

Abstract
Polymorphonuclear neutrophils (PMNs) produce and release copious amounts of reactive oxygen species (ROS) which target potential bacterial invaders but also contribute to the inflammation-associated organ injuries seen in sepsis. Calprotectin is an immune regulatory protein complex made of S100A8 and S100A9 that inhibits the oxidative metabolism of PMNs in vitro, an effect that can be potentiated by the controlled activation of the protease activated receptor-2 (PAR2). The aim of this study was to test the use of a dual strategy of calprotectin and PAR2 administration to mitigate the deleterious inflammation seen in sepsis. We hypothesized that exogenous calprotectin would protect against the injuries produced by lipopolysaccharides (LPS)-induced endotoxemia and that the controlled activation of PAR2 would potentiate this beneficial effect.
Exogenous S100A8 and/or a PAR2 activating peptide (PAR2 AP) were administered in a mouse model of LPS induced endotoxemia. The survival rates as well as markers of inflammation and oxidative damage were measured in the lungs, kidneys, and livers of endotoxemic mice.
Mice treated with S100A8 following LPS had less PMN infiltration and less severe histological changes in their lungs, kidneys, and livers. A significantly lower score of oxidative damage in the livers and lungs of S100A8/LPS treated mice was also noted when compared to mice treated with LPS alone. This protective and anti-inflammatory effect of S100A8 was potentiated by the controlled activation of PAR2. Finally, in further support to our hypothesis, the survival rate was almost doubled from 33% to 65% and 63% in mice treated by, respectively, S100A8 and PAR2 AP, whereas 85% of the mice treated with both PAR2 AP and S100A8 survived, a statistically significant higher rate.
These results support an anti-inflammatory, anti-oxidative, and protective effect of S100A8 in sepsis, and warrant further studies on the role of PAR2.
Keywords: Sepsis, Endotoxemia, Calprotectin, Inflammation, Oxidation, Neutrophils
1. Introduction
In the United States, approximately 750,000 cases of sepsis are diagnosed yearly. Most are caused by Gram-negative bacterial infection. This results in more than 175,000 deaths (Angus et al., 2001; Martin et al., 2003). Initially, septic patients display exaggerated inflammation which involves the widespread activation and margination of leukocytes from the circulation into visceral organs. The first immune competent cells activated during sepsis are PMNs; the lungs are the most intensely targeted organs (Kollef and Schuster, 1995). The overwhelming margination of PMNs results in their sequestration and may partially explain the switch from a hyper- to hypo-inflammatory immunological state (Sato et al, 1998; van Eeden et al., 1997).
When activated, PMNs copiously produce and release reactive oxygen species (ROS), which target potential bacterial invaders but cause further tissue injury. Thus, the oxidative metabolism of PMNs is thought to contribute to the pathophysiology of sepsis (Kovach and Standiford, 2012). Oxidative stress, an imbalance between the production of ROS and the activity of scavenger systems, is linked to a poorer prognosis in septic patients (Cowley et al., 1996; Guerreiro et al., 2010; Ritter et al., 2003). Interventions targeting redox mechanisms have been shown to reduce organ injury and death in animal models of sepsis (Ritter et al., 2004; Biswal and Remick, 2007; Albuszies and Bruckner, 2003) and experimental neutropenia reduces tissue injury in models of endotoxemia (Molnar et al., 1997; Sato et al., 1993).
Calprotectin is a heterocomplex formed by S100A8 and S100A9, two immune-regulatory calcium-binding proteins which together represent 45% of human PMN cytosolic proteins (by weight) (Edgeworth et al., 1991). Circulating levels of calprotectin in healthy subjects range between 0.1 and 0.6 mg/L, and may increase 20-fold in inflammatory conditions (Strasser et al., 1997; Schulze zur Wiesch et al., 2004) including sepsis (Rau et al., 2010). Elevated calprotectin levels in the circulation have been suggested as a potential diagnostic marker of sepsis (Terrin et al., 2011). Calprotectin is also present in cerebrospinal fluid, saliva, intestinal mucosa (Bjerke et al., 1993), and in fecal matter in association with intestinal inflammation (van den Bergh et al., 2003). Chronic and extreme elevations of serum concentrations of calprotectin (1.4–6.5 g/L) are prominent features of a familial syndrome which includes recurrent infections (Sampson et al., 2002). In models of endotoxemia, calprotectin exerts conflicting activities. Exogenously introduced calprotectin has resulted in either a significant increase in mortality rate (Vogl et al., 2007) or a protective effect on hepatic injuries and functions being reported (Ikemoto et al., 2007; Namura et al., 2010). This may be due to differences in experimental conditions or to the dual, temporal, and contextually dependent nature of the role of calprotectin in inflammation.
Calprotectin modulates a number of critical immune elements, including the receptor for advanced glycosylated end products (RAGE) (Boyd et al., 2008), the B7-costimulatory molecule (Shimizu et al., 2012), and the Toll-like receptor-4 (TLR4) (Vogl et al., 2007), a key-signaling molecule in Gram-negative sepsis which is activated by LPS. We have previously shown in vitro that calprotectin inhibits the oxidative metabolism of LPS-activated PMNs (Sroussi et al., 2010). Furthermore, we have demonstrated that the protease-activated receptor 2 (PAR2) has a key function in the regulation and modulation of this anti-oxidative effect (Sroussi et al., 2012).
PAR2 is a member of a family of seven-transmembrane G-protein-coupled receptors comprising 4 receptors and designated as PAR1 to PAR4 (Macfarlane et al., 2001; Ossovskaya and Bunnett, 2004). The activation of PARs is achieved by the cleavage of the N-terminus of the receptor exodomain. This can be accomplished by PMN-derived serine proteases and by enzymes of the coagulation cascade known to be activated in sepsis (Camerer et al., 2000; Borensztajn et al., 2008). The dysregulation of PMN-derived proteases known to activate or inhibit PAR2 produces diseases of dysregulated inflammation (i.e. Wegener's granulomatosis) (Jiang et al., 2010; Kallenberg, 2010), supporting a critical role for PAR2 in immunity and health.
In sepsis, PAR2 plays two roles: it potentiates the initial inflammatory response to LPS (Lindner et al., 2000), and it turns on compensatory anti-inflammatory mechanisms which limit the scope of inflammation (Kaneider et al., 2007). As a result, inhibiting PAR2 with a blocking peptide ameliorates LPS-induced nephrotoxicity (Jesmin et al., 2009). Conversely, the controlled activation of PAR2 results in reduced LPS-induced lung neutrophilia (Moffatt et al., 2002), in fewer PMNs recovered from bronchoalveolar lavage fluid in mice challenged with intranasal LPS (Peters et al., 2010), in improved bronchorelaxation in LPS-treated rats (Morello et al., 2005), and in protection of cardiac functions following ischemia/reperfusion injury (Zhong and Wang, 2009) (an oxidative stress and PMN associated phenomenon; Kaminski et al., 2002). Thus, similarly to that of calprotectin, the role of PAR2 in sepsis maybe both protective and harmful.
In this study, we evaluated the use of a calprotectin and PAR2 dual strategy to mitigate the deleterious inflammation seen in sepsis. We hypothesized that exogenous administration of a component of calprotectin (S100A8) would somewhat protect against organ injuries caused by LPS-induced endotoxemia, and that a combination of both S100A8 and a PAR2 activating peptide (AP) would exhibit an enhanced protective effect.
2. Materials and methods
2.1. Animals
All animal experiments were performed in accordance with protocols approved by the University of Illinois at Chicago Animal Care and Use Committee. Female CD1 mice were purchased at the age of 6 weeks from Charles River Laboratories (Wilmington, MA) and maintained for 1 week at constant room temperature with a 12 h light/dark cycle (lights on at 0600) in the accredited animal facility at the University of Illinois at Chicago The mice were housed in groups of 5 per cage and given unrestricted access to food and water.
2.2. Experimental design
Experimental sepsis in female CD1 mice was induced by an intraperitoneal (IP) administration of a sub-lethal dose of LPS (7.5 mg/kg). Thirty minutes later, the following were administered IP: S100A8 (100 μg/mouse), PAR2 activating peptide (PAR2 AP, 500 μg/mouse); both S100A8 (100 μg/mouse) and PAR2 activating peptide (PAR2 AP, 500 μg/mouse); and saline to a control group. Two hours later, the mice were sacrificed and organs harvested for histology studies, myeloperoxidase (MPO) measurements, and thiobarbituric acid reactive species (TBARS) assays.
A similar protocol was repeated on a second group of mice that were challenged with a lethal IP dose (50 mg/kg) of LPS. Four hours later, the following were administered IP: S100A8 (100 μg/mouse), PAR2 activating peptide (PAR2 AP, 500 μg/mouse); both S100A8 (100 μg/mouse) and PAR2 activating peptide (PAR2 AP, 500 μg/mouse); and saline solution to a control group of mice.
2.3. Expression and purification of recombinant S100A8 and S100A9 proteins
Recombinant S100A8 was produced and purified based on standard methods and as previously described (Tugizov et al., 2005). Briefly, S100A8 was cloned in a pGEX-2T GST vector (Amersham, Piscataway, NJ). The protein was expressed in Escherichia coli as a GST fusion protein. The GST tag was cleaved during the purification process. Protein concentration was assessed through a Bradford protein assay.
2.4. Reagents
Lipopolysaccharide (LPS) from E. coli 0111:B4 (LOT# 030M114) was purchased from Sigma (St Louis, MO). PAR2 AP (SLIGKV-NH2) (Cicala et al., 1999) which activates PAR2 was synthesized and purified >95% by peptide 2.0 (Chantilly, VA). Quantichrom™ TBARS Assay kit (DTBA-100) was purchased from BioAssay Systems (Hay-ward, CA). Mouse monoclonal antibody to 8-hydroxyguanosine [15A3] and a rabbit polyclonal antibody to nitro tyrosine (Cat#AB5411) were purchased from Abcam (Cambridge, MA) and Millipore (Billerica, MA), respectively. Histostain-plus Bulk Kit and lipid DAB substrate kit were both purchased from Invitrogen (Grand Island, NY). Hexadecyltrimethylammonium bromide, o-dianisidine dihydrochloride and hydrogen peroxide solution 30% (w/w) were purchased from Sigma (St Louis, MO). 0.5 M EDTA (pH 8.0) was purchased from Cambrex (Charles City, IA).
2.5. Myeloperoxidase (MPO) activity assay
Lungs were homogenized in 1 mL of phosphate-buffered saline, pH 6, with 5% hexadecyltrimethylammonium bromide and 5 mM EDTA. The homogenates were centrifuged at 40,000 × g for 20 min, the supernatant was transferred to a new tube, and the re-suspended palette was stored in −80 °C overnight, i.e. run a freeze-thaw cycle. The samples were homogenized and centrifuged a second time. The supernatant from the two homogenizations was then collected and mixed with assay buffer (0.2 mg/mL o-dianisidine hydrochloride and 0.0005% H2O2). Absorbance change was measured at 460 nm for 3 min, and MPO activity was calculated as the change in absorbance over time. Neutrophil sequestration was quantified as MPO activity normalized by the final wet lung weight.
2.6. Thiobarbituric acid reactive species (TBARS) assay
As an index of the lipid peroxidation, we used the formation of TBARS during an acid-heating reaction (Draper and Hadley, 1990). Briefly, livers of 50 mg were homogenized in 500 μl ice-cold PBS containing a cocktail of protease inhibitors. An aliquot of 100 μl was mixed with 200 μl trichloroacetic acid 10% and incubated for 15 min on ice before being centrifuged 5 min at 14,000 rpm in an Eppendorf Centrifuge. Next, 200 μl of supernatant was mixed with an equal volume of thiobarbituric acid reagent. This system was heated in a boiling water bath for 1 h and the TBARS were determined by the absorbance at 535 nm. Results were expressed as μM TBARS of protein.
2.7. Immunohistochemistry and H&E staining
Saline- and formalin-perfused livers, lungs, and kidneys were paraffin-embedded and sectioned at 5 μm. Two types of staining were used: hematoxylin and eosin (H&E), or immuno-staining using primary antibodies, followed by biotinylated secondary antibody, and finally with streptavidin-peroxidase (HRP). Peroxidase activity was detected using a DAB kit. All images shown were visualized with a Nikon Plan Apo 20× 0.75 objective lens. A mouse monoclonal antibody to 8-hydroxyguanosine and a rabbit polyclonal antibody to nitro tyrosine were used to quantify, respectively, DNA and protein oxidation. Quantification of immune positivity was conducted by counting stained cells in 20 randomly chosen high power field per slide.
2.8. Terminal deoxynucleotidyl transferase (TUNEL) staining
TUNEL staining of liver tissue was accomplished with the help of the In Situ Cell Death Detection Kit, Fluorescein from Roche Applied Sciences (Indianapolis, IN) according to the manufacturer instructions. The slides were then mounted with ProLong® Gold Antifade Reagent with 4′,6-diamidino-2-phenylindole (DAPI) from Invitrogen (Grand Island, NY). TUNEL positive cells were then counted in 10 high power field (HPF) per slide.
2.9. Data analysis
Statistical significance was determined by two sample t-tests (SPSS Inc., Chicago, IL) and by analysis of variance. P-values <0.05 denoted the presence of a statistically significant difference. Comparison of survival curves was conducted with a Gehan–Breslow–Wilcoxon test.
3. Results
3.1. S100A8 attenuates LPS-associated tissue damage
The effects of LPS alone or followed by exogenous recombinant S100A8 were examined in histological sections of the livers, lungs, and kidneys and compared to saline-treated controls. The representative histopathology of the three organs is presented in Fig. 1. The lungs of LPS-treated mice exhibited edema and inflammation. The lungs in mice treated with LPS/S100A8 exhibited milder damage. The livers of LPS-treated mice showed widespread hepatocellular apoptosis with pyknotic nuclei and dilated sinusoids, whereas the livers of LPS/S100A8-treated animals showed milder dilation of sinusoids, and scattered apoptosis. Histological differences between saline, LPS, and LPS/S100A8 treated kidneys were not as marked as those in lung and liver. However, tubule dilation was most apparent in the LPS-treated group, and this dilation was attenuated in the LPS/S100A8-treated group (Fig. 1).
Fig. 1.
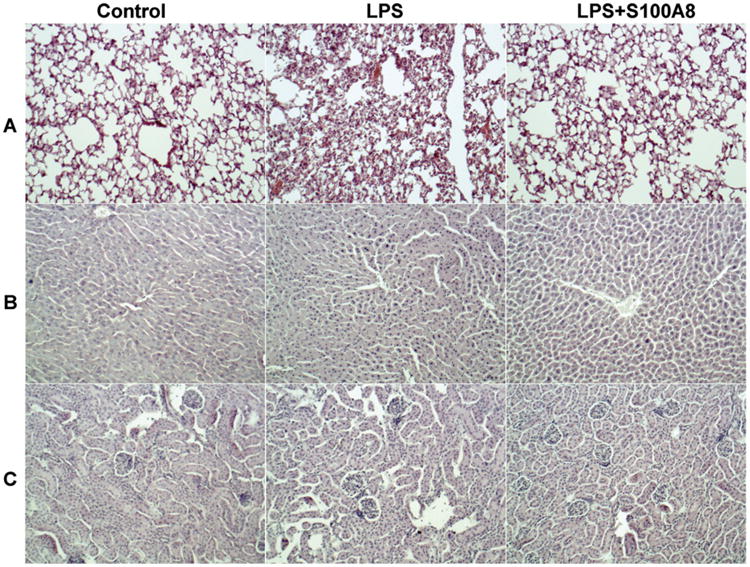
S100A8 mitigates the tissue damage associated with LPS induced endotoxemia. S100A8 (100 μg in 200 μl volume per mouse) or saline was given IP to mice 0.5 h after a 7.5 mg/kg LPS challenge (IP). 2 h later, the mice were sacrificed and organs were harvested. Representative histological sections stained with H&E are presented. The histology demonstrated PMNs infiltration and disruption of structures in lungs (A), livers (B), and kidneys (C) in LPS treated mice when compared to saline treated control mice. The effects of LPS were reduced by the administration of S100A8. Magnification: 200×.
3.2. Lung neutrophilia is attenuated by S100A8
Quantification of lung neutrophilia indicated that LPS increased the mean number of PMNs almost 4-folds from 84.2 ± 17.4 cells (mean ± SD) per high power field (HPF) in the control group injected with saline, to 341.6 ± 40.6 cells/HPF in the LPS group. The LPS treated mice that also received S100A8 had a significantly lower (P < 0.05) number of PMNs (168.2 ± 24.8 cells/HPF) when compared to mice treated with LPS (Fig. 2).
Fig. 2.

Quantification of PMNs in lungs. PMNs were identified morphologically in H&E sections. Value is shown as number (mean ± SEM) per high power field (HPF). The results presented are the product of the aggregation of three independent experiments conducted each with 5 mice per group. The data indicated a statically significant difference when comparing the LPS and LPS/S100A8 groups (*P < 0.01) and the LPS to the control saline treated group (#P < 0.01).
3.3. The oxidative damage to the liver caused by LPS is mitigated by S100A8
We next quantified the oxidative damage by measuring the oxidation of DNA and proteins in the liver. This was done by quantifying cells stained with antibodies for 8-hydroxydeoxyguanosine (8-OHdG) and nitro tyrosine. LPS-treated mice demonstrated a significantly higher number (34.3 ± 7.2 cells/HPF) of 8-OHdG positive cells when compare to control saline-treated mice (4.5 ± 2.3 cells/HPF) (P < 0.01). Mice treated with LPS and S100A8 exhibited a smaller number of 8-OHdG positive cells (13.7 ± 5.6 cells HPF) when compared with the LPS group (P < 0.01) (Fig. 3A and B). Similarly livers of mice treated with LPS had a higher number of cells positive for nitro tyrosine (21.9 ± 4.3 cells/HPF) when compared to controls (1.8 ± 1.0 cells/HPF). The administration of S100A8 after LPS significantly reduced the number of nitro tyrosine positive cells to 10.8 ± 3.6 cells/HPF (P < 0.01) (Fig. 3C and D).
Fig. 3.

S100A8 significantly reduced oxidative damage in the liver of mice challenged with LPS. (A and C) Immunohistochemistry demonstrated more 8-hydroxydeoxyguanasine (8-OHdG) and nitro tyrosine positive cells in a liver section of LPS treated mice compared to control saline treated mice, an effect mitigated by S100A8. (B and D) Quantification of 8-OHdG and nitro tyrosine positive cells per high power field. Results are mean ± SEM of three independent experiments conducted with 5 mice in each group. The data indicated a statically significant difference when comparing the LPS and LPS/S100A8 groups (*P < 0.01) and the LPS to the control saline treated group (#P < 0.01). Magnification: 200×.
3.4. LPS induces apoptosis in liver cells, an effect partially lessened by S100A8
As an additional marker of hepatic injury resulting from endotoxemia, we quantified DNA fragmentation in histological slides prepared from control, LPS and LPS/S100A8 treated mice. This quantification was achieved with TUNEL staining. The data indicated that LPS induced a more than 5-fold increase in the number of TUNEL stained cells when compared to control from 0.8 ± 0.8 cells/HPF in control to 4.8 ± 1.7 cells/HPF in LPS Fig. 4). The administration of S100A8 after LPS reduced this effect with an average 2.9 ± 1.1 cells/HPF, a number significantly lower than with LPS alone (P < 0.05).
Fig. 4.

S100A8 significantly reduced TUNEL staining in the liver of mice challenged with LPS. Representative fluorescent microscopy (magnification of 200×) in a liver section stained with TUNEL and DAPI and quantification of the number TUNEL stained cells per high power field (HPF).
3.5. Lung MPO activity is reduced by S100A8, an effect potentiated by PAR2 AP
MPO activity, a marker for PMN infiltration and oxidative activity, was next measured in lung tissue. We tested the effects of PAR2 AP alone, S100A8 alone, or both PAR2 AP and S100A8 in combination, in order to address possible synergy between these two molecules. LPS alone induced an almost 10-fold increase in lung MPO activity with 3.2 ± 0.4 (Δabsorbance/min/g) when compared to 0.33 ± 0.2 in control mice. This increase in MPO activity was significantly reduced by the administration of S100A8 (2.1 ± 0.4) or of PAR2 AP (2.5 ± 0.4) (P < 0.05). The administration of a combination of S100A8 and PAR2 AP further decreased lung MPO activity to 1.4 ± 0.2 when compared to S100A8 alone (P < 0.01) (Fig. 5).
Fig. 5.

MPO activity in the lungs of endotoxemic mice as a measure of PMN infiltration and oxidative metabolism. LPS increased lung MPO activity, an effect that is lessened by S100A8 and further significantly reduced by a combination of S100A8 and PAR2AP. Results are mean of three independent experiments ± SEM with groups of 5 mice each. #P < 0.01 when comparing LPS to saline treated controls. *P < 0.05 comparing LPS to LPS/S100A8 or LPS/PAR2 AP. **P < 0.05 comparing LPS/S100A8 to LPS/S100A8/PAR2 AP.
3.6. PAR2 AP potentiates the protective effect of S100A8 on liver oxidative damage
We next measured TBARS in the liver as an indicator of lipid peroxidation. LPS alone raised the TBARS score from 76.2 ± 16.4 μM/g (in controls) to 177.7 ± 16.4 μM/g. Livers of mice treated with LPS and S100A8 had a significantly (P < 0.05) lower TBARS score (107.9 ± 16.3 μM/g), as did the livers of LPS/PAR2 AP treated mice (117.3 ± 21.7 μM/g), when compared to the LPS only group. The administration of both S100A8 and PAR2 AP resulted in further protection of the liver from oxidative damage with a TBARS score similar to that of the control mice (78.1 ± 15.2 μM/g) (Fig. 6).
Fig. 6.

Measurement of TBARS in livers of mice challenged with LPS. TBARS values are increased by LPS, an effect significantly mitigated by S100A8 or PAR2 AP. Further statistically significant reduction in TBARS values can be achieved with a combination of PAR2 AP and S100A8. Results are mean of three independent experiments ± SEM with groups of 5 mice each. #P < 0.01 when comparing LPS to saline treated controls. *P < 0.05 comparing LPS to LPS/S100A8 or LPS/PAR2 AP. **P < 0.05 comparing LPS/S100A8 to LPS/S100A8/PAR2 AP.
3.7. Dual S100A8/PAR2 AP treatment improves the survival rate of endotoxemic mice
We tested the effect of S100A8 and/or PAR2 AP on the survival rates of endotoxemic mice that were given a lethal dose of LPS. A 50 mg/kg IP administration of LPS resulted in 33% overall survival at 72 h. We next tested the administration of S100A8 alone, PAR2 AP alone, or S100A8 and PAR2 AP in combination. The survival rate of the endotoxemic mice was raised significantly to 65% and 63% by S100A8 and PAR2 AP, respectively (P < 0.01 for each). The combined administration of both S100A8 and PAR2 AP resulted in 85% survival, a rate significantly greater than obtained with either molecule separately (P < 0.05) (Fig. 7). The combined use of S100A8 and PAR2 AP not only increased the number of mice that survived, but also prolonged the survival time of those mice that died. All death in the S100A8/PAR2 AP treated mice occurred 48 h after the administration of LPS, whereas half or more of the death in the other groups occurred within the first 48 h.
Fig. 7.

Survival curves of mice challenged with a lethal dose of LPS. S100A8 (n = 40) or PAR2 AP (n = 40) alone significantly increased the survival when administered intraperitoneally 4h after LPS (n = 40) from 33% to 65% and 63%, respectively (P < 0.01). The combination of PAR2 AP and S100A8 (n = 20) resulted in a survival rate of 85%. Which was statistically higher than with each alone (P < 0.05).
4. Discussion
Despite their critical scientific and medical importance in sepsis and other diseases, mechanisms regulating PMN oxidative metabolism are not fully understood and there is no clinically feasible and reliable method to specifically restrain PMNs. We have previously shown that calprotectin inhibits the oxidative metabolism of LPS-activated PMNs in vitro (Sroussi et al., 2010, 2012; Schwartz et al., 2010). In the present study, we show that a component of calprotectin (S100A8) can be exogenously introduced to reduce tissue damage, inflammation, and oxidative injuries to major organ systems in a model of LPS-induced endotoxemia. The anti-inflammatory and anti-oxidative effects of S100A8 resulted in a significant increase in the survival of mice challenged with a lethal dose of LPS. The data also indicated that the controlled activation of PAR2 enhanced the protective effect of S100A8, resulting in further reduction in oxidative damage and a further increase in the survival rate of mice challenged by a lethal dose of LPS.
The mechanisms of the protective effect of calprotectin during sepsis remain elusive. Others have shown that the activation of PAR2 results in a physical interaction between PAR2 and TLR4, and a potentiation of TLR4 activation by LPS (Rallabhandi et al., 2008; Nhu et al., 2010). It is tempting to hypothesize that calprotectin, which binds to TLR4 (Vogl et al., 2007), may interfere in the PAR2/TLR4 receptor cooperativity scheme. Accordingly, although calprotectin can activate TLR4 (Vogl et al., 2007; Yonekawa et al., 2011), it may also interfere in the potentiation of TLR4 signaling by PAR2. Alternatively, the beneficial effect of calprotectin may relate to other compensating anti-inflammatory signals known to be activated by PAR2 (Kaneider et al., 2007). Whether PAR2 and its activation, known to occur during sepsis (Jesmin et al., 2009; Pawlinski et al., 2004), are required for the protective effect of calprotectin must be addressed in additional studies. Independent of its mechanism of action, calprotectin, like PAR2, may exert temporally sensitive and opposite effects on the inflammatory response in the course of sepsis. The overall function of calprotectin in sepsis, like that of PAR2, may therefore not conform to a simple pro-or anti-inflammatory effect but rather to a complex, conjunctural, and temporally dependent immune regulatory task.
Circulating concentrations of calprotectin increase with acute inflammation and during sepsis (Rau et al., 2010; Terrin et al., 2011), and a gradual decrease in S100A8 is seen in patients recovering from septic shock (Payen et al., 2008). Data from others have identified calprotectin deficiency as a characteristic of non-healing chronic venous wounds (Trostrup et al., 2011) which feature persistent PMNs and innate immune activation (Pukstad et al., 2010). The notion that the functions of calprotectin in sepsis include an inherent anti-inflammatory effect is in accordance with a clinical report indicating that a higher gene expression of S100A9 in peripheral leukocytes, 7–10 days after a diagnosis of sepsis, correlated with a heightened risk of hospital-acquired infection at a later time point (Fontaine et al., 2011). The hyper- and hypo-inflammatory states of sepsis, and the dual protective and detrimental role played by PMNs in models of sepsis (Hoesel et al., 2005), require careful consideration before our data can be applied to clinical sepsis.
The injection of LPS into an animal model is sufficient to trigger signs of sepsis. Elevated LPS concentrations in the circulation of septic patients correlates with poorer prognosis in these patients (Marshall et al., 2004; Davies and Cohen, 2011). However, LPS-induced endotoxemia does not fully represent the complexity of this disease. A Gram-negative infection or an infection with multiple microbial agents may result in a different physiological response. The mice we used were young and otherwise healthy with no complicating and coexisting conditions (i.e. diabetes, obesity, etc.). Furthermore, the mice did not have access to antimicrobial treatment or organ supportive interventions (i.e. respiratory or renal). It is therefore difficult to directly apply the results of our studies to sepsis in realistic clinical settings. Future clinical studies dissecting calprotectinemia, the activation of PAR2, and a switch from a hyper- to hypo-inflammatory state during the course of sepsis will help clarify the clinical significance of the data presented herein.
Finally, the magnitude of the protective effect afforded by calprotectin against LPS-induced ailments in this study, and the potential clinical significance of these findings, encourage the continuation of these translationally minded investigations. This report strongly supports further examination of the role of calprotectin in the modulation of inflammation and its interplay with PAR2 in septic patients.
Acknowledgments
This study was funded by a National Institute of Health (NIH) grant from the National Institute of Dental and Craniofacial Research (NIDCR), grant numbers: 5K22DE017161-04 and 5K22DE017161-04S1.
References
- Albuszies G, Bruckner UB. Antioxidant therapy in sepsis. Intensive Care Medicine. 2003;29(10):1632–1636. doi: 10.1007/s00134-003-1861-5. [DOI] [PubMed] [Google Scholar]
- Angus DC, et al. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Critical Care Medicine. 2001;29(7):1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- Biswal S, Remick DG. Sepsis: redox mechanisms and therapeutic opportunities. Antioxidants and Redox Signalling. 2007;9(11):1959–1961. doi: 10.1089/ars.2007.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerke K, et al. Distribution of macrophages and granulocytes expressing L1 protein (calprotectin) in human Peyer's patches compared with normal ileal lamina propria and mesenteric lymph nodes. Gut. 1993;34(10):1357–1363. doi: 10.1136/gut.34.10.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borensztajn K, et al. Factor Xa stimulates proinflammatory and profibrotic responses in fibroblasts via protease-activated receptor-2 activation. American Journal of Pathology. 2008;172(2):309–320. doi: 10.2353/ajpath.2008.070347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JH, et al. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circulation Research. 2008;102(10):1239–1246. doi: 10.1161/CIRCRESAHA.107.167544. [DOI] [PubMed] [Google Scholar]
- Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(10):5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicala C, et al. Protease-activated receptor-2 involvement in hypotension in normal and endotoxemic rats in vivo. Circulation. 1999;99(19):2590–2597. doi: 10.1161/01.cir.99.19.2590. [DOI] [PubMed] [Google Scholar]
- Cowley HC, et al. Plasma antioxidant potential in severe sepsis: a comparison of survivors and nonsurvivors. Critical Care Medicine. 1996;24(7):1179–1183. doi: 10.1097/00003246-199607000-00019. [DOI] [PubMed] [Google Scholar]
- Davies B, Cohen J. Endotoxin removal devices for the treatment of sepsis and septic shock. Lancet Infectious Diseases. 2011;11(1):65–71. doi: 10.1016/S1473-3099(10)70220-6. [DOI] [PubMed] [Google Scholar]
- Draper HH, Hadley M. Malondialdehyde determination as index of lipid peroxidation. Methods in Enzymology. 1990;186:421–431. doi: 10.1016/0076-6879(90)86135-i. [DOI] [PubMed] [Google Scholar]
- Edgeworth J, et al. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. Journal of Biological Chemistry. 1991;266(12):7706–7713. [PubMed] [Google Scholar]
- Fontaine M, et al. Delayed increase of S100A9 messenger RNA predicts hospital-acquired infection after septic shock. Critical Care Medicine. 2011;39(12):2684–2690. doi: 10.1097/CCM.0b013e3182282a40. [DOI] [PubMed] [Google Scholar]
- Guerreiro MO, et al. Plasma superoxide dismutase activity and mortality in patients with septic. Journal of Trauma. 2010;69(6):E102–E106. doi: 10.1097/TA.0b013e3181dbb289. [DOI] [PubMed] [Google Scholar]
- Hoesel LM, et al. Harmful and protective roles of neutrophils in sepsis. Shock. 2005;24(1):40–47. doi: 10.1097/01.shk.0000170353.80318.d5. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, et al. Intrinsic function of S100A8/A9 complex as an anti-inflammatory protein in liver injury induced by lipopolysaccharide in rats. Clinica Chimica Acta. 2007;376(1–2):197–204. doi: 10.1016/j.cca.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Jesmin S, et al. Protease-activated receptor 2 blocking peptide counteracts endotoxin-induced inflammation and coagulation and ameliorates renal fibrin deposition in a rat model of acute renal failure. Shock. 2009;32(6):626–632. doi: 10.1097/SHK.0b013e3181a5359c. [DOI] [PubMed] [Google Scholar]
- Jiang B, et al. The role of proteinase 3 (PR3) and the protease-activated receptor-2 (PAR-2) pathway in dendritic cell (DC) maturation of human-DC-like monocytes and murine DC. Clinical and Experimental Rheumatology. 2010;28(1 Suppl. 57):56–61. [PubMed] [Google Scholar]
- Kallenberg CG. Pathophysiology of ANCA-associated small vessel vasculitis. Current Rheumatology Reports. 2010;12(6):399–405. doi: 10.1007/s11926-010-0138-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski KA, et al. Oxidative stress and neutrophil activation–the two keystones of ischemia/reperfusion injury. International Journal of Cardiology. 2002;86(1):41–59. doi: 10.1016/s0167-5273(02)00189-4. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, et al. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nature Immunology. 2007;8(12):1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollef MH, Schuster DP. The acute respiratory distress syndrome. New England Journal of Medicine. 1995;332(1):27–37. doi: 10.1056/NEJM199501053320106. [DOI] [PubMed] [Google Scholar]
- Kovach MA, Standiford TJ. The function of neutrophils in sepsis. Current Opinion in Infectious Diseases. 2012;25(3):321–327. doi: 10.1097/QCO.0b013e3283528c9b. [DOI] [PubMed] [Google Scholar]
- Lindner JR, et al. Delayed onset of inflammation in protease-activated receptor-2-deficient mice. Journal of Immunology. 2000;165(11):6504–6510. doi: 10.4049/jimmunol.165.11.6504. [DOI] [PubMed] [Google Scholar]
- Macfarlane SR, et al. Proteinase-activated receptors. Pharmacological Reviews. 2001;53(2):245–282. [PubMed] [Google Scholar]
- Marshall JC, et al. Diagnostic and prognostic implications of endotoxemia in critical illness: results of the MEDIC study. Journal of Infectious Diseases. 2004;190(3):527–534. doi: 10.1086/422254. [DOI] [PubMed] [Google Scholar]
- Martin GS, et al. The epidemiology of sepsis in the United States from 1979 through 2000. New England Journal of Medicine. 2003;348(16):1546–1554. doi: 10.1056/NEJMoa022139. [DOI] [PubMed] [Google Scholar]
- Moffatt JD, Jeffrey KL, Cocks TM. Protease-activated receptor-2 activating peptide SLIGRL inhibits bacterial lipopolysaccharide-induced recruitment of polymorphonuclear leukocytes into the airways of mice. American Journal of Respiratory Cell and Molecular Biology. 2002;26(6):680–684. doi: 10.1165/ajrcmb.26.6.4693. [DOI] [PubMed] [Google Scholar]
- Molnar RG, et al. The role of neutrophils in producing hepatocellular dysfunction during the hyperdynamic stage of sepsis in rats. Journal of Surgical Research. 1997;73(2):117–122. doi: 10.1006/jsre.1997.5216. [DOI] [PubMed] [Google Scholar]
- Morello S, et al. A protective role for proteinase activated receptor 2 in airways of lipopolysaccharide-treated rats. Biochemical Pharmacology. 2005;71(1–2):223–230. doi: 10.1016/j.bcp.2005.10.016. [DOI] [PubMed] [Google Scholar]
- Namura T, et al. Possible mechanism for regulation of inflammatory responses with the S100A8/A9 protein. Rinsho Byori. 2010;58(7):651–657. [PubMed] [Google Scholar]
- Nhu QM, et al. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunology. 2010;3(1):29–39. doi: 10.1038/mi.2009.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiological Reviews. 2004;84(2):579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, et al. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood. 2004;103(4):1342–1347. doi: 10.1182/blood-2003-09-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen D, et al. Gene profiling in human blood leucocytes during recovery from septic shock. Intensive Care Medicine. 2008;34(8):1371–1376. doi: 10.1007/s00134-008-1048-1. [DOI] [PubMed] [Google Scholar]
- Peters T, Mann TS, Henry PJ. Inhibitory influence of protease-activated receptor 2 and E-prostanoid receptor stimulants in lipopolysaccharide models of acute airway inflammation. Journal of Pharmacology and Experimental Therapeutics. 2010;335(2):424–433. doi: 10.1124/jpet.109.163253. [DOI] [PubMed] [Google Scholar]
- Pukstad BS, et al. Non-healing is associated with persistent stimulation of the innate immune response in chronic venous leg ulcers. Journal of Dermatological Science. 2010;59(2):115–122. doi: 10.1016/j.jdermsci.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Rallabhandi P, et al. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction: a novel paradigm for receptor cooperativity. Journal of Biological Chemistry. 2008;283(36):24314–24325. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau M, et al. Clinical manifestations but not cytokine profiles differentiate adult-onset Still's disease and sepsis. Journal of Rheumatology. 2010;37(11):2369–2376. doi: 10.3899/jrheum.100247. [DOI] [PubMed] [Google Scholar]
- Ritter C, et al. Plasma oxidative parameters and mortality in patients with severe burn injury. Intensive Care Medicine. 2003;29(8):1380–1383. doi: 10.1007/s00134-003-1833-9. [DOI] [PubMed] [Google Scholar]
- Ritter C, et al. Drug intervention trials in sepsis. Lancet. 2004;364(9433):498. doi: 10.1016/S0140-6736(04)16799-8. [DOI] [PubMed] [Google Scholar]
- Sampson B, et al. Hyperzincaemia and hypercalprotectinaemia: a new disorder of zinc metabolism. Lancet. 2002;360(9347):1742–1745. doi: 10.1016/S0140-6736(02)11683-7. [DOI] [PubMed] [Google Scholar]
- Sato T, et al. Inhibition of Corynebacterium parvum-primed and lipopolysaccharide-induced hepatic necrosis in rats by selective depletion of neutrophils using a monoclonal antibody. Journal of Leukocyte Biology. 1993;53(2):144–150. doi: 10.1002/jlb.53.2.144. [DOI] [PubMed] [Google Scholar]
- Sato Y, et al. Pulmonary sequestration of polymorphonuclear leukocytes released from bone marrow in bacteremic infection. American Journal of Physiology. 1998;275(2 Pt 1):L255–L261. doi: 10.1152/ajplung.1998.275.2.L255. [DOI] [PubMed] [Google Scholar]
- Schulze zur Wiesch A, et al. Myeloid related proteins MRP8/MRP14 may predict disease flares in juvenile idiopathic arthritis. Clinical and Experimental Rheumatology. 2004;22(3):368–373. [PubMed] [Google Scholar]
- Schwartz R, et al. Effect of human immunodeficiency virus infection on S100A8/A9 inhibition of peripheral neutrophils oxidative metabolism. Biomedicine and Pharmacotherapy. 2010;64(8):572–575. doi: 10.1016/j.biopha.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, et al. Loss of myeloid related protein-8/14 exacerbates cardiac allograft rejection. Circulation. 2012;124(25):2920–2932. doi: 10.1161/CIRCULATIONAHA.110.009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroussi HY, et al. S100A8 and S100A9 inhibit neutrophil oxidative metabolism in vitro: involvement of adenosine metabolites. Free Radical Research. 2010;44(4):389–396. doi: 10.3109/10715760903431434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroussi HY, et al. The down regulation of neutrophil oxidative metabolism by S100A8 and S100A9: implication of the protease-activated receptor-2. Molecular Immunology. 2011;50(1–2):42–48. doi: 10.1016/j.molimm.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser F, Gowland PL, Ruef C. Elevated serum macrophage inhibitory factor-related protein (MRP) 8/14 levels in advanced HIV infection and during disease exacerbation. Journal of Acquired Immune Deficiency Syndromes and Human Retrovirology. 1997;16(4):230–238. doi: 10.1097/00042560-199712010-00002. [DOI] [PubMed] [Google Scholar]
- Terrin G, et al. Serum calprotectin: an antimicrobial peptide as a new marker for the diagnosis of sepsis in very low birth weight newborns. Clinical and Developmental Immunology. 2011;2011:291085. doi: 10.1155/2011/291085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trostrup H, et al. S100A8/A9 deficiency in nonhealing venous leg ulcers uncovered by multiplexed antibody microarray profiling. British Journal of Dermatology. 2011;165(2):292–301. doi: 10.1111/j.1365-2133.2011.10384.x. [DOI] [PubMed] [Google Scholar]
- Tugizov S, et al. Inhibition of human papillomavirus type 16 E7 phosphorylation by the S100 MRP-8/14 protein complex. Journal of Virology. 2005;79(2):1099–1112. doi: 10.1128/JVI.79.2.1099-1112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bergh FA, et al. Calprotectin: a fecal marker for diagnosis and follow-up in patients with chronic inflammatory bowel disease. Nederlands Tijdschrift Voor Geneeskunde. 2003;147(48):2360–2365. [PubMed] [Google Scholar]
- van Eeden SF, et al. Polymorphonuclear leukocytes released from the bone marrow preferentially sequester in lung microvessels. Microcirculation. 1997;4(3):369–380. doi: 10.3109/10739689709146801. [DOI] [PubMed] [Google Scholar]
- Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nature Medicine. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Yonekawa K, et al. Myeloid related proteins activate Toll-like receptor 4 in human acute coronary syndromes. Atherosclerosis. 2011;218(2):486–492. doi: 10.1016/j.atherosclerosis.2011.06.020. [DOI] [PubMed] [Google Scholar]
- Zhong B, Wang DH. Protease-activated receptor 2-mediated protection of myocardial ischemia–reperfusion injury: role of transient receptor potential vanilloid receptors. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2009;297(6):R1681–R1690. doi: 10.1152/ajpregu.90746.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]