 7.4, 20 mM MgCl2, 1 mM dithiothreitol (DTT), 0.1 mM sodium orthovanadate, 100 mM β-glycerolphosphate, and 10 mM ATP, for three hours at 30 °C. JNK was removed from bound DCX with high salt washes and DCX was subsequently subjected to GSK3β (New England Biolabs). The GSK3β reaction was carried out under the following conditions: 20 mM Tris-HCl, 10 mM MgCl2, 5 mM DTT, 200 μM ATP for 30 minutes at 30 °C. Approximately 500 ng of substrate was incubated with 250 U of the GSK3β kinase. A typical experiment for the differential labeling experiment (Fig. 2a) would entail, for example, JNK1 priming of the light labeled DCX, whereas the heavy labeled DCX would be subjected to JNK1 followed by GSK3β treatment, or vice versa. Previous work by Bilimoria and colleagues10 confirm that kinase-dead JNK eliminates GSK3β activity, thus phosphorylation events are specific to JNK-GSK3β.
Mass spectrometry
Kinase-treated GST-DCX was purified using glutathione sepharose beads by heating to 100°C in Laemmli buffer supplemented with β-mercaptoethanol. Light DCX sample (treated with JNK1 only) was combined with heavy DCX (JNK1 and GSK3β) right before SDS-PAGE. A molecular weight (MW) window corresponding to GST-DCX and potential phospho-shifted MW species was excised and in-gel trypsinized using standard protocols. Samples were analyzed with an LTQ-Orbitrap Classic mass spectrometer (ThermoFisher Scientific, resolution setting: 30.000) linked to a micro-autosampler and a nanoflow HPLC pump (both: Eksigent). The reversed phase columns were packed in-house by using Magic C18 particles (3 μm, 200 Å; Michrom Bioresource) and PicoTip Emitters (New Objective, Woburn, MA). The peptides were eluted with a 30 minute linear gradient and data acquired in a data dependent fashion, fragmenting the 6 most abundant peptide species. Buffer A was 0.2 % formic acid, buffer B was acetonitrile/0.2 % formic acid, and loading buffer was 5 % formic acid/5 % acetonitrile.
Spectral Analysis
The low resolution ion trap MS/MS data were queried against the IPI Human 3.69 database or an in-house Custom database (for all recombinant DCX sequences) using MASCOT (Matrix Science). For MS1 quantification, the area under the appropriate selected ion chromatogram for each light and heavy monoisotopic peak was determined using Xcalibur (Thermo Scientific).
Supplementary Material
Acknowledgements
We thank J. Gunawardena for his thorough reading of our manuscript and for his advice. This work is funded in part by NIH grants NS066973 (J.S.), GM094844, GM096319 and GM081578 (H.S.), NS041021 and NS047188 (A.B.), and German Academic Exchange Service (D.W.).
Footnotes
Author Contributions
S.S. contributed the qualitative and quantitative MS experiments, and the manuscript; D.W. contributed qualitative MS analysis; P.B contributed the in vitro kinase assays and revisions to the manuscript; A.B., J.S. and H.S. contributed support, experimental design and revisions to the manuscript.
Dr. Singh’s current affiliation is Division of Cardiovascular Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, USA. Dr. Winter’s current affiliation is Institute of Biochemistry and Molecular Biology, University of Bonn, Bonn, Germany
References
- 1.Hastie CJ, McLauchlan HJ, Cohen P. Assay of protein kinases using radiolabeled ATP: a protocol. Nat Protoc. 2006;1:968–971. doi: 10.1038/nprot.2006.149. [DOI] [PubMed] [Google Scholar]
- 2.Mok J, Im H, Snyder M. Global identification of protein kinase substrates by protein microarray analysis. Nat Protoc. 2009;4:1820–1827. doi: 10.1038/nprot.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia BA, et al. Chemical derivatization of histones for facilitated analysis by mass spectrometry. Nat Protoc. 2007;2:933–938. doi: 10.1038/nprot.2007.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabakaran S, et al. Comparative analysis of Erk phosphorylation suggests a mixed strategy for measuring phospho-form distributions. Mol Syst Biol. 2011;7:482. doi: 10.1038/msb.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kubota K, et al. Sensitive multiplexed analysis of kinase activities and activity-based kinase identification. Nat Biotechnol. 2009;27:933–940. doi: 10.1038/nbt.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu Y, et al. A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc Natl Acad Sci U S A. 2009;106:11606–11611. doi: 10.1073/pnas.0905165106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh S, Springer M, Steen J, Kirschner MW, Steen H. FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J Proteome Res. 2009;8:2201–2210. doi: 10.1021/pr800654s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickson C. Protein techniques: immunoprecipitation, in vitro kinase assays, and Western blotting. Methods Mol Biol. 2008;461:735–744. doi: 10.1007/978-1-60327-483-8_53. [DOI] [PubMed] [Google Scholar]
- 9.Reiner O, Coquelle FM. Missense mutations resulting in type 1 lissencephaly. Cell Mol Life Sci. 2005;62:425–434. doi: 10.1007/s00018-004-4344-0. [DOI] [PubMed] [Google Scholar]
- 10.Bilimoria PM, et al. A JIP3-regulated GSK3beta/DCX signaling pathway restricts axon branching. J Neurosci. 2010;30:16766–16776. doi: 10.1523/JNEUROSCI.1362-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gdalyahu A, et al. DCX, a new mediator of the JNK pathway. EMBO J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forde JE, Dale TC. Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci. 2007;64:1930–1944. doi: 10.1007/s00018-007-7045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 14.Olsen JV, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 15.Steen H, Jebanathirajah JA, Springer M, Kirschner MW. Stable isotope-free relative and absolute quantitation of protein phosphorylation stoichiometry by MS. Proc Natl Acad Sci U S A. 2005;102:3948–3953. doi: 10.1073/pnas.0409536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 17.Cutillas PR, et al. Ultrasensitive and absolute quantification of the phosphoinositide 3-kinase/Akt signal transduction pathway by mass spectrometry. Proc Natl Acad Sci U S A. 2006;103:8959–8964. doi: 10.1073/pnas.0602101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hur EM, Zhou FQ. GSK3 signalling in neural development. Nat Rev Neurosci. 2010;11:539–551. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.