

Abstract
While cognitive deficits are increasingly recognized as common symptoms in amyotrophic lateral sclerosis (ALS), the underlying histopathologic basis for this is not known, nor has the relevance of neuroinflammatory mechanisms and microglial activation to cognitive impairment (CI) in ALS been systematically analyzed. Staining for neurodegenerative disease pathology, TDP-43, and microglial activation markers (CD68, Ibal) was performed in 102 autopsy cases of ALS, and neuropathology data were related to clinical and neuropsychological measures. ALS with dementia (ALS-D) and ALS with impaired executive function (ALS-Ex) patients showed significant microglial activation in middle frontal and superior or middle temporal (SMT) gyrus regions, as well as significant neuronal loss and TDP-43 pathology in these regions. Microglial activation and TDP-43 pathology in middle frontal and superior or middle temporal regions were highly correlated with measures of executive impairment, but not with the MMSE. In contrast, only one ALS-D patient showed moderate Alzheimer's disease (AD) pathology. Tau and Aβ pathology increased with age. A lower MMSE score correlated with tau pathology in hippocampus and SMT gyrus, and with Aβ pathology in limbic and most cortical regions. Tau and Aβ pathology did not correlate with executive measures. We conclude that microglial activation and TDP-43 pathology in frontotemporal areas are determinants of FTLD spectrum dementia in ALS and correlate with neuropsychological measures of executive dysfunction. In contrast, AD pathology in ALS is primarily related to increasing age and associated with a poorer performance on the MMSE.
Keywords: Amyotrophic lateral sclerosis, Frontotemporal lobar degeneration, Cognitive impairment, TDP-43, Microglia
Introduction
Amyotrophic lateral sclerosis (ALS) is the most frequent adult onset motor neuron disease, leading to death after a mean survival of approximately 3 years [33]. While ALS is primarily characterized by motor symptoms, cognitive deficits are increasingly recognized as common symptoms of disease in these patients [38, 52, 53]. Neuropsychological testing frequently reveals impairments in executive function, including planning, working memory and attention. Moreover, a large autopsy series of 87 ALS patients with or without frontotemporal lobar degeneration (FTLD) showed that 12.5% of patients with ALS fulfilled criteria for probable or definite FTLD [25]. Imaging studies of cognitive decline in ALS show atrophy in prefrontal regions anterior to the motor cortex and cortical hypometabolism in the frontotemporal regions [1, 4, 18]. The link between FTLD and ALS was further strengthened by the discovery of TAR DNA-binding protein of 43 kDa (TDP-43) as a major component of ubiquitinated inclusions in both ALS and the largest subset of FTLD cases now known as FTLD-TDP [47]. This suggests that ALS could be a manifestation of FTLD within a continuum of TDP-43 proteinopathies [19]. Abnormal cytoplasmic accumulation of TDP-43 has been observed in neurons and glial cells of the motor cortex, brainstem, and spinal cord in ALS [39, 47]. These neuronal cytoplasmic inclusions have been found to vary in morphology from small granules and compact Lewy body-like inclusions to filamentous skeins. Several studies suggest that neuronal cell death in ALS is non-cell autonomous with an important role played by microglial cells [2, 23, 31]. Neuroimaging demonstrated in vivo microglial activation not only in the motor cortex, but also in the prefrontal areas of the brain [2, 23, 57]. However, direct demonstration of the relevance of microglial activation to non-motor symptoms like cognitive impairment (CI) in ALS has not been studied. Indeed, few studies have systematically analyzed neuropathological correlates of cognitive decline in ALS [24, 52, 54, 59]. Here, we describe the neuropathological findings in a large and clinically well-defined cohort of ALS autopsy cases and evaluate the relevance of TDP-43 pathology, microglial activation, and co-morbidity associated with Alzheimer's disease (AD) pathology for Cl in ALS.
Methods
Autopsy cohort
Individuals who underwent autopsy in the Center for Neurodegenerative Disease Research at the University of Pennsylvania from 1985 to 2010 were enrolled. Our cohort included 102 cases with a clinical diagnosis of ALS in accordance with llie modified El Escorial Criteria [9] and a neuropathological diagnosis of ALS (Table I). Detailed clinical characteristics (age at onset, age at death, site of onset, disease duration. ALS global disease severity as measured by a functional rating score (ALSFRS-R) [11]. gender, performance on cognitive tests) were ascertained by retrospective chart review of clinical visits from 1985 through 2010 within the University of Pennsylvania Health System; the vast majority of patients were seen by one neurologist (L.M.). Unless otherwise specified, results of testing used in this study were from the visit most proximate to death, occurring within 12 months of death. ALS patients with dementia (ALS-D) were identified (n = 12) by the clinical assessments of two experienced neurologists (L.M., L.E.) based on the history of the patient's caregivers and a detailed neurological examination. Of the ALS cases included in this study, 18 (18.6%) had a family history of ALS (fALS), while 79 cases were sporadic (sALS). For five cases, data were missing or not unequivocal. Three of the fALS cases showed an SODI mutation, while none showed a TARDP gene mutation.
Tahle 1.
Demographic and neuropsychological data of ALS autopsy cohort
| ALS-nD (n = 88) | ALS-D (n = 12) | ALS cognitive status unknown (n = 2) | p value | |
|---|---|---|---|---|
| Gender (male) | 59.1% | 58.3% | 100% | 0.50 |
| Age of onseta | 59.6 (11.8) | 51.5 (20.7) | – | 0.15 |
| Disease duration (years)b | 2 (1–4) | 3 (1–7) | – | 0.6 |
| Age at death (years)a | 63.3 (11.2) | 59.6 (9.0) | 49 and 72 | 0.32 |
| Brain weight (mg)a | 1,333 (139) | 1,253 (128) | 1,435 and 1,500 | 0.07 |
| Braak staging | 40% No NFT | 33.3% No NFT | – | 0.67 |
| 32% I–II | 50% I–II | |||
| 16% III–IV | 16.7% III–IV | |||
| 12% V–VI (85%) | 0% V–VI (100%) | |||
| CERAD staging | 77.0% No plaq. | 80.0% No plaq. | – | 0.98 |
| 6.8% A | 10.0% A | |||
| 9.5% B | 10.0% B | |||
| 6.7% C | 0% C | |||
| APOE (1 or more ε4) | 18.5% | 50% | – | 0.21 |
| ALSFRS-Rb | 21 (17.8–25) | 19 (14–25) | – | |
| Years of educationb | 15 (12–16) (48%) | 16 (12–22)(33%) | – | 0.60 |
| F-wordsa | 11.0 (4.0) (57%) | 4.3 (2.5) (33%) | – | 0.002 |
| Oral trailsb | 51 (50–52) (48%) | 47.5 (47–50) (33%) | – | 0.16 |
| MMSEb | 30 (29–30) (30%) | 27.5 (13.5–29) (33%) | – | 0.04 |
| FBI caregivera | 41.4 (9.6) (22%) | 28 (8%) | – | 0.2 |
ALS-D ALS with dementia, ALSFRS-R ALS Functional raring scale. ALS-nD ALS without dementia, CERAD Consortium to Establish a Registry For Alzheimer's Disease, FBI Frontal Behavioral Inventory, F-words F-words test, Interq. range interquartile range, oral trails Oral Trail Making Test, MMSE Mini-Mental State Examination, NFT neurofibrillary tangles
Mean (standard deviation)
Median (interquartile range)
Basic neuropathological characterization
Pathology was examined in six regions of the ceniral nervous system (CNS); amygdala, hippocampus (CA1/subiculum), middle frontal gyrus, superior or middle temporal gyrus (SMT), motor cortex (precentral gyrus), and cervical spinal cord (CSC). While many more regions are routinely examined for diagnostic assessment, we chose these brain regions for detailed examination here because they are among the most consistently affected in both FTLD and ALS [20, 62].
Sections were fixed and cut into 6–10 μm sections, stained with hematoxylin and eosin (HE) and thioflavin S, and immunohistochemistry (IHC) was performed with antibodies to tau, α-synuclein, ubiquitin, and TDP-43 as previously described in detail [20, 46]. Sections were reviewed blind to the results of cognitive testing, and the extent of TDP-43, tau, and Aβ pathology as well as the extent of neuron loss (as monitored by HE) were rated for each region on a 4-poim ordinal scale (0, none; 1, mild; 2, moderate; 3. severe/numerous) as previously described [21, 58] (Fig. 1). Assessment of tau neurofibrillary tangle pathology was performed according to revised Braak criteria [7, 8, 21, 58]. The extent of Aβ pathology was described using Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria [43, 55] and their combination using NIA-Reagan criteria [12, 27].
Fig. 1.
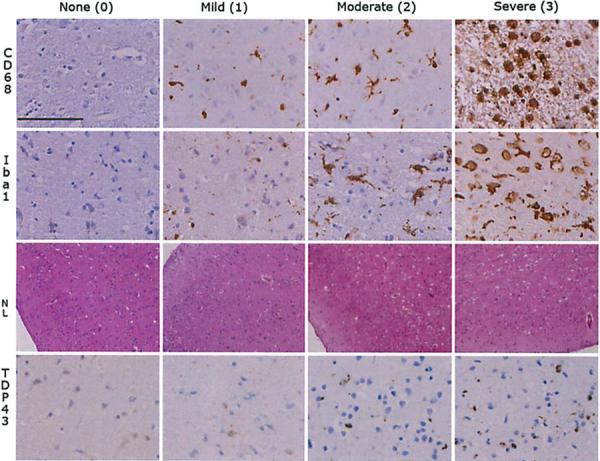
Semiquantitative staging of pathology in ALS. The figure illustrates the semiquantitative staging used to grade the extent of microglial activation (CD68, Ibal, images taken with ×40 objective, scale bar 100 μm), neuronal loss (NL, images taken with ×10, and TDP-43 pathology (images taken with ×40 objective)
Analysis of microglial activation
Sections of 6-μm thickness were cut from paraffin-embedded specimens. For IHC, all slides were deparaffinized and rehydrated in a series of xylene and graded ethanol. After immersion in methanol/H202 for 30 min, slides were washed in 0.1 M Tris buffer (pH 7.6) and blocked in 0.1 M Tris/2% FBS. Sections were stained using polyclonal rabbit anti-Ibal antibody (Wako Chemicals, Richmond, VA) at 1:1.000 and incubated overnight at 4°C. Sections were then rinsed and washed in Tris and incubated with Vector biotinylated anti-rabbit IgG (Vector Laboratories Inc., Burlingame, Ca) at 1:1.000 for 1 h. After rinsing again, the ICH reaction was visualized using 3,3′-diaminobenzidine (DAB) and the sections were dehydrated through graded ethanol, cleared in xylene, and coverslipped in Cytoseal 60 mounting medium. Sections were stained for CD68 using mouse anti-human CD68 (Dako, Carpinteria, CA) at 1:1.000. The extent of microglial activation was rated on a 4-point ordinal scale combining gray and white matter pathology for each region (Fig. 1).
Neuropsychological testing
Ante-mortem cognitive testing was performed at 3- to 6-month intervals during routine clinic visits. Data for three cognitive tests were available for a subset of the autopsy cohort within 12 months of death. These included a test of letter-guided verbal fluency (F-words test, n = 54 patients) [36], a test of frontal executive function (Oral Trail Making Test, n = 47 patients) [37] assessing information processing speed and the capacity to maintain a complex mental set, and the Mini-Mental State Examination (MMSE, n = 31) [17]. For the letter fluency test, patients without significant dysarthria were asked to generate as many unique words (proper nouns and numbers excluded) beginning with the letter “F” in 1 min. Patients with dysarthria, but showing preserved limb and hand function, were asked to write the words; 90 s were allotted to these patients. The total number of unique words produced was recorded. For the Oral Trail Making Test, patients without significant dysarthria were asked to sequentially say letters of the alphabet, beginning with A, alternating with numbers, beginning with 1, in an ascending order (i.e., A-l, B-2) and ending at Z-26. Each error was subtracted from the maximum score of 52. Patients with significant dysarthria, but with relatively preserved hand and limb ability, were asked to perform the same task in writing. We included the Frontal Behavioural Inventory (FBI), a 24-item caregiver-based behavioral questionnaire designed for the diagnosis and quantification of FTLD symptoms [32].
Statistical analysis
Normal distribution of the variables was tested and accordingly mean and standard deviation (SD) were used in case of normal distribution, and median and first and third quartiles were used in case of non-normally distributed data. For the analyses presented in Table 1, independent sample t test and Mann–Whitney U test were used for normally and non-normally distributed variables, respectively. For the purpose of multivariate analyses, a power transformation was applied to non-normally distributed variables. To quantify non-normally distributed variables across three or more groups, Kruskal–Wallis analysis of variance on ranks was applied followed in case of significance by Dunn's Method. All correlations were studied using Spearman's rank order correlation coefficient. Bonferroni correction for multiple testing was applied when contrasts were not driven by a specific hypothesis. For all other testing, we set significance level to 0.01 to reduce the likelihood of false positive discovery findings. All statistical tests were two sided. Data analysis was performed using SPSS (Version 17.0 SPSS Inc., Chicago, IL, USA) and R 2.13.1.
Results
Gross pathology findings
Demented patients had a lower brain weight, although the difference was not statistically significant (Table 1). Mild to moderate gross brain atrophy in frontal regions (other than the primary motor cortex) was observed in 12 cases (11.7%).
Distribution of pathology across different CNS regions in the entire cohort
TDP-43 pathology was most extensive in the CSC and the motor cortex as illustrated in Fig. 2. Similarly, neuronal loss was most extensive in the CSC and motor cortex, whereas considerably lesser degrees of neuronal loss were observed in other cortical and subcortical areas (Fig. 2). Staining for both Ibal and CD68 showed microglial activation to be most pronounced in CSC and motor cortex, with a tendency to show more pronounced expression in the white matter (Fig. 3). The extent and anatomical distribution of tau tangle and Aβ pathology in ALS autopsy cases are shown in Table 1 and Fig. 2, respectively.
Fig. 2.
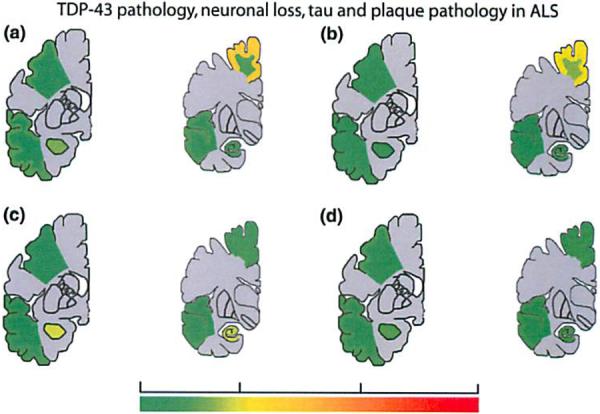
Subtypes of pathology in ALS. Anatomical heal map shows extern of TDP-43 pathology (a), neuronal loss (b), tau pathology (c). and Aβ pathology (d) in different regions of the central nervous system in ALS
Fig. 3.
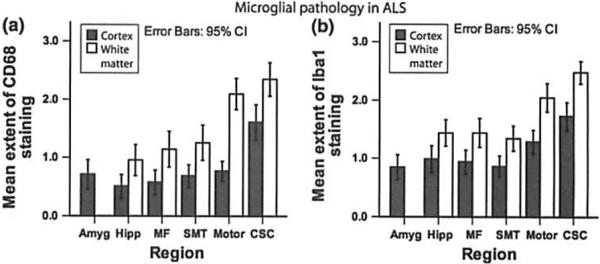
Microglial pathology in ALS. Bar plot shows microglial activation in the cortex and White matter Of different regions of the central nervous system of ALS as delected by staining for CD68 (a) and Ibal (b). Whiskers in bar plot indicate 95% confidence interval of mean. Amyg amygdala, CSC cervical spinal cord. Hipp hippocampus (CA1, subiculum), MF middle frontal gyrus, Mot motor conex, NL neuronal loss. SMT superior or middle temporal gyrus
Regional correlation of pathology in the entire cohort
The extent of TDP-43 pathology correlated with neuronal loss in all regions (with the exception of motor cortex and CSC), and both were most severe in the CSC and the motor cortex. Microglial activation as shown by staining for Ibal correlated with neuronal loss in the middle frontal gyms, motor cortex, and CSC. Neither tau nor Aβ pathology correlated with neuronal loss in ALS. An overview on the correlation between different types of pathology in different regions of the CNS is provided in Table 2.
Table 2.
Correlation between different types of pathology in ALS
| Tau | Aβ | NL | TDP-43 | Ibal | CD68 | |
|---|---|---|---|---|---|---|
| Amyg | ||||||
| Tau | ρ = 0.44 | n.s. | n.s. | n.s. | n.s. | |
| Aβ | ρ = 0.44 | n.s. | n.s. | n.s. | n.s. | |
| NL | n.s. | n.s. | ρ = 0.47 | n.s. | n.s. | |
| TDP-43 | n.s. | n.s. | ρ = 0.47 | n.s. | n.s. | |
| Ibal | n.s. | n.s. | n.s. | n.s. | ρ = 0.51 | |
| CD68 | n.s. | n.s. | n.s. | n.s. | ρ = 0.51 | |
| Hipp | ||||||
| Tau | ρ = 0.51 | ρ = 0.34 | n.s. | n.s. | n.s. | |
| Aβ | ρ = 0.5l | n.s. | n.s. | n.s. | n.s. | |
| NL | n.s. | n.s. | ρ = 0.55 | n.s. | n.s. | |
| TDP-43 | n.s. | n.s. | ρ = 0.55 | n.s. | n.s. | |
| Ibal | n.s. | n.s. | n.s. | n.s. | ρ = 0.46 | |
| CD68 | n.s. | n.s. | n.s. | n.s. | ρ = 0.46 | |
| MF | ||||||
| Tau | ρ = 0.51 | n.s. | n.s. | n.s. | n.s. | |
| Aβ | ρ = 0.51 | n.s. | n.s. | n.s. | n.s. | |
| NL | n.s. | n.s. | ρ = 0.47 | ρ = 0.46 | n.s. | |
| TDP-43 | n.s. | n.s. | ρ = 0.47 | n.s. | n.s. | |
| Ibal | n.s. | n.s. | ρ = 0.46 | n.s. | ρ = 0.52 | |
| CD68 | n.s. | n.s. | n.s. | n.s. | ρ = 0.52 | |
| SMT | ||||||
| Tau | ρ = 0.41 | n.s. | n.s. | n.s. | n.s. | |
| Aβ | ρ = 0.41 | n.s. | n.s. | n.s. | n.s. | |
| NL | n.s. | n.s. | ρ = 0.45 | n.s. | n.s. | |
| TDP-43 | n.s. | n.s. | ρ = 0.45 | n.s. | n.s. | |
| Ibal | n.s. | n.s. | n.s. | n.s. | n.s. | |
| CD68 | n.s. | n.s. | n.s. | n.s. | n.s. | |
| Mot | ||||||
| Tau | n.s. | n.s. | n.s. | n.s. | n.s. | |
| Aβ | n.s. | n.s. | n.s. | n.s. | n.s. | |
| NL | n.s. | n.s. | n.s. | ρ = 0.43 | n.s. | |
| TDP-43 | n.s. | n.s. | n.s. | n.s. | n.s. | |
| Ibal | n.s. | n.s. | ρ = 0.43 | n.s. | ρ = 0.40 | |
| CD68 | n.s. | n.s. | n.s. | n.s. | ρ = 0.40 | |
| CSC | ||||||
| Tau | n.s. | n.s. | n.s. | n.s. | n.s. | |
| Aβ | n.s. | n.s. | n.s. | n.s. | n.s. | |
| NL | n.s. | n.s. | n.s. | ρ = 0.41 | n.s. | |
| TDP-43 | n.s. | n.s. | n.s. | n.s. | n.s. | |
| Ibal | n.s. | n.s. | ρ = 0.41 | n.s. | ρ = 0.53 | |
| CD68 | n.s. | n.s. | n.s. | n.s. | ρ = 0.53 |
All correlations are Spearman rank order correlation (level of significance p < 0.001 after Bonferroni correction)
Aβ amyloid beta, Amyg amygdala, CSC cervical spinal cord, Hipp hippocampus (CA1, subiculum), MF middle frontal gyrus, Mot motor cortex, NL neuronal loss, n.s. not significant, SMT superior or middle temporal gyrus
Relation of pathology to age, disease duration, and clinical phenotype in the entire cohort
We observed both tau and Aβ pathology to increase significantly with age (both age at onset of disease as well as age at death) in all cortical and limbic regions (p < 0.01, ρ > 0.4 in each region). Neither tau nor Aβ pathology was correlated with duration of disease in any of the investigated areas. By comparison, TDP-43 pathology did not correlate with disease duration or age in the brain regions we examined. Microglial activation as detected by staining for Ibal and CD68 tended to decrease in motor cortex and CSC with ongoing disease, perhaps indicative of a burned out or end stage of disease with few remaining neurons and less active disease, but this did not reach statistical significance. No correlation of any type of pathology with the ALSFRS-R was observed (data not shown). The extent of microglial activation, or TDP-43, tau or Aβ pathology did not show a significant difference in any of the regions analyzed here between patients with sALS and fALS.
Pathology in the ALS subgroup with ALS-D
Of the 102 autopsy cases, 12 had a clinical history of dementia (ALS-D), 88 patients were not demented (ALS-nD), and for two patients, data were missing or equivocal. Within the ALS-D group, all but one patient met criteria for FTLD [45] and NIA-Reagan criteria [27] of no or low probability of AD. One patient showed tau pathology consistent with Braak stage III–IV [7, 8] and Aβ pathology with a CERAD score B [43, 55]. Thus, we identified one patient in our cohort who had co-occurring AD.
We observed different anatomic distributions of disease for TDP-43, neuronal loss, and microglial activation compared to tau and Aβ pathology. Cases with ALS-D thus showed more extensive TDP-43 pathology than ALS-nD in the middle frontal cortex, SMT cortex, and the hippocampus (Fig. 4). Furthermore, neuronal loss was more extensive in ALS-D than ALS-nD in the middle frontal cortex and the SMT gyrus. Likewise, microglial activation as detected by staining for CD68 was significantly more extensive in the middle frontal cortex, SMT gyrus, and motor cortex in ALS-D as compared to ALS-nD patients (Fig. 4). For Ibal, this difference reached statistical significance only in the SMT and middle frontal gyrus. By comparison, no significant differences with regard to tau or Aβ pathology were observed (p > 0.01 for tau and Aβ pathology in each area).
Fig. 4.
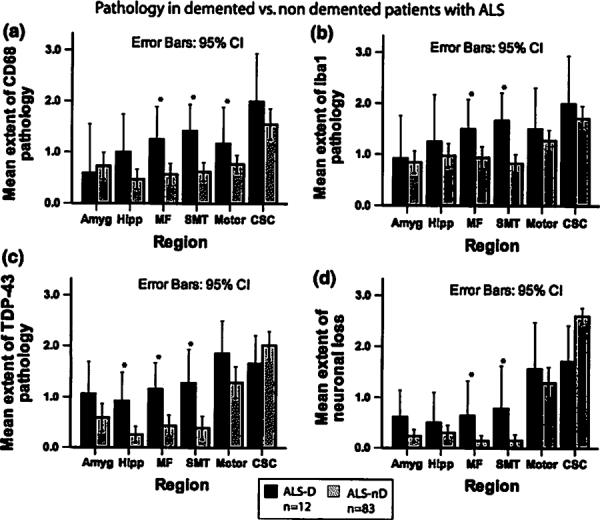
Pathology in ALS with and without dementia. Bar plots show extent of microglial activation (a, b), TDP-43 pathology (c), and neuronal loss (d) in ALS patients with (ALS-D) and without dementia (ALS-nD). Whiskers in bar plot indicate 95% confidence interval of the mean
Relation between pathology in ALS and impaired executive function
Within the autopsy cohort, 13 (32.5%) of 40 patients with available data showed a score below the median for the fluency words and Oral Trail Making Tests. These patients were defined as having an impaired executive function (ALS-Ex). In 27 cases, scores were above/equal to the median for either both tests or one of the tests (“executive function not/slightly impaired”).
As in the ALS-D cases above, we observed different anatomic distributions of TDP-43 pathology and microglial activation in ALS-Ex compared to ALS patients without executive dysfunction. ALS-Ex cases thus showed more extensive TDP-43 pathology in the middle frontal cortex and the SMT gyrus compared to cases in which executive function was not/slightly impaired (p < 0.01 each, Fig. 5). Microglial activation also was more extensive in ALS-Ex compared to ALS patients with minimal executive dysfunction in the middle frontal gyrus (Fig. 6), SMT gyrus, and motor cortex as seen by staining with CD68 compared to cases in which executive function was not/slightly impaired (Fig. 5). Staining for Ibal was significantly more extensive in ALS-Ex in the SMT gyrus, while the difference did not reach significance for the other regions.
Fig. 5.
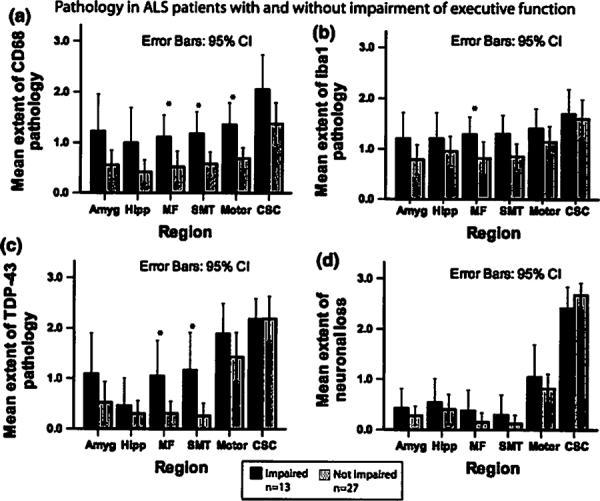
Pathology in ALS with and without impaired executive function. Bar plots shows extent of micgroglial activation (a, b), TDP-43 pathology (c), and neuronal loss (d) in ALS patients with and without impairment of executive function. Whiskers in bar plot indicate 95% confidence interval of the mean. Impairment of executive function was defined by scores below the median in both F-words test and Oral Trail Making Test
Fig. 6.
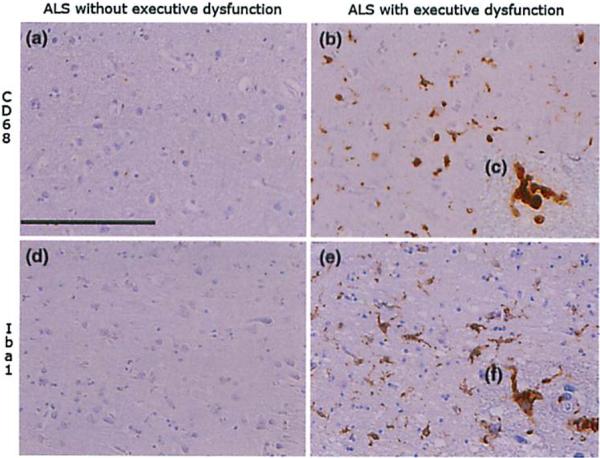
Microglial activation in ALS with impairment of executive function. Images illustrate microglial aclivalion as seen by staining for CD68 and Iba I in the middle frontal gyrus of exemplary cases of ALS with (b, c, e, f) and without (a, d) impairment of executive function. Images a, b, d, and e taken wilh ×20 objective, scale bar 200μm. Images c and f show higher resolution (×60) of activated microglial cells
Reduced output on the letter fluency test was correlated with greater TDP-43 pathology (r = −0.43, p = 0.005) and microglial activation as seen by staining for Ibal (ρ = −0.42, p = 0.003) in the middle frontal gyrus. Furthermore, reduced scores on the Oral Trail Making Test were correlated with greater middle frontal microglial activation as seen by staining for CD68 (ρ = −0.43, p = 0.006).
This contrasts with the findings for tau and Aβ pathology. No significant differences between ALS-Ex and ALS patients with minimal executive dysfunction were observed in any region for tau and Aβ pathology or for neuronal loss (p > 0.01 each). There were no significant correlations between tau/Aβ pathology and measures of executive functioning.
Pathology in ALS with low MMSE
In this autopsy cohort, eight (25.8%) of 31 patients with available data showed an MMSE score lower than the median of 27 points (“low MMSE”), while 23 patients had a score higher or equal to the median (“high MMSE”). Low MMSE cases showed more extensive tau pathology in the hippocampus and SMT gyrus as compared to high MMSE cases. They showed more extensive Aβ pathology in all the investigated limbic and cortical regions. Furthermore, neuronal loss was more extensive in the low MMSE subgroup in the amygdala, and middle frontal cortex. By comparison, microglial activation as seen by staining for both CD68 and Ibal was more extensive in the amygdala of the low MMSE cases as compared to the high MMSE cases (Fig. 7). MMSE did not correlate with TDP-43 pathology or microglial activation in the frontotemporal areas.
Fig. 7.
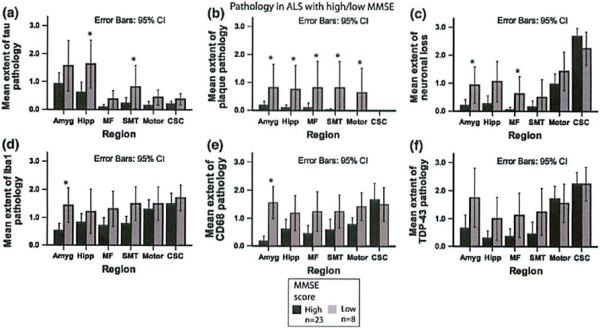
Pathology in ALS related to MMSB. Bar plots Shaw extern of tau (a) and Aβ putholugy (b), as well as ncuronal loss (c), microglial activation (d, e) and TDP-43 patholopy (f) in ALS patients with MMSE above or equal to the median (MMSE high) and MMSE below the median (MMSE low). Whiskers in bar plot indicate 95% confidence Interval of the mean
Scores on the MMSE decreased with increasing extent of Aβ pathology in the amygdala and hippocampus (ρ = −0.65, p < 0.001 each), middle frontal cortex (ρ = −0.66, p < 0.001), SMT cortex (ρ = −0.67, p < 0.001), and motor cortex (ρ = −0.60, p < 0.001). Furthermore, the MMSE decreased with increasing tau pathology in the hippocampus (ρ = −0.46, p = 0.009). The extent of neuronal loss correlated inversely with the MMSE score in the amygdala (ρ = −0.56, p = 0.002), hippocampus (ρ = −0.51, p = 0.003), and middle frontal cortex (ρ = −0.50, p = 0.006).
Discussion
To the best of our knowledge, this is so far the largest study that systematically analyzed pathological correlates of dementia and CI in an autopsy cohort of ALS. While the clinical picture of ALS is dominated by progressive deterioration of motor function, CI and dementia are increasingly recognized [52]. Indeed, CI could compromise patients' capacity to participant in health-care decision making such as end-of-life decisions, as well as reduce compliance with supportive and potentially life-prolonging therapies like non-invasive ventilation [51]. Thus, CI may have major clinical implications in ALS.
Among the subgroup of patients with ALS-D [45], the pattern of dementia resembled that seen in FTLD and was associated with more extensive histopathologic abnormalities due lo TDP-43, while only one ALS-D patient had AD pathology Of intermediate severity. In the entire ALS cohort examined in this study, we found neuropsychological tests of executive function to be related to TDP-43 pathology and microglial activation in middle frontal and SMT regions, while tau pathology was related to poorer performance on the MMSE. These findings confirm the relevance of TDP-43 pathology in the cognitive deficits of ALS patients and suggest that this could be mediated in pan by an inflammatory process. By comparison, cooccurring AD rarely causes dementia in ALS, and when Aβ and tangle pathology becomes severe enough to impact cognitive performance, this appears to compromise cognitive measures sensitive lo AD pathology such as the MMSE, but not performance on executive measures.
Further, TDP-43 pathology was related to neuronal loss in many of the investigated areas, which could be due to a deleterious loss of TDP-43 nuclear function [3, 5, 14, 28, 29, 47–49]. Interestingly, and for reasons which are unclear, the correlation of TDP-43 pathology to neuronal loss could not be shown for the motor cortex and the CSC, but this may reflect clearance of TDP-43 from the extracellular space following its release from degenerating neurons harboring TDP-43 inclusions.
Amyoirophic lateral sclerosis with dementia patients were distinguished from ALS-nD patients by widespread and extensive TDP-43 pathology in frontotemponil conical areas (Fig. 4) and the pattern of the distribution of TDP-43 pathology was similar to that observed in FTLD. In contrast, neither tau nor Aβ pathology showed any difference regarding non-demented patients with ALS. More specifically, we found the burden of TDP-43 pathology to be related to impaired executive functions (Fig. 5), including direct correlations between TDP-43 density in frorntotemporal regions and executive test performance. These correlations were not observed in other brain regions, and correlations were not observed between executive measures and the burden of tau and Aβ pathology. This emphasizes the specificity of the role of TDP-43 in the cognitive deficits of ALS patients. Moreover, executive tests such as these are likely to be useful in neuropsychological assessments to reflect TDP-43-related CI in ALS.
Although microglial activation is a known feature of ALS and FTLD pathology [6, 16, 40, 64], this has not been systematically studied histopathologically before. Furthermore, our data demonstrate for the first time that microglial activation correlates with dementia and CI in ALS, and is related to neuropsychological measures of executive dysfunction.
CD68 is a predominantly intracellular molecule associated with the endosomal compartment and is a pan-macrophage marker also expressed in microglia [34, 42]. The calcium binding protein Iba I is believed to be involved in membrane ruffling related to cell motility and phagocytosis by microglia/macrophages, and is considered a specific marker for microglial activation [30, 50].
We observed extensive microglial activation in the same frontotemporal regions of ALS-D and ALS-Ex with extensive TDP-43 pathology (Figs. 4, 5, 6). Moreover, these regions showed significant correlations between TDP-43 markers of disease and the executive deficits causing cognitive difficulty in ALS. The specificity of these findings is emphasized by the absence of correlations between the extent of microglial activation and TDP-43 pathology in other brain regions, and the absence of correlations with other histopathologic features such as tau and Aβ density thereby suggesting that microglial activation is a key pathological correlate of CI and dementia in ALS (Figs. 4, 5). Our finding that microglial activation also correlates with neuronal loss supports the notion that inflammatory mechanisms could be important mediators of neuronal dysfunction and death in ALS [15, 35]. Taken together with a previous study [61], these data suggest that microglial activation is deleterious and contributes to non-cell autonomous neuron dysfunction and death. Thus, if confirmed, these findings imply that treatment with anti-inflammatory agents may attenuate non-motor symptoms of ALS.
Tau and Aβ pathology were found to have a very different role in ALS. First, we observed both tau and Aβ pathology in ALS, but this was found to be of low abundance (Fig. 2). Indeed, the distribution of these histopathologic features of AD followed the pattern observed in normal aging [19, 43, 46, 55, 56, 58], and only-one ALS-D patient showed intermediate probability for AD [12, 27]. This observation is in accordance with previous Studies that observed AD pathology in ALS to be both infrequent and scarce [26, 44, 60, 63]. A study of 30 autopsy cases with ALS [24] reported a higher proportion of AD pathology, but the mean age of this cohort was considerably older (66 years) than in our study, and age was observed to be an important determinant of AD pathology in ALS. Further, neither tau nor Aβ pathology correlated with executive measures, but instead correlated with a measure commonly used to track AD—the MMSE. This is in line with previous literature that observed the MMSE to be useful to support a diagnosis of AD, but to be of limited value for identifying FTLD cases [22, 41]. MMSE did not correlate with TDP-43 or microglial activation. Therefore, our data indicate that AD-related pathology in ALS is comparatively rare and of minor clinical relevance to CI or dementia in ALS.
Taken together, we observed a double dissociation: measures of executive functioning and FTLD spectrum dementia were related most closely to TDP-43 pathology and microglial activation in frontotemporal regions, while MMSE and dementia of the Alzheimer type reflected AD-related pathology such as tau burden that increased with age and was most prominent in the medial temporal regions. Our observations thus emphasize the potential value of more extensive neuropsychological assessments in all ALS patients, but the extent of CI in the entire cohort was mild [10, 13], suggesting that the motor component of these tests had minimal impact on cognitive performance. Furthermore, and in contrast to the large overall size of the cohort, the number of cases with ALS-D was rather small, so that our results on this group should be regarded as preliminary and additional follow-up studies are needed. Despite these caveats, our data indicate that TDP-43 pathology and microglial activation in the frontotemporal areas are important determinants of CI in ALS, and our study emphasizes the need for increased research on non-motor symptoms of ALS and the mechanisms that underlie them.
Acknowledgments
We thank the many patients who contributed samples for this study. We thank John Robinson and Terry Schuck for their help in sorting out and reviewing slides. This work was supported by the NIH (AG033101, AG17586, AG10124, AG32953), the Wyncote Foundation, and the Koller Family Foundation. VMYL is the John H. Ware, 3rd, Professor of Alzheimer's Disease Research. JQT is the William Maul Measey-Truman G. Schnabel, Jr., Professor of Geriatric Medicine and Gerontology. JB was supported by a grant of the Deutsche Forschungsgemeinschaft DFG (AOBJ586910). JBT is supported by a grant of the Fundación Alfonso Martin Escudero.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Abrahams S, Goldstein LH, Suckling J, Ng V, Simmons A, Chitnis X, Atkins L, Williams SC, Leigh PN. Frontotemporal white matter changes in amyotrophic lateral sclerosis. J Neural. 2005;252:321–331. doi: 10.1007/s00415-005-0646-x. [DOI] [PubMed] [Google Scholar]
- 2.Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- 3.Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. Phosphorated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30:170–181. doi: 10.1111/j.1440-1789.2009.01089.x. [DOI] [PubMed] [Google Scholar]
- 4.Avants B, Khan A, McCluskey L, Elman L, Grossman M. Longitudinal cortical atrophy in amyotrophic lateral sclerosis with frontotemporal dementia. Arch Neurol. 2009;66:138–139. doi: 10.1001/archneurol.2008.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci USA. 2008;105:3785–3789. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bellucci A, Bugiani O, Ghetti B, Spillantini MG. Presence of reactive microglia and neuroinflammatory mediators in a case of frontotemporal dementia with P301S mutation. Neurodegener Dis. 2011;8:221–229. doi: 10.1159/000322228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 9.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 10.Canning SJ, Leach L, Stuss D, Ngo L, Black SE. Diagnostic utility of abbreviated fluency measures in Alzheimer disease and vascular dementia. Neurology. 2004;62:556–562. doi: 10.1212/wnl.62.4.556. [DOI] [PubMed] [Google Scholar]
- 11.Cedarbaum JM, Stambler N, Malta E, Fuller C, Hilt D, Thurmond B, Nakanishi A. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III) J Neurol Sci. 1999;169:13–21. doi: 10.1016/s0022-510x(99)00210-5. [DOI] [PubMed] [Google Scholar]
- 12.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 13.Crum RM, Anthony JC, Bassett SS, Folstein MF. Population-based norms for the Mini-Mental State Examination by age and educational level. JAMA. 1993;269:2386–2391. [PubMed] [Google Scholar]
- 14.Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113:521–533. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- 15.Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD. Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol. 2002;52:771–778. doi: 10.1002/ana.10374. [DOI] [PubMed] [Google Scholar]
- 16.Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol. 1993;50:30–36. doi: 10.1001/archneur.1993.00540010026013. [DOI] [PubMed] [Google Scholar]
- 17.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 18.Garraux G, Salmon E, Degueldre C, Lemaire C, Franck G. Medial temporal lobe metabolic impairment in dementia associated with motor neuron disease. J Neurol Sci. 1999;168:145–150. doi: 10.1016/s0022-510x(99)00188-4. [DOI] [PubMed] [Google Scholar]
- 19.Geser F, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis and frontotemporal lobar degeneration: a spectrum of TDP-43 proteinopathies. Neuropathology. 2010;30:103–112. doi: 10.1111/j.1440-1789.2009.01091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geser F, Martinez-Lage M, Robinson J, Uryu K, Neumann M, Brandmeir NJ, Xie SX, Kwong LK, Elman L, McCluskey L, Clark CM, Malunda J, Miller BL, Zimmerman EA, Qian J, Van Deerlin V, Grossman M, Lee VM, Trojanowski JQ. Clinical and pathological continuum of multisystem TDP-43 proteinopathies. Arch Neurol. 2009;66:180–189. doi: 10.1001/archneurol.2008.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geser F, Robinson JL, Malunda JA, Xie SX, Clark CM, Kwong LK, Moberg PJ, Moore EM, Van Deerlin VM, Lee VM, Arnold SE, Trojanowski JQ. Pathological 43-kDa transactivation response DNA-binding protein in older adults with and without severe mental illness. Arch Neurol. 2010;67:1238–1250. doi: 10.1001/archneurol.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregory CA, Orrell M, Sahakian B, Hodges JR. Can frontotemporal dementia and Alzheimer's disease be differentiated using a brief battery of tests? Int J Geriatr Psychiatry. 1997;12:375–383. doi: 10.1002/(sici)1099-1166(199703)12:3<375::aid-gps518>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 23.Hall ED, Oostveen JA, Gumey ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 24.Hamilton RL, Bowser R. Alzheimer disease pathology in amyotrophic lateral sclerosis. Acta Neuropathol. 2004;107:515–522. doi: 10.1007/s00401-004-0843-1. [DOI] [PubMed] [Google Scholar]
- 25.Hu WT, Seelaar H, Josephs KA, Knopman DS, Boeve BF, Sorenson EJ, McCluskey L, Elman L, Schelhaas HJ, Parisi JE, Kuesters B, Lee VM, Trojanowski JQ, Petersen RC, van Swieten JC, Grossman M. Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neural. 2009;66:1359–1364. doi: 10.1001/archneurol.2009.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudson AJ. Amyotrophic lateral sclerosis and its association with dementia, parkinsonism and other neurological disorders: a review. Brain. 1981;104:217–247. doi: 10.1093/brain/104.2.217. [DOI] [PubMed] [Google Scholar]
- 27.Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neural. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- 28.Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, Neumann M, Trojanowski JQ, Lee VM. Expression of TDP-43 C-terminal fragments in vitro recapitulates pathological features of TDP-43 proteinopathies. J Biol Chem. 2009;284:8516–8524. doi: 10.1074/jbc.M809462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iguchi Y, Katsuno M, Niwa J, Yamada S, Sone J, Waza M, Adachi H, Tanaka F, Nagata K, Arimura N, Watanabe T, Kaibuchi K, Sobue G. TDP-43 depletion induces neuronal cell damage through dysregulation of Rho family GTPases. J Biol Chem. 2009;284:22059–22066. doi: 10.1074/jbc.M109.012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Ibal. Glia. 2002;40:164–174. doi: 10.1002/glia.10149. [DOI] [PubMed] [Google Scholar]
- 31.Ince PG, Highley JR, Kirby J, Wharton SB, Takahashi H, Strong MJ, Shaw PJ. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol. 2011;122:657–671. doi: 10.1007/s00401-011-0913-0. [DOI] [PubMed] [Google Scholar]
- 32.Kertesz A, Davidson W, Fox H. Frontal behavioral inventory: diagnostic criteria for frontal lobe dementia. Can J Neurol Sci. 1997;24:29–36. doi: 10.1017/s0317167100021053. [DOI] [PubMed] [Google Scholar]
- 33.Kieran MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 34.Kim SH, Shi Y, Hanson KA, Williams LM, Sakasai R, Bowler MJ, Tibbetts RS. Potentiation of amyotrophic lateral sclerosis (ALS)-associatcd TDP-43 aggregation by the protea-some-targeting factor, ubiquilin 1. J Biol Chem. 2009;284:8083–8092. doi: 10.1074/jbc.M808064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kriz J, Nguyen MD, Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–278. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- 36.Lezak M. Neuropsychological assessment. Oxford University Press; New York: 1983. [Google Scholar]
- 37.Libon DJ, Massimo L, Moore P, Coslett HB, Chatterjee A, Aguirre GK, Rice A, Vesely L, Grossman M. Screening for frontotemporal dementias and Alzheimer's disease with the Philadelphia Brief Assessment of Cognition: a preliminary analysis. Dement Geriatr Cogn Disord. 2007;24:441–447. doi: 10.1159/000110577. [DOI] [PubMed] [Google Scholar]
- 38.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60:1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- 39.Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, Eisen A, McClusky L, Kretzschmar HA, Monoranu CM, Highley JR, Kirby J, Siddique T, Shaw PJ, Lee VM, Trojanowski JQ. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 40.McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- 41.Miller BL, Cummings JL, Villanueva-Meyer J, Boone K, Mehringer CM, Lesser IM, Mena I. Frontal lobe degeneration: clinical, neuropsychological, and SPECT characteristics. Neurology. 1991;41:1374–1382. doi: 10.1212/wnl.41.9.1374. [DOI] [PubMed] [Google Scholar]
- 42.Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The consortium to establish a registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 44.Morita K, Kaiya H, Ikceda T, Namba M. Presenile dementia combined with amyotrophy: a review of 34 Japanese cases. Arch Gerontol Geriatr. 1987;6:263–277. doi: 10.1016/0167-4943(87)90026-4. [DOI] [PubMed] [Google Scholar]
- 45.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 46.Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, Forman MS, Troost D, Kretzschmar HA, Trojanowski JQ, Lee VM. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117:137–149. doi: 10.1007/s00401-008-0477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 48.Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobagyi T, Shaw CE, Rogelj B. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763–1771. doi: 10.1093/brain/awq111. [DOI] [PubMed] [Google Scholar]
- 49.Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M. Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet. 2009;18:3353–3364. doi: 10.1093/hmg/ddp275. [DOI] [PubMed] [Google Scholar]
- 50.Ohsawa K, Imai Y, Kanazawa H, Sasaki Y, Kohsaka S. Involvement of Ibal in membrane ruffling and phagocytosis of macrophages/microglia. J Cell Sci. 2000;113:3073–3084. doi: 10.1242/jcs.113.17.3073. [DOI] [PubMed] [Google Scholar]
- 51.Olney RK, Murphy J, Forshew D, Garwood E, Miller BL, Langmore S, Kohn MA, Lomen-Hoerth C. The effects of executive and behavioral dysfunction on the course of ALS. Neurology. 2005;65:1774–1777. doi: 10.1212/01.wnl.0000188759.87240.8b. [DOI] [PubMed] [Google Scholar]
- 52.Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6:994–1003. doi: 10.1016/S1474-4422(07)70265-X. [DOI] [PubMed] [Google Scholar]
- 53.Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology. 1999;53:1665–1670. doi: 10.1212/wnl.53.8.1665. [DOI] [PubMed] [Google Scholar]
- 54.Takeda T, Uchihara T, Arai N, Mizutani T, Iwata M. Progression of hippocampal degeneration in amyotrophic lateral sclerosis with or without memory impairment: distinction from Alzheimer disease. Acta Neuropathol. 2009;117:35–44. doi: 10.1007/s00401-008-0447-2. [DOI] [PubMed] [Google Scholar]
- 55.Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 56.Thal DR, Rub U, Schultz C, Sassin I, Ghebremedhin E, Del Tredici K, Braak E, Braak H. Sequence of Abeta-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59:733–748. doi: 10.1093/jnen/59.8.733. [DOI] [PubMed] [Google Scholar]
- 57.Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banali RB. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PKI1195 positron emission tomography study. Neurobiol Dis. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, Miller BL, Kretzschmar HA, Lee VM, Trojanowski JQ, Neumann M. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neural. 2008;67:555–564. doi: 10.1097/NEN.0b013e31817713b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Van Deerlin VM, Trojanowski JQ, Chen-Plotkin AS. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121:373–380. doi: 10.1007/s00401-010-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wikstrom J, Paetau A, Palo J, Sulkava R, Haltia M. Classic amyotrophic lateral sclerosis with dementia. Arch Neurol. 1982;39:681–683. doi: 10.1001/archneur.1982.00510230007002. [DOI] [PubMed] [Google Scholar]
- 61.Wilson CM, Grace GM, Munoz DC, He BP, Strong MJ. Cognitive impairment in sporadic ALS: a pathologic continuum underlying a multisystem disorder. Neurology. 2001;57:651–657. doi: 10.1212/wnl.57.4.651. [DOI] [PubMed] [Google Scholar]
- 62.Yoshida M. Amyotrophic lateral sclerosis with dementia: the clinicopathological spectrum. Neuropathology. 2004;24:87–102. doi: 10.1111/j.1440-1789.2003.00544.x. [DOI] [PubMed] [Google Scholar]
- 63.Yoshida M, Murakami N, Hashizume Y, Takahashi A. A clinicopathological study on 13 cases of motor neuron disease with dementia. Rinsho Shinkeigaku. 1992;32:1193–1202. [PubMed] [Google Scholar]
- 64.Zhu X, Hill RA, Nishiyama A. NG2 cells generate oligodendrocytes and gray matter astrocytes in the spinal cord. Neuron Glia Biol. 2008;4:19–26. doi: 10.1017/S1740925X09000015. [DOI] [PubMed] [Google Scholar]