

Abstract
We characterized in this study the pharmacokinetics and antitumor efficacy of histidine-lysine (HK):siRNA nanoplexes modified with PEG and a cyclic RGD (cRGD) ligand targeting αvβ3 and αvβ5 integrins. With noninvasive imaging, systemically administered surface-modified HK:siRNA nanoplexes showed nearly 4-fold greater blood levels, 40% higher accumulation in tumor tissue, and 60% lower luciferase activity than unmodified HK:siRNA nanoplexes. We then determined whether the surface-modified HK:siRNA nanoplex carrier was more effective in reducing MDA-MB-435 tumor growth with an siRNA targeting Raf-1. Repeated systemic administration of the selected surface modified HK:siRNA nanoplexes targeting Raf-1 showed 35% greater inhibition of tumor growth than unmodified HK:siRNA nanoplexes and 60% greater inhibition of tumor growth than untreated mice. The improved blood pharmacokinetic results and tumor localization observed with the integrin-targeting surface modification of HK:siRNA nanoplexes correlated with greater tumor growth inhibition. This investigation reveals that through control of targeting ligand surface display in association with a steric PEG layer, modified HK: siRNA nanoplexes show promise to advance RNAi therapeutics in oncology and potentially other critical diseases.
Introduction
RNAi silencing of oncogenes via small dsRNA (siRNA) has a great potential for cancer treatment, but is limited by several substantial obstacles. For example, necessary advances include avoiding siRNA degradation by nucleases in blood and tissues, minimizing side effects of the siRNA or delivery system, transport of the highly negative charged siRNA to target tissue and then across cellular membranes, and shifting intracellular trafficking away from lysosomal degradation to endosomal lysis. After considerable exploration of a wide range of approaches, including chemical protective analogues alone, antibody-carrier chimera, and cell-penetrating peptide conjugates, most efforts to achieve these milestones are now focused on developing target-specific and biologically metastable nanoparticle carriers for siRNA oligonucleotides.1−4
Whereas there have been no FDA-approved siRNA carriers for systemic treatment, a few have advanced to clinical trials (clinicaltrials.gov).5−8 These early clinical trials with different carriers encourage testing other preclinical siRNA carriers that have therapeutic efficacy in animal models, including synthetic polymers, peptides, siRNA aptamers, neutral and cationic liposomes (see review9). To investigate siRNA-mediated gene silencing in tumor cells, our lab has synthesized a number of branched histidine-lysine (HK)-rich peptides.10 While lysines presumably bind and protect siRNA via electrostatic interaction, pH-sensitive histidines play an important role in buffering the acidic endosomes and may interact with the endosomal membrane, aiding the endosomal escape of siRNA. Many other investigators have reported on the use of histidine-containing peptides for DNA plasmid or siRNA delivery.11−14 By varying the amino acid sequence and number of branches, a four-branched polymer, H3K(+H)4b, with repeating patterns primarily of −HHHK–, was found to be an effective carrier of siRNA with low toxicity in vivo.15
Although in vitro and in vivo tumor growth inhibition indicative of therapeutic efficacy has been achieved with nonligand targeted HK nanoplexes, a more stable and targeted delivery system is thought necessary to improve the therapeutic index and range of siRNA gene targets. To improve the stability of the siRNA nanoparticle, surface coatings with hydrophilic polymers such as polyethylene glycol (PEG) or carbohydrates such as hyaluronic acid and oligo-maltose have been studied.16,17 Such modifications shield the nanoplex surface to prevent protein binding, reduce reticuloendothelial uptake, and/or extend the circulation time in vivo. Nevertheless, such hydrophilic coated nanoparticles can also exhibit a decrease in cellular uptake due to steric hindrance of the surface layer.
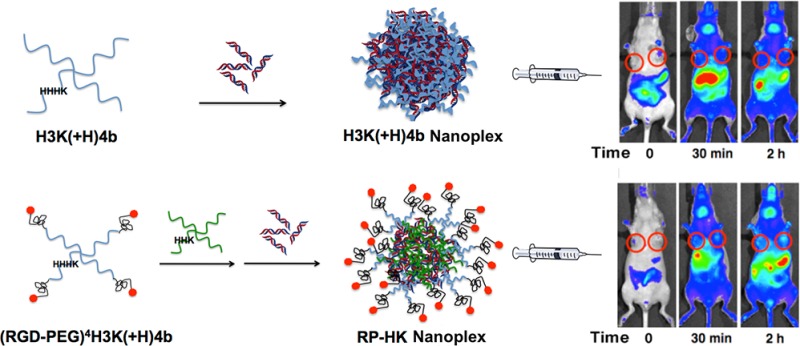
To circumvent steric hindrance of decreased cellular uptake, ligands for receptors can be conjugated to the nanoplex and displayed on its surface with improvement in a wide array of delivery systems.16,18−21 In the present study, the cRGD-PEG conjugate was attached to the N-terminal lysine on each branch of H3K(+H)4b to target selectively αvβ3 and αvβ5 integrins that are overexpressed on the cell surfaces of MDA-MB-435 tumor xenografts (Figure 1).
Figure 1.
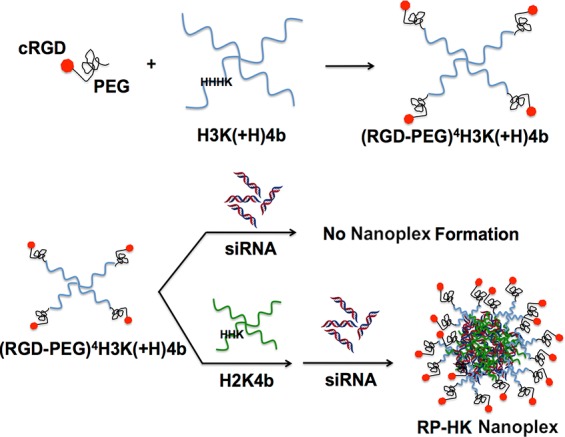
Schematic illustration of (RGD-PEG) modification patterns and HK siRNA nanoplex formation. Each of four N-terminal branches was conjugated to a single (RGD-PEG). A two-step mixing was utilized to prepare modified nanoplexes: (RGD-PEG)4H3K(+H)4b was first added to a helper peptide, H2K4b. The resulting mixture was then mixed with siRNA at room temperature for 40 min to form HK siRNA nanoplexes.
We previously determined the optimal cyclic RGD and PEGylation patterns on H3K(+H)4b (Figure 1) by systemically administered siRNA targeting the luciferase gene in a bioluminescence mouse model. To increase binding with siRNA and form stable siRNA nanoplexes, an unmodified H2K4b (H2K indicating that the predominant repeating groups −HHK−) was combined with ligand-PEG-modified HK peptide, (RGD-PEG)4H3K(+H)4b, and siRNA, resulting in HK:siRNA nanoplexes that provided maximal gene silencing without cytokine induction in vivo.22 In the current study, we further investigate the cRGD-targeted HK:siRNA nanoplexes in terms of pharmacokinetics, biodistribution, and antitumor efficacy and with comparison to unmodified HK:siRNA nanoplexes. The cRGD-PEG modification of HK:siRNA nanoplexes extended the half-life in the blood and showed greater tumor accumulation, resulting in reduction of luciferase activity by more than 70%. Consistent with these effects on pharmacokinetics and biodistribution, the ligand-targeted HK:siRNA nanoparticle showed enhanced antitumor efficacy with an siRNA targeting the Raf-1 oncogene, showing reduced target gene mRNA and protein expression and suppressing growth of MDA-MB-435 subcutaneous xenograft tumors in mice.
Materials and Methods
Animals
Female athymic mice (4–8 weeks old, body weight ∼20 g) were purchased from NCI Frederick. All experiments were performed in accordance with regulations by the Institutional Animal Care and Use Committee of the University of Maryland School of Medicine.
Cell Line
A human malignant cell line MDA-MB-435, stably expressing Firefly luciferase (stable transformation by electroporation with the linearized pCpG-Luc plasmid), was cultured and maintained in Dulbecco’s minimal essential medium (DMEM) containing 10% fetal calf serum (FCS) and 20 mM glutamine. The pCpG-Luc plasmid was made by ligating the luciferase gene (digested from pMOD-Luc plasmid, InvivoGen, San Diego, CA) into the multiple cloning site of pCpG-mcs (InvivoGen).
Peptides
Branched peptides, H2K4b and H3K(+H)4b, with predominant repeating groups, −HHK– and −HHHK–, respectively, were synthesized on a Rainin Voyager synthesizer (PTI, Tucson, AZ) by the biopolymer core facility at the University of Maryland, as previously described.23 Each of four terminal branches emanates from the three-lysine core: for H3K(+H)4b, the branch sequence is KHHHKHHHKHHHHKHHHK; for H2K4b, the branch sequence is KHKHHKHHKHHKHHKH-HKHK. For modified H3K(+H)4b, cyclic (c)RGD and PEG (3.4 kD) were conjugated to each N-terminal branch as described previously, resulting in four (RGD-PEG) conjugates per HK peptide, (RGD-PEG)4H3K(+H)4b.22
siRNA
Sequences of siRNA targeting luciferase (siLuc) were as follows: sense, 5′-CUG-CAC- AAG-GCC-AUG-AAG- A-dTdT-3′; antisense, 5′-UCU-UCA-UGG-CCU-UGU-GCA-G-dTdT-3′; targeting, 5′-CTG-CAC-AAG-GCC-ATG-AAG-A-3′. siLuc was also used as the control siRNA for studying inhibition of tumor growth. Raf-1 siRNA duplex (siRaf-1) was as follows: sense, 5′-GCA-UCA- GAU-GAU-GGC-AAA- C-dTdT-3′; antisense, 5′-GUU-UGC-CAU-CAU-CUG-AUG-CdTdT-3′; targeting, 5′-GCA-TCA- GAT-GAT-GGC-AAA- C-3′ (Dharmacon, Lafayette, CO.). Luciferase siRNA was conjugated to Alexa Fluor 546 (Dharmacon) or a near-infrared (NIR) dye, Cy5.5 (Sigma-Aldrich, St. Louis, MO) on the 5′ end of the sense strand. Each siRNA was maintained in siRNA buffer (Dharmacon) for 30 min at RT for annealing the duplex.
Preparation and Biophysical Properties of HK Nanoplexes
The ternary (RGD-PEG)4H3K(+H)4b/H2K4b/siRNA and H3K(+H)4b nanoplexes were prepared as previously described for experiments in vivo.22 As defined by gel retardation assays and the biophysical characteristics, the optimal N/P ratios for H3K(+H)4b/siRNA and (RGD-PEG)4H3K(+H)4b/H2K4b/siRNA were about 2.2:1 (w/w, N/P ratio 2.7/1) and 4.0:0.8:1 (w/w/w, N/N/P ratio 2.7/1/1) used in this study.
For these ratios of HK:siRNA nanoplexes, the size and zeta potential were determined with the Zetasizer (Malvern, Westborough, Mass.) prior to their injection. The size is reported as the average size obtained from unimodal analysis of dynamic light scattering of the particles at a 90° angle carried out with software provided by the instrument manufacturer. Each particle size and zeta potential data point represents the mean ± SD of four measurements.
Fluorescence Quenching Assay
The relative binding affinity between various HK polymers and siRNA was studied with Alexa 546-labeled siRNA (λex = 550 nm, λem = 570 nm, Dharmacon, Lafayette, CO). Fluorescence-labeled siRNA (0.14 μM, 100 μL) in phosphate buffer (10 mM, pH 7) was added to a 96-well plate, and changes in fluorescence were then measured by adding 0.033 μM increments of H3K(+H)4b, H2K4b, (RGD-PEG)4H3K(+H)4b, or (RGD-PEG)4H3K(+H)4b/H2K4b. After incubation of HK and siRNA at room temperature for 20 min, the fluorescence intensity was measured by a Wallac Victor 3 1420 multilabel counter (Perkin-Elmer, Turku, Finland), and the relative fluorescence intensity was normalized by subtracting background levels.
Preparation of HK:siRNA Nanoplexes for Treatment of Tumor Xenografts
HK:siRNA nanoplexes were prepared as previously described.22 Briefly, for unmodified nanoplexes, H3K(+H)4b peptide (88 μg in 150 μL of water) was rapidly added to the siRNA (40 μg in 150 μL water) and mixed with a Vortex mixer. For the modified HK combination, the (RGD-PEG)4H3K(+H)4b peptide (160 μg in 150 μL of water) or PEG4H3K(+H)4b (152 μg in 150 μL of water) was mixed with unmodified H2K4b (32 μg in 75 μL of water) and maintained for 30 min at room temperature before siRNA was added. The nanoplexes were allowed to form for 40 min at room temperature.
Biodistribution, Pharmacokinetics, and Bioluminescence
MDA-MB-435 cells (2 × 106 cells) expressing luciferase were injected into the midclavicular line of female nude mice (NCI Frederick). After tumors grew to about 250 mm3, mice were separated into three siRNA treatment groups: aqueous siLuc, H3K(+H)4b/siLuc (w/w, 2.2:1), and (RGD-PEG)4H3K(+H)4b/H2K4b/siLuc (w/w/w, 4:0.8:1). The aqueous siRNA or HK:siRNA nanoplexes containing 40 μg of siLuc (including 5% (2 μg) of Cy5.5-labeled siLuc) were administered in the tail vein. For biodistribution studies, the mice were imaged by the IVIS-200 optical imaging system (Xenogen Corp., Almeda, CA) at 0, 15, 30, and 60 min, and 2, 4, 6, and 24 h postinjection. Fluorescence emission at the tumor location, given as relative units after normalization of maximal and minimal intensities,24 was measured with regions of interest of equal size at specified time points. Biodistribution imaging and tumor luciferase activity were measured by IVIS-200 before and 48 h after siRNA treatment targeting luciferase.22
Similarly, to determine the effects of PEG and cRGD modifications, the PEG4H3K(+H)4b/H2K4b/siLuc nanoplexes (w/w/w, 3.8:0.8:1) were compared with (RGD-PEG)4H3K(+H)4b/H2K4b and H3K(+H)4b nanoplexes for their ability to silence luciferase in tumor xenografts of about 60 mm3. Tumor imaging with IVIS-200 imaging was done 48 h after injection of the nanoplexes.
For pharmacokinetic experiments, tumor-bearing mice were treated with Cy5.5-labeled Luc siRNA (5% of total Luc siRNA) in complex with different formulations of HK peptides as described above for the biodistribution study. At several time points (2, 5, 10, 15, 25, 45, 60, and 120 min), blood (100 μL) was collected and serum was isolated.25 The serum (30 μL) was then mixed with phosphate-buffered saline (PBS) (20 μL) in a 96-well clear bottom plate, and the fluorescence signal was imaged by the IVIS-200 system. The concentration of the nanoplexes were calculated from a standard curve of fresh aqueous siRNA or nanoplexes.24 As described previously,26,27 the pharmacokinetic analysis of the data was performed using a two-compartment model (f = A·exp(−αt) + B·exp(−βt)) with first-order adsorption and elimination from the central compartment by using Sigma Plot, 11.0. After curve fitting of the data, the PK parameters were calculated based on the expressions given in the Supporting Information To avoid negative values, asymmetric error bars were used in Figures 3 and 4B.
Figure 3.
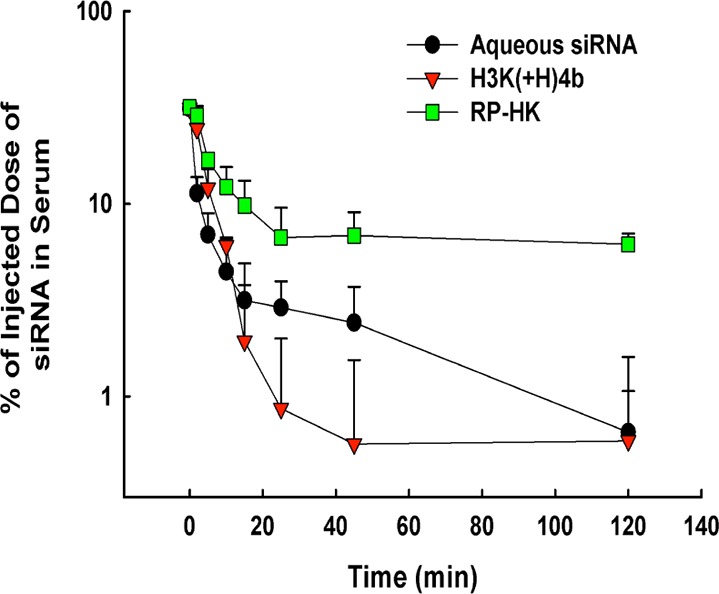
Serum pharmacokinetics of Cy5.5-labeled siRNA and nanoplexes. A semilogarithmic plot of serum concentration versus time for up to 2 h after intravenous injection of siRNA alone or in complex with the unmodified HK or modified RP-HK peptides. The data represent the mean ± SD of %ID/mL (n = 4 per carrier), where % ID/mL is the percentage of injected dose per milliliter of serum.
Figure 4.

Real-time biodistribution of Cy5.5-labeled siRNA and nanoplexes. (A) Representative biodistribution of Cy5.5-labeled siRNA in a mouse, with the siRNA administered as an aqueous solution, unmodified HK nanoplex or RP-HK nanoplex. Times 15, 30, 60, 120 min, and 4, 6, and 24 h are shown. Red circles encircle the tumors. (B) Fluorescence emission localized at the tumor was measured for the different siRNA groups. The data represent the mean ± SD of fluorescence of four determinations for each carrier. *, P < 0.05; **, P < 0.01; RP-HK carrier versus unmodified HK carriers and control groups.
Inhibition of Tumor Growth and Raf-1 Expression
Mice with MDA-MB-435 xenografts (about 25 mm3 in size) were separated into four treatment groups: untreated, H3K(+H)4b/Control siLuc, H3K(+H)4b/siRaf-1, and (RGD-PEG)4H3K(+H)4b/H2K4b/siRaf-1. HK:siRNA nanoplexes with the same w/w ratios as described above were injected intravenously three times a week for a total of six or seven treatments depending on the experiment. Tumor volume was determined before each treatment with skin calipers by using the formula 1/2 × length × width2. Two days after the last injection, the mice were euthanized and total RNA was isolated from excised tumor xenografts using the RNeasy mini kit (Qiagen, Hilden, Germany). Expression of Raf-1 (368 nt) and β-actin (294 nt) mRNA was assessed by reverse transcriptase-polymerase chain reaction (RT-PCR) as described previously.28 RT-PCR products were then loaded onto a 3% agarose and subjected to electrophoresis at a constant voltage of 100 V for 90 min in TBE buffer containing ethidium bromide. The band density was visualized on a UV transilluminator with a wavelength of 365 nm, and the image was digitized and analyzed by UN-SCAN-IT (Silk Scientific, Orem, UT).
Immunohistochemical Detection of Raf-1 and Ki67, and TUNEL Assay
Tumors were fixed in 10% formalin for 24 h and processed as paraffin-embedded tissue sections. Immunostaining was performed according to the manufacturer’s protocol (Vector, Versatile ABC, Burlingame, CA). Briefly, tumor sections were deparaffinized in xylene and rehydrated in graded ethanol. Antigens were retrieved by maintaining tissue in 10 mM citrate buffer, pH 6.0, at boiling point for 40 min. Endogenous peroxidase and nonspecific binding were blocked by 3% H2O2 for 10 min and 5% goat serum for 30 min, respectively. For detection of Raf-1 and Ki67, tissue sections were incubated with rabbit antihuman polyclonal antibody (Raf-1: 1:50 dilution, Ab-259, Genscript, Piscataway, NJ, USA; Ki67: 1:50 dilution, Chemicon, Ramona, CA, USA) at 4 °C overnight, and secondary horseradish peroxidase-labeled antibody was added for 30 min. The chromogen diaminobenzidine (DAB) was applied for 5 min to permit color development. Finally tissue was dehydrated and mounted with glass coverslips. Four randomly picked brightfield images were converted to normalized blue images29 allowing automatic classification (quantification) of positive DAB staining. The TUNEL assay was performed on paraffin-embedded tissue according to the manufacturer’s instruction (FragEL DNA Fragmentation Detection Kit, Calbiochem, San Diego, CA). The tissue sections were incubated with deoxynucleotidyl terminal transferase for 90 min after specimen permeabilization and endogenous peroxidases inactivation. The labeling reaction was then terminated, and tumor sections were stained with DAB substrate.
Results
Nanoplexes Formed with Modified and Unmodified HK Peptides
Relative binding affinities of HK peptides to siRNA were determined by titration of Alexa 546-labeled siRNA with different peptides. When the peptide binds siRNA, fluorescence intensity is attenuated due to nanoplex formation. These results of the quenching studies corroborated a previous study that used a gel retardation assay,22 confirming that a combination of unmodified H2K4b and (RGD-PEG)4H3K(+H)4b was required to form the ternary nanoplexes (Figures 1 and 2). The unmodified HK peptides, H2K4b and H3K(+H)4b, had high affinity for siRNA to form nanoplexes (as evidenced by decreased fluorescence at N/P ∼ 2), the modified (RGD-PEG)4H3K(+H)4b peptide alone showed negligible binding to siRNA. A combination of unmodified H2K4b peptide, with a greater charge density, and the RGD-PEG modified H3K(+H)4b peptide showed quenching of the siRNA that was intermediate between the modified alone and unmodified peptides, suggesting that both peptides are present in the nanoplex (Figure 2). The size and zeta potential of the unmodified or combined (RGD-PEG)4H3K(+H)4b/H2K4b siRNA nanoplexes used for in vivo experiments are shown in Table 1. To simplify the nomenclature, the combination of (RGD-PEG)4H3K(+H)4b and H2K4b will be designated RP-HK.
Figure 2.
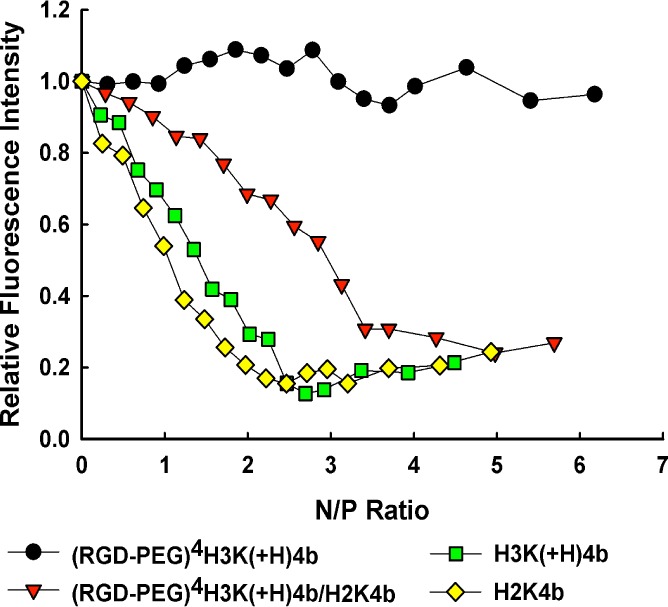
Relative binding affinity of HK peptide formulations with siRNA. Relative binding affinity of four HK peptide formulations (H3K(+H)4b; H2K4b; (RGD-PEG)4H3K(+H)4b; (RGD-PEG)4H3K(+H)4b/H2K4b (RP-HK)) for Alexa 546-labeled siRNA was studied in 10 mM phosphate buffer, pH 7.0. Relative fluorescence intensity is the percentage of unbound siRNA after subtraction of background fluorescence. See Results section for further details.
Table 1. Size and Zeta Potential of HK siRNA Nanoplexes.
| peptide | size (nm)a | zeta potential (mV)a |
|---|---|---|
| H3K(+H)4b | 166.7 ± 24.8b | 41.5 ± 5.5 |
| PEG-HK | 68.6 ± 11.3 | 17.3 ± 1.3 |
| RP-HK | 82.4 ± 5.4 | 22.7 ± 4 |
The size and zeta potential of nanoplexes were measured before their systemic administration to mice.
Each data point represents the mean ± SD of four measurements.
PEGylation Prolonged the Circulation Time of Nanoplexes
The concentration of nanoplexes in blood was determined by measuring NIR fluorescence from Cy5.5-labeled siRNA, alone or incorporated within the nanoplexes (Figure 3). The advantage of the NIR-based methodology is its high sensitivity and low background, thereby requiring only small amounts of blood to detect the Cy5.5 signal. At indicated time points up to 2 h, blood was drawn, and the amount of siRNA in the serum was measured. Fifteen minutes after injection, the concentrations of the siRNA, alone or as nanoplexes, decreased to less than 10% of the injected dose. As shown in Figure 3, aqueous siRNA and unmodified HK:siRNA nanoplexes were eliminated substantially faster than the modified RP-HK:siRNA nanoplexes. The key pharmacokinetic parameters were determined by fitting the data with a bicompartment model (Table 2).26 The sterically stabilized and ligand-targeting nanoplexes had a 3-fold greater terminal half-life (t1/2β) and mean residence time (MRT), and a 4-fold increase in the area under the curve compared to unmodified HK nanoplexes. In addition, the surface modification of the nanoplexes markedly reduced elimination clearance (CL) by 10-fold. Thus, all pharmacokinetic parameters indicated that surface modification of the HK:siRNA nanoplexes greatly increased its residence time in the bloodstream, which suggests that the modified nanoplexes will have greater tumor uptake and siRNA silencing of the targeted oncogene.
Table 2. Pharmacokinetic Parameters for siRNA Formulations.
| t1/2βa (min) | CLb(mL/min/kg) | AUC2hc (μg/mL × min) | MRTd (min) | |
|---|---|---|---|---|
| aqueous siRNA | 27.7 | 18.50 | 126.3 ± 52.7 | 41.4 |
| H3K(+H)4b:siRNA | 86.6 | 16.62 | 98.0 ± 58.6 | 129.5 |
| RP-HK:siRNA | 256.7 | 1.57 | 385.0 ± 92.4 | 374.7 |
t1/2β, terminal half-life.
CL, clearance.
AUC2h, area under curve from 0 to 2 h.
MRT, mean residence time.
Tumor Localization Improved by Ligand Targeted HK:siRNA Nanoplexes
The tumor-specific targeting efficacy was assessed by NIR fluorescence imaging of Cy5.5-conjugated siRNA with a noninvasive imaging system, IVIS-200, before and at specified times after injection (Figure 4A). Autofluorescence was observed in the abdomen, but this did not interfere with measurement of the nanoplex levels within tumors. As soon as 15 min after injection, the modified RP-HK:siRNA nanoplexes gave higher fluorescence in tumor tissue than that of the unmodified HK:siRNA nanoplexes. By contrast, administration of aqueous siRNA resulted in minimal tumor fluorescence, indicative of negligible siRNA uptake. The amount of targeted RP-HK:siRNA nanoplexes within tumors was quantified by measuring intratumoral fluorescence (Figure 4B). While unmodified HK:siRNA nanoplexes rapidly reached maximal accumulation within the tumor 30 min after injection, the modified RP-HK:siRNA nanoplexes required 60 to 120 min after injection to achieve maximal levels. Compared to accumulation of unmodified HK nanoplexes within tumors, accumulation of modified RP-HK nanoplexes was 40% higher (P < 0.01) at 60 min. Although the nanoplex distribution in the major organs was difficult to assess due to autofluorescence in the abdominal region, significant accumulation of fluorescence with the aqueous siRNA-treated group occurred earlier in the spleen compared to the HK:siRNA nanoplex groups. Not surprisingly, fluorescence in aqueous siRNA-treated mice was observed in the bladder with the first image (15 min), whereas siRNA of the nanoplexes was not detected at a significant level in the bladder until 2 h after injection. There was little difference in organ fluorescence accumulation between the modified RP- and unmodified HK nanoplexes except that accumulation in the liver occurred earlier with unmodified nanoplexes.
Tumor Luciferase Silencing Was Consistent with PK and Biodistribution
The correlation between PK/biodistribution and target gene silencing of the different nanoplexes was determined with the same group of mice, which had tumors expressing luciferase. Luciferase activity was measured by the IVIS imaging system before and 48 h after treatment (Figure 5). Compared to aqueous siLuc, the modified RP-HK:siLuc nanoplexes inhibited luciferase expression by about 70%, whereas the unmodified HK: siLuc nanoplexes only inhibited luciferase activity by about 10%. These results with the labeled siRNA were consistent with our previous results with unlabeled siRNA.22 Thus, the labeled siRNA apparently does not interfere with the functional characteristics or activity of the HK nanoplexes in silencing its target. Consequently, down-regulation of luciferase activity correlated with increased blood circulation time and tumor tissue accumulation of nanoplexes.
Figure 5.
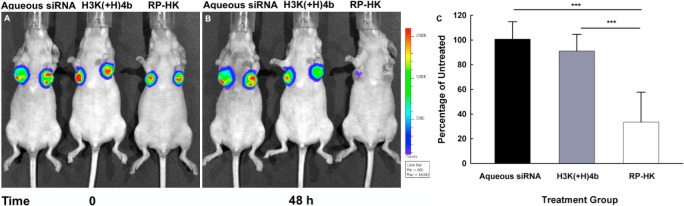
Bioluminescence assay for silencing of luciferase expression. The representative mice were from the same treatment groups used for the biodistribution study. Panels A and B show tumor bioluminescence images of representative mice taken before and 48 h after treatment, respectively. The treatment groups in A and B were as follows (from the left to right): aqueous siRNA, H3K(+H)4b, and RP-HK nanoplexes, respectively. (C) The percent silencing of luciferase activity by different treatment groups compared to the untreated control group represents the mean ± SD of fluorescence of four determinations. ***, P < 0.001.
We were particularly interested in determining the contribution of the cRGD targeting ligand to the tumor-tissue gene silencing by RP-HK nanoplexes. Consequently, the ability of RP-HK nanoplexes to reduce the luciferase activity of the tumor was compared to HK nanoplexes with the same amount of PEGylation but with the cRGD absent (Figure 6). The PEGylated alone HK (PEG-HK) nanoplexes silenced luciferase activity within tumor xenografts more effectively than unmodified HK but less effectively than RP-HK nanoplexes. Moreover, the PEG and cRGD appeared to have approximate additive contributions. The PEG-HK:siLuc nanoplexes were 60% more effective than the unmodified HK:siLuc nanoplexes (P < 0.05), whereas the RP-HK:siLuc nanoplexes were 50% more effective than PEG-HK:siLuc nanoplexes (P < 0.01).
Figure 6.

PEG and RGD modifications of HK enhance gene silencing in tumor xenografts. Luciferase siRNA nanoplexes with unmodified, PEG or RGD-PEG (RP) modified H3K(+H)4b were systemically administered to tumor xenografts bearing mice. Before and 48 h after treatment, luciferase activity was determined by an IVIS 200 system. The data represent the mean ± SD of luciferase of four determinations for each carrier. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Inhibition of Tumor Tissue Gene Expression and Growth by Targeting Raf-1
In addition to silencing the luciferase marker, we evaluated the ability of different carriers to deliver siRNA targeting the Raf-1 oncogene in vivo. We examined inhibition of MDA-MB-435 tumor growth by modified or unmodified HK siRNA nanoplexes with siRaf-1. Six systemic injections were given via tail vein, three times a week for 2 weeks. As early as the second injection, the modified RP-HK:siRaf-1 nanoplexes showed 30% greater reduction in tumor size compared to the untreated mice (P < 0.01) (Figure 7A). In contrast, there was no statistical difference in tumor growth for treatment with the unmodified HK:siRaf-1 nanoplex compared to the untreated groups. After the last injection, the modified RP-HK nanoplexes reduced tumor size 35% more effectively than the unmodified HK nanoplexes (P < 0.01) and nearly 60% more than tumors of untreated mice (P < 0.01). Tumor volumes of mice treated with the modified RP-HK carrier in complex with a negative control siLuc were similar to those of untreated mice.
Figure 7.
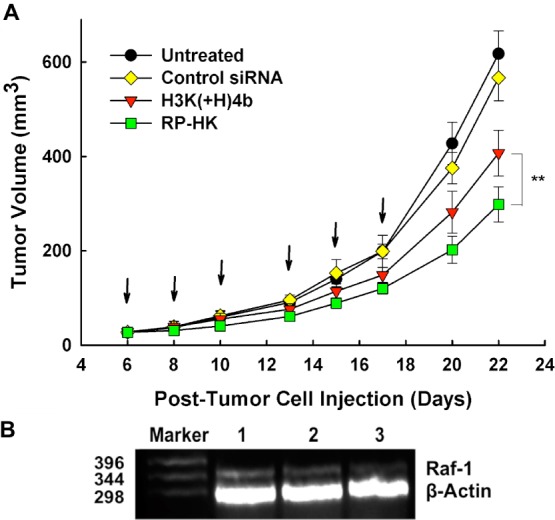
MDA-MB-435 tumor growth and Raf-1 mRNA inhibition. (A) MDA-MB-435 tumor-bearing mice were separated into four groups when tumor size was about 25 mm3: untreated, H3K(+H)4b/siLuc control, H3K(+H)4b/Raf-1 siRNA, and RP-HK/Raf-1 siRNA. Treatment was given three times a week for 2 weeks (arrows). Data for each time point represent the mean ± SD of tumor volume of five determinations for each treatment group. **, P < 0.01. (B) Raf-1 and β-actin mRNA expression was determined by RT-PCR analysis of RNA isolated from tumor tissue at day 22. Lanes 1–3 represents untreated, H3K(+H)4b/Raf-1 siRNA and RP-HK/Raf-1 siRNA respectively.
To evaluate whether reduction in tumor growth rate was dependent on reduction in the expression of the Raf-1 oncogene, we determined the Raf-1 mRNA in the various treatment groups by RT-PCR. Compared to the untreated group (Figure 7B), the modified HK nanoplexes down-regulated Raf-1 mRNA expression by 90% (lane 3), whereas the unmodified treatment group decreased the RNA by 60% (lane 2). The correlation between Raf-1 mRNA expression and luciferase activity clearly indicated that the target gene silencing was significantly enhanced by ligand-targeting modification of the HK peptides.
Targeted HK:siRNA Nanoplexes Induce Immunohistological Changes within Tumor Tissue
Down-regulation of Raf-1 and its downstream effects in tumor tissue of mice treated systemically with HK:siRaf-1 nanoplexes was evaluated by immunohistochemistry. Tumors from treated mice were sectioned and stained for Raf-1 and Ki67 as well as for apoptosis (with the TUNEL assay) (Figure 8A). Non-necrotic areas of tumor were examined histologically from mice 2 days after the last treatment. DAB-stained cells were identified (by brown staining) and quantified by analysis of four arbitrarily selected normalized blue images.29 In viable areas of tumors, Raf-1 protein was down-regulated by nearly 40% and 80% in the unmodified and modified HK:siRaf-1 nanoplex treatment groups, respectively, compared to untreated groups (Figure 8; P < 0.01, modified vs unmodified or untreated). Notably, reduction of Raf-1 protein with immunostaining was consistent with Raf-1 mRNA suppression determined by RT-PCR. Furthermore, as evidenced by the cell proliferation marker Ki67 (Figure 8), the modified nanoplex treatment reduced cell proliferation more effectively (50%) than did unmodified treatment or untreated (P < 0.01, modified vs unmodified, untreated). The TUNEL assay was used to assess cellular areas of apoptosis (Figure 8). Quantification of staining was not appropriate with TUNEL assay since the apoptosis region was sporadically distributed (Figure 8B). Qualitatively, modified RP-HK:siRaf-1 nanoplexes generally induced larger areas of apoptosis than unmodified HK:siRaf-1 nanoplexes. In conclusion, immunohistological analysis showed greater reduction of Raf-1 protein and cellular proliferation as well as increased apoptosis with the RP-HK treatment, consistent with enhanced antitumor efficacy.
Figure 8.

Histochemical analysis of Raf-1, Ki67, and apoptosis. (A) Scale bars equal 10 μm. (B) Quantification of cells staining positive for Raf-1 and Ki67 with different treatments is shown, expressed as the percentage of untreated control. The data represent the mean ± SD of percentage of four images for each treatment. ***, P < 0.001.
Discussion
The potential of RNAi to provide a substantial advance in therapeutics, especially for cancer treatment, has been limited due to lack of an efficient carrier. Our group has methodically investigated improvements in a family of branched histidine-lysine peptides carriers of siRNA by altering their sequence and number of branches.15 While the H3K(+H)4b peptide was demonstrated to be an effective carrier of siRNA both in vitro and in vivo, modification of H3K(+H)4b with a cRGD targeting ligand, in concert with PEGylation, was found to provide a more effective carrier, using an in vivo bioluminescence assay.22 Although PEGylation and addition of a tumor-specific ligand were expected to increase accumulation of the nanoplexes within the tumor, the mechanisms for the improved carrier were never investigated, nor whether the tumor targeting activity of the RP-HK:siRNA nanoplexes could translate into greater tumor reduction. We established in this study that the half-life in the bloodstream correlates with increased accumulation of the modified HK nanoplexes within the tumor. The improved pharmacokinetics of the modified HK nanoplexes resulted in significantly greater reduction of targeted oncogene expression with a marked decrease in tumor size.
PEGylation can provide tumor targeting by facilitating the enhanced permeability and retention (EPR) effect due to an increased blood circulation half-life.30 We show here that PEGylation alone of HK:siRNA nanoplexes enhances siRNA activity in tumor tissue. The reduction of intratumoral luciferase was further enhanced using targeted RP-HK:siRNA nanoplexes (Figure 6). Thus, it appears that the cRGD and PEG modifications have important and additive contributions. PEG has been used in numerous studies to increase the hydrophilicity of nanoparticles including nanoplexes, minimize clearance by phagocytic cells, and increase the half-life of the nanoparticle in vivo.31 The improved tumor tissue siRNA activity by the PEGylated-only HK:siRNA nanoplexes was likely the result of surface steric stabilization by PEG, which reduced the zeta potential due to an increase in hydrodynamic radius.32 The reduction in zeta potential (Table 1) by nearly 50% for RP-HK:siRNA nanoplexes (23 mV) compared to unmodified HK:siRNA nanoplexes (41 mV) is attributable to PEG, since the zwitterionic cRGD peptide is not known to alter the hydrodynamic surface properties. Reduction of the surface charge with sterically stabilized nanoplexes would be expected to inhibit their nonspecific attachment and internalization by cells, prolong their half-life in the circulation, and importantly enhance their ligand receptor-mediated cellular uptake.33,34 Without steric stabilization, nonselective attachment to cell surfaces by highly positively charged unmodified HK:siRNA nanoplexes may contribute to a larger fractional α clearance and thus slightly lower blood levels (AUC) than that of aqueous siRNA.
It may be possible to increase blood circulation and EPR tumor targeting of HK:siRNA nanoplexes further by conjugation with even higher molecular weight PEG (e.g., 5.1 kDa). For example, Sato and colleagues showed that the molecular weight of PEG as well as the degree of PEGylation significantly influenced the circulation time of polylysine siRNA nanoplexes. By increasing the weight ratio of PEG (to polylysine) from 70% to 90%, the circulatory half-life of nanoplexes in the bloodstream was enhanced by 100-fold.35 Consequently, increasing or modifying the PEGylation of HK:siRNA nanoplexes might further improve the pharmacokinetic properties and therapeutic window by this mechanism.
An important contribution to tumor tissue targeting appears to be provided by the cyclic RGD peptide ligand, attributable to selective binding to the αvβ3 and αvβ5 integrins that are overexpressed and activated on cell surfaces of both MDA-MB-435 cancer cells and tumor tissue neovasculature.19,36 The NIR optical fluorescence biodistribution studies on RP-HK:siRNA demonstrated that ligand-targeted nanoplexes accumulated at higher levels in the tumor tissue than unmodified nanoplexes (Figure 4). With combined cRGD ligand and PEGylation, the RP-HK:siRNA nanoplexes gave the greatest improvement in pharmacology, biodistribution, and tumor tissue targeting siRNA activity, both for a constitutively expressed reporter gene and a therapeutic gene, Raf-1. Perhaps surprisingly, the modified RP-HK:siRNA nanoplexes resulted in an improved pharmacokinetic profile distinct from that observed with a transferrin targeted cyclodextrin-based siRNA nanoplexes. In those studies, the blood circulation half-life of aqueous siRNA and targeted cyclodextrin:siRNA nanoplexes were similar.37 Moreover, accumulation of the targeted cyclodextrin:siRNA nanoplexes occurred in kidneys within 20 min of intravenous administration, whereas targeted RP-HK:siRNA nanoplexes showed a substantial delay: fluorescent siRNA was not observed in the bladder until 2 h after administration. It is unclear why this difference in renal clearance between the two nanoplexes occurred, since the particle size of the targeted RP-HK:siRNA nanoplexes should enable their passing readily through the endothelial fenestrations of the glomerulus. Although several studies, including our results with unmodified HK:siRNA nanoplexes, have demonstrated a rapid decrease in blood levels of polymeric nanoplexes within 20 min of intravenous administration,27,33 the targeted RP-HK:siRNA nanoplexes were detected in the blood for up to 2 h. The prolonged β-phase by which serum levels of RP-HK polyplex nearly levels off between 20 min and 2 hours is intriguing and may in part represent polyplex absorption to serum proteins. However, no reported studies of PEGylated polyplexes have achieved the prolonged blood circulation observed with PEGylated liposomes,38 such as wrapsomes39 in which PEG extended the blood half-life in mice up to 17 h.
Inhibition of the target gene was consistent with pharmacokinetics and distribution in that luciferase expression was markedly down-regulated by the modified nanoplexes. Nevertheless, it was still surprising that a single treatment would give rise to over 70% of gene silencing of luciferase, particularly since many cells expressing luciferase are several layers removed from the vasculature. We corroborated this striking down-regulation of luciferase activity with both xenogen imaging and measurement of the luciferase activity from extracts of tumor xenografts. This silencing effect by RP-HK siRNA polyplexes was nearly identical over a wide range of tumor sizes (50 to 250 mm3) (Figure 5),22 indicating that targeting mechanisms such as EPR remained similar.40 Moreover, the decrease in luciferase activity was homogeneous throughout the tumor, further validating the results. Perhaps more importantly, the reduction of Raf-1 mRNA and protein expression by 90% and 80%, respectively, further confirms the results of luciferase activity. Several other investigators have also found that nonviral carriers of siRNA have unexpectedly high rates of silencing their targets.1,41−43 Taken together, these results strongly suggest that the siRNA can selectively silence their targets in the tumor xenograft very efficiently, but the mechanisms for this high efficiency remain to be elucidated.
This high efficiency of siRNA delivery to subcutaneous xenograft tumors, observed with the RP-HK:siRNA nanoplexes, is in marked contrast to other stabilized and targeted nucleic acid delivery systems. It has been suggested that effective siRNA delivery to tumor xenografts may be aided by membranous exosomal vesicles, which are derived from multivesicular bodies (MVB) and subsequently released from the cell.44,45 The 40 to 100 nm exosomes, which contain specific proteins and RNA including RNA-induced silencing complex (RISC), P body component GW182, mRNA, and microRNA, have been speculated to have an important role in intercellular communication between tumor cells.46,47 Importantly, the exosomal mRNA retains its function after transfer to the recipient cell. Although there is yet no evidence that siRNA entrapped within exosomes can shuttle intercellularly to target mRNA, exosome-mediated gene transfer has been exploited for targeted siRNA delivery across the bloodbrain barrier resulting in 60% gene inhibition.48 Thus, exosome-mediated intercellular communication within tumor cells may have a role in efficient luciferase or Raf-1 knockdown within the tumor. Despite the possibility of a biologically mediated mechanism having a role in the knock-down of the targeted gene, the importance of increasing transfection efficiency of nanoplexes and the underpinning mechanisms that increase this efficiency should not be minimized.
Although tumor growth was inhibited more effectively with the modified HK nanoplexes, reduction in tumor size was not equivalent to Raf-1 gene inhibition; that is, we did not observe a 90% reduction in tumor size corresponding to the 90% knockdown we observed with the Raf-1 gene. Because tumor proliferation and apoptosis are mediated by multiple and complex signal transduction pathways regardless of the oncogene targeted, there may be cross-talk and compensatory responses between pathways that facilitate the development of resistance to therapeutics. For example, Hoeflich and colleagues have indicated that diminished mitogen-activated protein kinase (MEK), downstream of Raf-1, activates PI3K pathway driving tumor development in basal-cell like breast cancers.49 Depending on the malignant cell and its compensatory responses, oncogenes such as survivin can be targeted, which may result in greater tumor inhibition; for example, preliminary data has indicated that siRNA targeting survivin decreased MDA-MB-435 cellular growth significantly more than targeting Raf-1. Regardless of the oncogene targeted, it will be important to block additional signal pathways to suppress tumor growth more effectively. Moreover, there have been a number of studies showing that blocking multiple pathways in malignant tumor cells gives a synergistic reduction in tumor size.50−52 Consequently, future work in our laboratory will focus on enhancing siRNA delivery systems in combination with synergistic inhibitory therapies that target interactive signal pathways.
Conclusions
We investigated the pharmacokinetics, biodistribution, and therapeutic activity of RP-HK:siRNA nanoplexes targeting Raf-1 gene expression in a subcutaneous xenograft tumor model in mice. Consistent with greater silencing of the reporter gene in tumor xenografts, the pharmacokinetics showed that RP-HK:siRNA nanoplexes have an increased half-life in the bloodstream and greater accumulation of siRNA within the tumor. Consequently, translational studies were undertaken with a therapeutic siRNA candidate targeting Raf-1. The results show that targeted HK:siRNA nanoplexes gave a significantly greater reduction of the targeted tumor tissue gene expression, at both mRNA and protein levels. This resulted in a marked decrease in tumor growth rate, with histochemistry measurements of downstream effects supporting an siRNA-mediated mechanism of action for the observed efficacy.
Acknowledgments
The authors thank Dr. Pamela Talalay for her helpful suggestions and careful reading of the manuscript. We also thank the Biopolymer Lab at the University of Maryland School of Medicine for synthesizing the peptides in this study. This work was supported by the National Institutes of Health (R01-CA136938).
Glossary
ABBREVIATIONS
- AUC2h
area under the curve from 0 to 2 h
- DAB
3,3′-diaminobenzidine
- CL
clearance
- cRGD
cyclic peptide containing arginine-glycine-aspartic acid motif
- HK
generic term for histidine-lysine peptides
- H3K(+H)4b and H2K4b
two unmodified four-branched peptides that differ in their histidine and lysine content
- IHC
immunohistochemistry
- NIR
near-infrared
- RP-HK
(cRGD-PEG)4- H3K(+H)4b/H2K4b; modified HK alone (cRGD-PEG)4- H3K(+H)4b
- PEG-HK
(PEG)4-H3K(+H)4b/H2K4b
- MRT
mean residence time
- PEG
polyethylene glycol
- siLuc
siRNA that targets luciferase
- siRaf-1
siRNA that targets Raf-1
- siRNA
small interfering RNA
- t1/2β
terminal half-life
Supporting Information Available
Further details on the formulas for PK parameters are given. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yang X. Z.; Dou S.; Sun T. M.; Mao C. Q.; Wang H. X.; Wang J. J. Controlled Release 2011, 156, 203–11. [DOI] [PubMed] [Google Scholar]
- Bartlett D. W.; Davis M. E. Biotechnol. Bioeng. 2008, 99, 975–85. [DOI] [PubMed] [Google Scholar]
- Peer D.; Park E. J.; Morishita Y.; Carman C. V.; Shimaoka M. Science 2008, 319, 627–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E.; Zhu P.; Lee S. K.; Chowdhury D.; Kussman S.; Dykxhoorn D. M.; Feng Y.; Palliser D.; Weiner D. B.; Shankar P.; Marasco W. A.; Lieberman J. Nat. Biotechnol. 2005, 23, 709–17. [DOI] [PubMed] [Google Scholar]
- Davis M. E.; Zuckerman J. E.; Choi C. H.; Seligson D.; Tolcher A.; Alabi C. A.; Yen Y.; Heidel J. D.; Ribas A. Nature 2010, 464, 1067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumberg D.; Schultheis B.; Traugott U.; Vank C.; Santel A.; Keil O.; Giese K.; Kaufmann J.; Drevs J. Int. J. Clin. Pharmacol. Ther. 2012, 50, 76–8. [DOI] [PubMed] [Google Scholar]
- Alsina M.; Tabernero J.; Shapiro G.; Burris H.; Infante J. R.; Weiss G. J.; Cervantes-Ruiperez C.; Gounder M. M.; Paz-Ares L.; Falzone R.; Hill J.; Cehelsky J.; Vaishnaw A.; Gollob J.; LoRusso P.. Open-Label Extension Study of the RNAi Therapeutic ALN-VSP02 in Cancer Patients Responding to Therapy; 2012. ASCO Annual Meeting, Chicago, IL, June 1–5, 2012; American Society of Clinical Oncology: Chicago, IL, 2012; p 3062. [Google Scholar]
- Brower V. J. Natl. Cancer Inst. 2010, 102, 1459–61. [DOI] [PubMed] [Google Scholar]
- Leng Q.; Woodle M. C.; Lu P. Y.; Mixson A. J. Drugs Future 2009, 34, 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Q.; Scaria P.; Zhu J.; Ambulos N.; Campbell P.; Mixson A. J. J. Gene Med. 2005, 7, 977–86. [DOI] [PubMed] [Google Scholar]
- Putnam D.; Gentry C. A.; Pack D. W.; Langer R. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 1200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kichler A.; Mason A. J.; Bechinger B. Biochim. Biophys. Acta 2006, 1758, 301–7. [DOI] [PubMed] [Google Scholar]
- Midoux P.; Monsigny M. Bioconjug. Chem. 1999, 10, 406–11. [DOI] [PubMed] [Google Scholar]
- Hatefi A.; Megeed Z.; Ghandehari H. J. Gene Med. 2006, 8, 468–76. [DOI] [PubMed] [Google Scholar]
- Leng Q.; Scaria P.; Lu P.; Woodle M. C.; Mixson A. J. Cancer Gene Ther. 2008, 15, 485–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. D.; Huang L. Mol. Pharmaceutics 2008, 5, 496–504. [DOI] [PubMed] [Google Scholar]
- Jiang G.; Park K.; Kim J.; Kim K. S.; Oh E. J.; Kang H.; Han S. E.; Oh Y. K.; Park T. G.; Kwang Hahn S. Biopolymers 2008, 89, 635–42. [DOI] [PubMed] [Google Scholar]
- Schiffelers R. M.; Ansari A.; Xu J.; Zhou Q.; Tang Q.; Storm G.; Molema G.; Lu P. Y.; Scaria P. V.; Woodle M. C. Nucleic Acids Res. 2004, 32, e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood J. D.; Bednarski M.; Frausto R.; Guccione S.; Reisfeld R. A.; Xiang R.; Cheresh D. A. Science 2002, 296, 2404–7. [DOI] [PubMed] [Google Scholar]
- Kircheis R.; Wightman L.; Schreiber A.; Robitza B.; Rossler V.; Kursa M.; Wagner E. Gene Ther. 2001, 8, 28–40. [DOI] [PubMed] [Google Scholar]
- Thiel K. W.; Hernandez L. I.; Dassie J. P.; Thiel W. H.; Liu X.; Stockdale K. R.; Rothman A. M.; Hernandez F. J.; McNamara J. O. 2nd; Giangrande P. H. Nucleic Acids Res. 2012, 40, 6319–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S. T.; Leng Q.; Scaria P.; Woodle M.; Mixson A. J. Cancer Gene Ther. 2011, 18, 707–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Q.; Mixson A. J. Nucleic Acids Res. 2005, 33, e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. J.; Veiseh O.; Bhattarai N.; Sun C.; Hansen S. J.; Ditzler S.; Knoblaugh S.; Lee D.; Ellenbogen R.; Zhang M.; Olson J. M. PLoS One 2010, 5, e9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. D.; Chen Y. C.; Hackett M. J.; Huang L. Mol. Ther. 2008, 16, 163–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland M.; Tozer T. N., Clinical Pharmacokinetics and Pharmacodynamics: Concepts and Applications, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2011; pp 571–602. [Google Scholar]
- Merkel O. M.; Librizzi D.; Pfestroff A.; Schurrat T.; Buyens K.; Sanders N. N.; De Smedt S. C.; Behe M.; Kissel T. J. Controlled Release 2009, 138, 148–59. [DOI] [PubMed] [Google Scholar]
- Leng Q.; Mixson A. J. Cancer Gene Ther. 2005, 12, 682–90. [DOI] [PubMed] [Google Scholar]
- Brey E. M.; Lalani Z.; Johnston C.; Wong M.; McIntire L. V.; Duke P. J.; Patrick C. W. Jr. J. Histochem. Cytochem. 2003, 51, 575–84. [DOI] [PubMed] [Google Scholar]
- Tanaka T.; Shiramoto S.; Miyashita M.; Fujishima Y.; Kaneo Y. Int. J. Pharm. 2004, 277, 39–61. [DOI] [PubMed] [Google Scholar]
- Brannon-Peppas L.; Blanchette J. O. Adv. Drug Delivery Rev. 2004, 56, 1649–59. [DOI] [PubMed] [Google Scholar]
- Woodle M. C.; Collins L. R.; Sponsler E.; Kossovsky N.; Papahadjopoulos D.; Martin F. J. Biophys. J. 1992, 61, 902–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett D. W.; Su H.; Hildebrandt I. J.; Weber W. A.; Davis M. E. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 15549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko T. S.; Rammohan R.; Lukyanov A. N.; Whiteman K. R.; Torchilin V. P. Int. J. Pharm. 2002, 240, 95–102. [DOI] [PubMed] [Google Scholar]
- Sato A.; Choi S. W.; Hirai M.; Yamayoshi A.; Moriyama R.; Yamano T.; Takagi M.; Kano A.; Shimamoto A.; Maruyama A. J. Controlled Release 2007, 122, 209–16. [DOI] [PubMed] [Google Scholar]
- Wong N. C.; Mueller B. M.; Barbas C. F.; Ruminski P.; Quaranta V.; Lin E. C.; Smith J. W. Clin. Exp. Metastasis 1998, 16, 50–61. [DOI] [PubMed] [Google Scholar]
- Zuckerman J. E.; Choi C. H.; Han H.; Davis M. E. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 3137–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodle M. C.; Lasic D. D. Biochim. Biophys. Acta 1992, 1113, 171–99. [DOI] [PubMed] [Google Scholar]
- Yagi N.; Manabe I.; Tottori T.; Ishihara A.; Ogata F.; Kim J. H.; Nishimura S.; Fujiu K.; Oishi Y.; Itaka K.; Kato Y.; Yamauchi M.; Nagai R. Cancer Res. 2009, 69, 6531–8. [DOI] [PubMed] [Google Scholar]
- Heneweer C.; Holland J. P.; Divilov V.; Carlin S.; Lewis J. S. J. Nucl. Med. 2011, 52, 625–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett D. W.; Davis M. E. Nucleic Acids Res. 2006, 34, 322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita F.; Minakuchi Y.; Nagahara S.; Honma K.; Sasaki H.; Hirai K.; Teratani T.; Namatame N.; Yamamoto Y.; Hanai K.; Kato T.; Sano A.; Ochiya T. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 12177–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofek P.; Fischer W.; Calderon M.; Haag R.; Satchi-Fainaro R. FASEB J. 2010, 24, 3122–34. [DOI] [PubMed] [Google Scholar]
- Meckes D. G. Jr.; Shair K. H.; Marquitz A. R.; Kung C. P.; Edwards R. H.; Raab-Traub N. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears R.; Craven R. A.; Hanrahan S.; Totty N.; Upton C.; Young S. L.; Patel P.; Selby P. J.; Banks R. E. Proteomics 2004, 4, 4019–31. [DOI] [PubMed] [Google Scholar]
- Lakhal S.; Wood M. J. Bioessays 2011, 33, 737–41. [DOI] [PubMed] [Google Scholar]
- Valadi H.; Ekstrom K.; Bossios A.; Sjostrand M.; Lee J. J.; Lotvall J. O. Nat. Cell Biol. 2007, 9, 654–9. [DOI] [PubMed] [Google Scholar]
- Alvarez-Erviti L.; Seow Y.; Yin H.; Betts C.; Lakhal S.; Wood M. J. Nat. Biotechnol. 2011, 29, 341–5. [DOI] [PubMed] [Google Scholar]
- Hoeflich K. P.; O’Brien C.; Boyd Z.; Cavet G.; Guerrero S.; Jung K.; Januario T.; Savage H.; Punnoose E.; Truong T.; Zhou W.; Berry L.; Murray L.; Amler L.; Belvin M.; Friedman L. S.; Lackner M. R. Clin. Cancer Res. 2009, 15, 4649–64. [DOI] [PubMed] [Google Scholar]
- Spankuch B.; Kurunci-Csacsko E.; Kaufmann M.; Strebhardt K. Oncogene 2007, 26, 5793–807. [DOI] [PubMed] [Google Scholar]
- Sun T. M.; Du J. Z.; Yao Y. D.; Mao C. Q.; Dou S.; Huang S. Y.; Zhang P. Z.; Leong K. W.; Song E. W.; Wang J. ACS Nano 2011, 5, 1483–94. [DOI] [PubMed] [Google Scholar]
- Xiong X. B.; Lavasanifar A. ACS Nano 2011, 5, 5202–13. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.