

Abstract
Alternate prime/boost vaccination regimens employing recombinant replication-deficient adenovirus or MVA, expressing Influenza A virus nucleoprotein and matrix protein 1, induced antigen-specific T cell responses in intradermally (ID) vaccinated mice; with the strongest responses resulting from Ad/MVA immunization. In BALB/C mice the immunodominant response was shifted from the previously identified immunodominant epitope to a novel epitope when the antigen was derived from A/Panama/2007/1999 rather than A/PR/8. Alternate immunization routes did not affect the magnitude of antigen-specific systemic IFN-γ response, but higher CD8+ T-cell IFN-γ immune responses were seen in the bronchoalveolar lavage following intransal (IN) boosting after intramuscular (IM) priming, whilst higher splenic antigen-specific CD8+ T cell IFN-γ was seen following IM boosting. Partial protection against heterologous influenza virus challenge was achieved following either IM/IM or IM/IN but not ID/ID immunization. These data may be of relevance for the design of optimal immunization regimens for human influenza vaccines, especially for influenza-naïve infants.
Influenza vaccines in current use induce protective antibodies against the highly polymorphic external viral glycoprotein haemagglutinin (HA). However frequent changes in composition and annual revaccination are required to maintain effective immunity because of the constant genetic drift in HA sequences of seasonal influenza viruses. In addition, seasonal influenza vaccinations are not effective against pandemic influenza, and there is now some evidence that recent seasonal influenza infection, rather than vaccination, may result in some protection against pandemic influenza1,2.
A vaccine that could protect against all subtypes of influenza A virus, with the same or greater efficacy against seasonal influenza as currently licensed vaccines has been the focus of much research effort3,4. The availability of such vaccines could bring about a major improvement in protection of the population from both seasonal and pandemic influenza5 with considerable economic benefits. To achieve this will require a fundamental change in the composition and mode of action of influenza vaccines. Much pre-clinical research has focussed on protective T cell responses to internal influenza antigens such as nucleoprotein (NP) and matrix protein 1 (M1). Compared to the external viral glycoproteins, the conservation between these antigens derived from influenza A viruses of different subtypes is generally high; typically over 90% identical at the amino acid level. Human T cells specific for these antigens, and others, acquired following natural exposure to seasonal influenza have been shown to cross-react with H5N1 antigens6. In pre-clinical studies, vaccination with DNA or adenovirus vectors expressing influenza NP induced NP-specific T cell responses, and a high level of protection was seen after challenge with a heterosubtypic virus, although the immunodominant epitope (NP147–155) in the BALB/c mouse strain that was studied is completely conserved between the vaccine and challenge virus7. In a further study, heterosubtypic protection was demonstrated in C57BL/6 mice despite 2 amino acid differences in the immunodominant NP366–374 between the adenovirus-vectored vaccine and challenge virus antigen (Table 1) thereby suggesting that heterosubtypic protection is achievable in this model.
Table 1. Sequences of T cell epitopes in vaccine and PR8 challenge virus.
| Epitope | Epitope Name | MHC Restriction | Strain | Sequence | Accession No. or Reference |
| NP147–155 | NP147–155 | H-2Kd, Ld | A/PR/8/34 | TYQRTRALV | AAM75159 |
| NP366–374 | NP (PR8) | H-2Kb, Db | A/PR/8/34 | ASNENMETM | AAM75159 |
| NP366–374 | NP (Pan) | H-2Kb, Db | Panama | ASNENMDNM | ADG21462.1 |
| NP366–374 | NP (H17) | H-2Kb, Db | H17 | ASNENMDAM | ACF54415.137 |
| NP335-352 | NPpep36-PR8 | H-2Kb, Db | A/PR/8/34 | SAAFEDLRVLSFIKGTKV | AAM75159 |
| NP335-352 | NPpep36-Pan | H-2Kb, Db | Panama | SAAFEDLRLLSFIRGTKV | ADG21462.1 |
| M1 | Pep60 | Not determined | Panama | SPLTKGILGFVFTLTVPSER | ABG91472.1 |
Recombinant replication-deficient viral vectors are potent immunogens capable of both priming and boosting T cell responses against the recombinant antigens they encode. They are highly immunogenic in humans, and this, combined with their excellent safety profile, makes them ideal vectors for inducing protective T cell responses to influenza antigens. In clinical studies they have been administered by intradermal or intramuscular administration8, but pre-clinical studies have assessed mucosal immunization, including via intranasal administration, and demonstrated higher immune responses in the respiratory tract and greater protection against virus challenge following intranasal delivery9. Intranasal immunization is used for licensed live attenuated influenza vaccines (LAIV), and could potentially be used for recombinant viral vectors. However, the delivery devices required for intranasal or aerosolised immunization are more expensive to produce than needles and syringes used for intramuscular or intradermal vaccination. Despite being less invasive to use, the device used for LAIV administration generates large particle sizes that are less effective in vaccine delivery and can cause vaccine to drip out of the nose or roll back into the pharynx, reducing vaccine acceptability and efficacy10. LAIV is licensed for use in children over the age of two years, but in infants aged between 6 months and two years use of intranasal LAIV resulted in a greater number of hospitalizations due to wheezing11. However, pre-clinical studies have indicated that intramuscular vaccination can prime strong mucosal responses12,13, and this route of vaccination may therefore allow safe priming of mucosal responses in infants without the need for delivery of vaccine directly to the respiratory tract. As infants are among the most susceptible members of the population to influenza virus infection, we wished to test a vaccination regimen that could be safely and effectively used in infants, whilst still inducing protective immune responses. We therefore employed alternate vaccination regimens, including intradermal prime-boost or intramuscular priming followed by either intranasal or intramuscular boosting. Intradermal administration of seasonal influenza subunit vaccine has been shown to result in significantly higher immune responses (HI titres) than intramuscular administration in elderly adults14 and may also be an appropriate immunization route for infants.
In this pre-clinical study we have tested the use of recombinant replication-deficient viral-vectored vaccines that are suitable for use in humans to induce protective T cell responses against influenza NP and M1. We found a high level of protection against influenza virus challenge despite differences in the immunodominant epitope. Systemic influenza-directed T cell responses were similar in the intramuscular and intradermal vaccination regimens but improved survival following heterologous influenza challenge was seen following intramuscular but not intradermal vaccination. There were differences in CD8+ T cell responses in the systemic (spleen) and local (lungs and bronchoalveolar lavage (BAL)) immune responses within days following intranasal vaccination when compared to intramuscular or intradermal administration. However, similar levels of protection following influenza virus challenge were achieved following intranasal or intramuscular boosting, despite the increased influenza-directed T cell response in the BAL following intranasal boosting and lower splenic T cell responses.
Results
The vaccine regimen employing recombinant replication-deficient adenovirus (Ad) priming followed by recombinant Modified Vaccinia Ankara (MVA) boosting has been demonstrated to be highly immunogenic for the induction of both antigen-specific CD4+ and CD8+ T cells15,16. We tested four alternative prime-boost regimens employing these viral vectors expressing NP and M1 from A/Panama/2007/1999, administered intradermally (ID). In both the spleen and lymph nodes the Ad prime/MVA boost regimen was most immunogenic, inducing T cell responses more than three-fold higher than co-administration of the vaccines or homologous prime/boost with either vaccine when assayed by interferon–γ ELISpot (Figure 1). The Ad prime/MVA boost regimen with an eight week interval between prime and boost was then adopted for all further experiments.
Figure 1. Vaccination regimen can affect immunogenicity.

IFN-γ ELISpot responses, in (a) splenocytes or (b) lung draining lymph nodes, to the carboxy terminal of NP, two weeks post ID immunisation of C57BL/6 mice with 1.0 × 106 pfu MVA -NP+M1 or 5.0 × 109 vp Adhu5- NP+M1 or in combination, preceded 8 weeks earlier with an ID vaccination with 1.0 × 106 pfu MVA -NP+M1 or 5.0 × 109 vp Adhu5- NP+M1 or combination. Mean with SEM are depicted, as are results from one way Anova, assuming Gaussian distribution with Dunnett Post-test comparing mean response to Ad-MVA regimen; p-value *** = < 0.001.
We then determined the response to individual 18-20 mers spanning the NP+M1 antigen, 3 weeks following a single ID immunization with Ad-NP+M1. However, with respect to CD8+ T cell responses, from the initial peptide matrix study, we identified three peptides that induced IFN-γ+ responses from immunized mice (Figure 2). Interestingly, in C57BL/6 mice the immunodominant response was to NPpep36-Pan, representing sequence NP335–352 and a weaker response was observed to peptide 39, which contains the well documented H2Db immunodominant epitope NP366–374 “NP(Pan)” (Figure 2). Furthermore, no responses were observed to the epitope in M1, represented by peptide 60 (data not shown). The response was dominated by single cytokine producing CD8+IFN-γ+ T cells, with some dual cytokine a producing CD8+IFN-γ+TNF-á+ cells in both C57BL/6 and BALB/c mice (data not shown). As expected the dominant responses in BALB/c mice was to peptide 16 which contains the conserved epitope NP147–155 TYQRTRALV (Figure 2)17.
Figure 2. CD8+ T immunodominant response following Adhu5-NP+M1 prime.

C57BL/6 or BALB/c mice were immunized, intradermally, with indicated vp of Adhu5- NP+M1 and assessed 3 weeks later, by ICS, for antigen-specific cytokine+ splenic CD8+ T cell responses. Graph represents the frequency of antigen-specific cytokine+ CD8+ T following stimulation with indicated peptides. Mean with SEM are depicted.
A comparison of immunization routes was then carried out, with both prime and boost administered ID or intramuscularly (IM) or the adenovirus prime IM and the MVA boost intranasally (IN). No significant differences were detected in the immunogenicity of the three regimens through splenic ELISpot, although all were significantly more immunogenic than vaccination with vector controls expressing GFP instead of NP+M1 (Figure 3 left panel). The majority of the influenza-specific IFNγ+ response was directed toward the carboxy terminal of NP, but strong responses were also seen to the N-terminal half of NP and the M1 antigen (Figure 3 right panel).
Figure 3. Alternate vaccination routes generate similar levels of splenic immunogenicity.
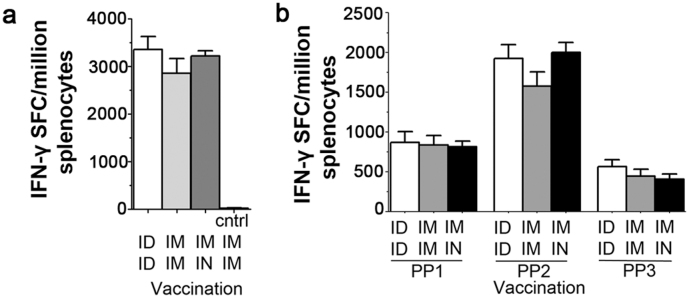
(a) Summed splenic IFN-γ ELISpot responses of C57BL/6 mice, in response to peptides spanning NP & M1, two weeks post vaccination, with 1.0 × 106 pfu MVA -NP+M1 preceded 8 weeks earlier with 5.0 × 109 vp Adhu5- NP+M1. Mean with SEM are depicted.
Examination of CD8+ T cell responses in the spleen using ICS instead of ELISpot demonstrated higher frequencies of antigen-specific CD8+ IFN γ+ T cells following ID/ID and IM/IM when compared to the IM/IN route (Figure 4).There were equivalent frequencies of antigen-specific CD8+ IFN γ+ T cells in the lungs of all immunised groups, however only the IM/IN route induced a strong CD8+ IFN γ+ T cell response in the BAL and the IM/IN route was the only regimen to induce a CD107ahi IFN γ− CD8+ T response above background in the lung (Figure 4 b–e). Thus the route of immunization was found to influence the immune response in the respiratory tract with IN boosting leading to increased responses in the BAL compared to IM boosting, following IM priming in both cases.
Figure 4. Antigen-specific CD8+ T cell response following Adhu5-NP+M1 and MVA -NP+M1 vaccination.

C57BL/6 mice were immunized, using alternate routes, with 5.0 × 109 vp Adhu5- NP+M1 and 8 weeks later with 1.0 × 106 pfu MVA -NP+M1. Intracellular cytokine staining was performed on (a) & (d) spleen or (b) & (e) lung or (c) Bronchoalveolar lavage (BAL) taken 2 weeks after the final vaccination. Graphs represent the frequency of IFN-γ producing (top) or CD107ahi IFN-γneg (bottom) CD8+ T cells in response to a single peptide pool spanning NP & M1. Mean with SEM are depicted, one way Anova, assuming Gaussian distribution with Bonferroni Post-test comparing all routes to each other was performed; p-value * = 0.01 to 0.05; p-value ** = 0.001 to 0.01; p-value *** = < 0.001.
We also analysed the quality of the CD8+ T cells induced by these different regimens before boosting and at short intervals after boosting mice by the IM or IN route with MVA-NP+M1 (Figure 5). In the lung, polyfunctional CD8+ T cells trended toward higher frequencies in the IN when compared to the IM boosting group at 2 days and 7 days following MVA boosting, and the proportion of effector and effector memory phenotype T cells also increased over this time (data not shown).
Figure 5. Quality of the antigen-specific CD8+ T cell response following Adhu5-NP+M1 and MVA -NP+M1 vaccination.

C57BL/6 mice were immunized, using alternate routes, with 5.0 × 109 vp Adhu5- NP+M1 and 8 weeks later with 1.0 × 106 pfu MVA -NP+M1. Intracellular cytokine staining was performed and bar charts represent the proportion of CD8+ T cells secreting 1, 2, or all 3 measured cytokines isolated from (a) spleen or (b) lung.
Having demonstrated higher CD8+ T cell IFN γ+ responses to NP and M1 in the BAL, following IN rather than IM boosting (Figure 4), we then conducted influenza virus challenge experiments to determine the relative protective efficacy of alternate immunization regimens. C57BL/6 mice were immunised as before, and subsequently challenged by intranasal administration of A/PR8 virus six week after MVA boosting (Figure 6a). Samples were taken from four of the animals in each group prior to challenge to assay peripheral blood mononuclear cells (PBMC) responses to NP and M1 by IFN γ+ ELISpot, which indicated that there were no statistically significant differences in peripheral PBMC responses by animals immunised by the ID/ID, IM/IM or IM/IN regimens, with no significant responses to the antigens in either naïve mice or those immunised with the same viral vectors expressing GFP or no recombinant antigen (Figure 6b).
Figure 6. Morbidity and mortality after heterologous influenza challenge following Adhu5-NP+M1 and MVA -NP+M1 vaccination.

Groups of C57BL/6 (a–f) or BALB/c (g & h) mice were immunized as in (a) or left unvaccinated (naïve). (b) Peripheral blood mononuclear cell IFN-γ ELISpot responses, to the carboxy terminal of NP, prior to influenza challenge are shown. Mean with SEM are depicted. Six weeks after final immunization, animals were challenged intranasally with 20–25 pfu PR8 (c–f) or 50 μl of X31 (104.5 TCID50/ml) or H17 (104.5 TCID50/ml) (g & h) and monitored for survival (c, e & f); survival curves offset by 0.2 units to allow ease of viewing) and weight loss (d, g & h).
Survival and weight loss curves following PR8 challenge are shown in Figure 6c & d. There were no significant differences in either survival or weight loss between the IM/IM and IM/IN groups, although both groups survived for longer (IM/IM (n = 20) vs naïve (n = 17) Log-rank (Mantel-Cox) Test; P value 0.0028: IM/IN (n = 8) vs naïve (n = 17) Log-rank (Mantel-Cox) Test P value 0.0273) and lost less bodyweight (IM/IM vs naïve unpaired t-test of area under the curve analysis of peak weight loss day 2–4; P value 0.0002) and (IM/IN vs naïve unpaired t-test of area under the curve analysis of peak weight loss day 2–4; P value 0.0001) than naïve mice.
There was no significant difference in survival of mice vaccinated ID/ID and the naïve control group.
In a repeat experiment employing a more virulent virus challenge, survival of the mice immunised by either IM/IM or IM/IN was again prolonged over naïve mice or those immunised by control vectors expressing GFP (IM/IM (n = 20) vs IM/IM GFP (n = 20) Log-rank (Mantel-Cox) Test; P value 0.0002: IM/IN (n = 19) vs IM/IN GFP (n = 17) Log-rank (Mantel-Cox) Test P value 0.0013) but there was no significant difference between the IM/IM and IM/IN groups immunised with NP+M1 (Figure 6e & f).
The IM/IM and IM/IN regimens were also compared in BALB/c mice challenged with either X31 or H17 influenza viruses (Figure 6g & h). The immunodominant epitope in BALB/c mice, NP147–155 TYQRTRALV is conserved in the vaccine and both challenge viruses, but there are other differences in the NP sequences. X31 has the same NP sequence as PR8 (H1N1) and there is 92% identity between the vaccine (A/Panama/2007/1999 H3N2) and challenge virus NP overall. H17 has an NP sequence derived from HK/8/68, an H3N2 virus with 96% identity to the vaccine NP overall. There were statistically significant differences in bodyweight loss between mice vaccinated with NP+M1 by either the IM/IM or IM/IN regimen and the control groups. For X31 (IM/IM vs IM/IM GFP unpaired t-test of area under the curve analysis of peak weight loss day 4–8; P value 0.0127) and (IM/IN vs IM/IN GFP unpaired t-test of area under the curve analysis of peak weight loss day 4–8; P value 0.0011), but no statistically significant difference between IM/IM and IM/IN. For H17 (IM/IM vs IM/IM GFP unpaired t-test of area under the curve analysis of peak weight loss day 4–8; P value 0.0021) and (IM/IN vs IM/IN GFP unpaired t-test of area under the curve analysis of peak weight loss day 4–8; P value < 0.0001), and a statistically significant difference between IM/IM and IM/IN (IM/IM vs IM/IN unpaired t-test of area under the curve analysis of peak weight loss day 4–8; P value 0.0140).
Discussion
There are numerous studies, supporting the role for both CD8+ and CD4+ T cells in combating influenza infection and subsequent illness (reviewed by Ref. 18,19,20,21), as such, there is a concerted drive to develop vaccines that can efficaciously induce influenza-directed T cell immunity. T cell responses specific for the relatively well conserved internal influenza antigens are an important component of naturally acquired immunity to influenza, especially against heterosubtypic viruses. Inactivated influenza vaccines have a minimal effect on boosting these responses22, and there is an increasing body of evidence that recent vaccination with TIV (Trivalent Influenza Vaccine) could increase susceptibility to infection with a virus not included in the vaccine, by preventing infection with the viruses matched to the vaccine and thereby preventing the acquisition of cellular immune responses to conserved antigens1,23,24,25,26. This heterosubtypic protection is not expected to completely prevent infection with influenza virus, but to result in a mild, possibly sub-clinical infection, reduction in virus shedding and rapid recovery rather than a severe illness or death. Combined with a humoral response, the breadth of which may be increased at least within subtype to allow recognition of drifted variants, this could result in complete protection against influenza subtypes included within the vaccine and additionally and importantly, partial protection against all other subtypes which might circulate in the event of a new pandemic.
The novel influenza vaccine MVA-NP+M1 has been tested for safety, immunogenicity and efficacy in clinical trials. Safety is as expected for MVA-vectored vaccines, which generally cause some mild or moderate adverse events within the first two days after vaccination, but these resolve quickly, and the complete inability of MVA to replicate after immunization makes the vaccines safe to use in all sections of the population. A single dose of MVA-NP+M1 resulted in a large expansion of NP and M1-specific T cells in volunteers, with responses still above pre-vaccination levels a year after vaccination27. In an influenza challenge trial, there was a significant reduction in the duration of virus shedding in vaccinated compared to unvaccinated volunteers, as well as a reduction of symptoms in the vaccinated group28.
However the vaccine has only been tested in volunteers over the age of 18 years, who all had pre-existing CD4+ and CD8+ responses to the vaccine antigens as a result of prior exposure to influenza virus. For infants and young children, a priming immunization will be required. The simian adenovirus vaccine vector ChAdOx129 is now being tested in a Phase I clinical trial, and this will be followed by ChAdOx1 and MVA prime boost clinical studies. However it is important to determine the best route for immunization to achieve the maximum level of protection with these viral vectored vaccines, and to consider whether, despite the high level of conservation of NP and M1, a small number of amino acid changes in key epitopes could result in diminished immunogenicity and protection.
In this pre-clinical study we have examined the effect of priming and boosting T cell responses to one naturally occurring NP sequence and challenging with a virus expressing a different one. We found that in C57BL/6 mice the immunodominant epitope within NP varied depending on the sequence that was used to immunise. By examining the response to peptides spanning the entire antigen rather than only the known immunodominant epitope we were able to identify a novel immunodominant epitope present in some NP sequences. The pattern of NP366–74 dominating after immunization with PR8 NP, but NP335–352 dominating after immunization with Panama NP was found in mice immunised with replication deficient adenovirus, or DNA vaccine (data not shown) priming, then MVA boosting, reinforcing the idea that this shift in immunodominance is a feature of the NP sequences themselves rather than the vectors used to deliver them.
We also examined the effect of immunization route on immunogenicity and protection. Following the discovery that ID administration of MVA was more protective in a mouse malaria challenge model than IM30, early clinical trials of MVA-vectored malaria vaccines31 and TB vaccines32 employed ID immunization. However this presents some practical difficulties; the volume that can be delivered by this route is only about 10% of that which can be delivered by IM immunization, and ID immunizations are considerably more technically demanding to perform. This led to a switch to IM immunizations for malaria vaccines16, and a comparison of the same dose of MVA-NP+M1 delivered by either route in a clinical trial concluded that there was no significant difference in immunogenicity as measured by the IFN– γ ELISpot assay in peripheral blood27.
Some pre-clinical studies have demonstrated that mucosal vaccination leads to higher mucosal immune responses33, and this is important for a respiratory tract pathogen such as influenza virus. However, despite the use of an intranasal spray to deliver LAIV vaccine, there are some disadvantages to this mode of administration in infants. We therefore wished to examine IM priming followed by IN boosting, as this has been used effectively in pre-clinical studies to induce high level mucosal responses12,13. This would allow infants to be vaccinated IM, which is easier to employ in that age group, followed by a subsequent IN boost in childhood, to enhance mucosal immunity further. We found that while responses in the spleen following IM/IN immunization were slightly lower than either ID/ID or IM/IM, responses in the lung and BAL were higher, confirming that it is sufficient for the boost to be given IN to achieve strong mucosal responses. In influenza virus challenge experiments, IM/IM was more protective than ID/ID, which was surprising given the similar level of systemic CD8+ T cell immunogenicity following both vaccination regimens. However, it has been demonstrated that environmental factors (antigen presenting cell, cytokines, chemokines and mode of antigen presentation) at the site of antigen exposure can induce differential expression of tissue-selective homing lymphocytes receptors34 and may therefore impact on sequential responses following repeat exposure to antigen. Indeed, direct antigen presentation appears to be more important for intramuscular, compared to the intradermal, induction of cytotoxic CD8+ T cells to Vaccina virus35, and different tissue-specific dendritic cells have also been found to lead to different outcomes in the induction of regulatory T cells36 which may in turn affect antigen-specific responses. There was no significant difference in protection against influenza virus challenge six weeks after either intranasal or intramuscular boosting following intramuscular priming despite the observed differences in mucosal immunity two weeks after boosting. This was also observed in a second mouse strain challenged with two different influenza viruses.
As has been seen in other studies of heterosubtypic rather than homosubtypic influenza virus challenge7,23, protection is not always complete, but is achieved despite sequence changes in the challenge virus in both the previously known immunodominant epitope in NP PR8 and the newly identified immunodominant epitope in NP Panama. Thus the level of polymorphism that is found in internal antigens of influenza A viruses capable of infecting humans should not present a barrier to achieving useful protection through T cell responses to these antigens.
The route of immunization is important for protection, with both IM/IN and IM/IM regimens providing a higher degree of protection than ID/ID. However, stronger immune responses in the BAL following IN rather than IM boosting with MVA perhaps surprisingly did not result in improved protection against IN virus challenge. This is an important finding, indicating that in clinical trials it will not be sufficient to assess mucosal immunity to predict vaccine efficacy. Further vaccine development (efficacy as well as immunogenicity) in relevant animal models (e.g. the pig) could provide a useful way of developing the most protective immunization regimens for use in infants, in which the aim will be to prime an immune response that forms the basis of lifelong broad immunity to influenza rather than providing short term protection against specific HA sequences.
Methods
Ethics statement
All mouse procedures were performed in strict accordance with the terms of licences from the UK Home Office, under the terms of the Animals (Scientific Procedures) Act 1986 (licence numbers 30/2889 and 30/241 and the Irish Department of Health and Children, under the Cruelty to Animals Act (licence numbers B100/4034 and B100/3157) and 4) and according to the approval of the UCC AECC and University of Oxford Animal Ethics Committees.
Vaccines
MVA-NP+M1 expresses NP and M1 from A/Panama/2007/1999 as a single fusion antigen and is described in27. A replication-deficient E1E3 deleted adenovirus Hu5 expressing the same antigen under the control of a CMV promoter was also used.
Animals and immunizations
Female Balb/c or C57BL/6 mice of 6 weeks of age or older (Harlan, UK) were immunized intramuscularly (IM) in the musculus tibialis, intradermally (ID) in the ear, or intranasally (IN) with a total volume of 50 μl of vaccine diluted in PBS. For all murine immunizations AdHu5 was administered at 5 × 109 vp and MVA was administered at 106 pfu unless otherwise indicated.
ELISpots
Murine spleens or peripheral blood samples were treated with ACK lysis buffer to remove RBCs prior to stimulation with the relevant peptides (final concentration of 5 μg/ml) on IPVH-membrane plates (Millipore) coated with 5 μg/ml anti-mouse IFN-γ (AN18). After 18–20 hours of stimulation, IFN-γ spot forming cells (SFC) were detected by staining membranes with either anti-mouse IFN-γ biotin (1 μg/ml) (R46A2) followed by streptavidin-Alkaline Phosphatase (1 μg/ml) and development with AP conjugate substrate kit (BioRad, UK).
Intracellular cytokine staining
Murine splenocytes were stimulated for a total of 2 hours with a single pool containing whole NP+M1 peptides (10 μg/ml final concentration) or media only (unstimulated control), with the addition of Golgi-Plug and Golgi-Stop (BD) (0.2 uL Golgi-Plus and 0.2 uL Golgi-stop per 1 × 106 splenocytes) for the final 4 hours. Following surface staining with CD4-efluor 650, CD8 PerCPCy5.5, CD62L-APC-ef780 and CD127-PE-CY7, cells were fixed with 4% paraformaldehyde and stained intracellularly with TNF-á FITC, IL-2 PE and IFN-ã APC (or in some instance, IFN-ã eflour 450) and diluted in Perm-Wash buffer (BD). Sample acquisition was performed on a LSR II and data analyzed in FlowJo (TreeStar).
Influenza virus challenge
Six weeks following the last immunization mice were challenged with A/PR/8/34, (PR8), A/X-31(H3N2) (X31) or E61-13-H17 (H17) influenza virus. Mice were anesthetized with 100 μL of ketamine/dormitor administered via intraperitoneal injection (i.p.). Virus was inoculated intranasally in a volume of 50 μL. PR8 was administered at 20–25 PFU, X31 at 106.5 TCID50/ml, an and H17 at 104.5 TCID50/ml. Mice were monitored daily for 10 days or more for clinical disease, symptoms including weight loss, piloerection, and reduced motility. A 20% reduction in bodyweight was defined as a humane endpoint and animals meeting this criterion were euthanized.
Author Contributions
Conceived and designed the experiments: T.L., J.B.C., A.C.M. & S.C.G. Performed the experiments: T.L., Y.L., C.E.M., A.v.L., J.B.C., A.V. & A.C.M. Analyzed the data: T.L., Y.L. & A.C.M. Wrote the paper: T.L., A.C.M. & S.C.G.
Acknowledgments
We wish to thank the Jenner Institute Viral Vector Core Facility for preparation of the viral vectored vaccines. We thank John McCauley, Awen Gallimore at the Cardiff Institute of Infection & Immunity and Michael Bennett at the National Institute of Medical Research, Mill Hill, for the influenza viruses used in challenges. AvL was funded by a fellowship of the Dr. Saal van Zwanenberg Stichting. Dr. Lambe is supported by the Oxford Martin School. SCG is a Jenner Investigator.
References
- Cowling B. J. et al. Protective efficacy of seasonal influenza vaccination against seasonal and pandemic influenza virus infection during 2009 in Hong Kong. Clinical infectious diseases 51, 1370–1379 (2010). [DOI] [PubMed] [Google Scholar]
- Mathews J. D., McBryde E. S., McVernon J., Pallaghy P. K. & McCaw J. M. Prior immunity helps to explain wave-like behaviour of pandemic influenza in 1918–9. BMC infectious diseases 10, 128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe T. Novel viral vectored vaccines for the prevention of influenza. Mol Med 18, 1153–1160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert S. C. Advances in the development of universal influenza vaccines. Influenza Other Respi Viruses (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arinaminpathy N. et al. Impact of cross-protective vaccines on epidemiological and evolutionary dynamics of influenza. Proceedings of the National Academy of Sciences of the United States of America 109, 3173–3177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee L. Y. et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J Clin Invest 118, 3478–3490 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein S. L. et al. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine (2005). [DOI] [PubMed] [Google Scholar]
- Berthoud T. K. et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clinical Infectious Diseases 52, 1–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price G. E. et al. Single-dose mucosal immunization with a candidate universal influenza vaccine provides rapid protection from virulent H5N1, H3N2 and H1N1 viruses. PLoS One 5, e13162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy C. J. et al. Aerosolized adenovirus-vectored vaccine as an alternative vaccine delivery method. Respir Res 12, 153 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshe R. B. et al. Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med 356, 685–696 (2007). [DOI] [PubMed] [Google Scholar]
- Patel V. et al. Long-lasting humoral and cellular immune responses and mucosal dissemination after intramuscular DNA immunization. Vaccine 28, 4827–4836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman D. R., Bivas-Benita M., Simmons N. L., Miller D. & Barouch D. H. Route of adenovirus-based HIV-1 vaccine delivery impacts the phenotype and trafficking of vaccine-elicited CD8+ T lymphocytes. J Virol 84, 5986–5996 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D. et al. Intradermal influenza vaccine administered using a new microinjection system produces superior immunogenicity in elderly adults: a randomized controlled trial. J Infect Dis 198, 650–658 (2008). [DOI] [PubMed] [Google Scholar]
- Draper S. J. et al. Enhancing blood-stage malaria subunit vaccine immunogenicity in rhesus macaques by combining adenovirus, poxvirus, and protein-in-adjuvant vaccines. J Immunol 185, 7583–7595 (2010). [DOI] [PubMed] [Google Scholar]
- Sheehy S. H. et al. Phase Ia Clinical Evaluation of the Safety and Immunogenicity of the Plasmodium falciparum Blood-Stage Antigen AMA1 in ChAd63 and MVA Vaccine Vectors. PLoS One 7, e31208 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodmer H. C., Pemberton R. M., Rothbard J. B. & Askonas B. A. Enhanced recognition of a modified peptide antigen by cytotoxic T cells specific for influenza nucleoprotein. Cell 52, 253–258 (1988). [DOI] [PubMed] [Google Scholar]
- Epstein S. L. & Price G. E. Cross-protective immunity to influenza A viruses. Expert review of vaccines 9, 1325–1341 (2010). [DOI] [PubMed] [Google Scholar]
- Stanekova Z. & Vareckova E. Conserved epitopes of influenza A virus inducing protective immunity and their prospects for universal vaccine development. Virol J 7, 351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotsaert M., Saelens X. & Leroux-Roels G. Influenza vaccines: T-cell responses deserve more attention. Expert Rev Vaccines 11, 949–962 (2012). [DOI] [PubMed] [Google Scholar]
- Hillaire M. L., Osterhaus A. D. & Rimmelzwaan G. F. Induction of virus-specific cytotoxic T lymphocytes as a basis for the development of broadly protective influenza vaccines. Journal of biomedicine & biotechnology 2011, 939860 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe T. et al. T-Cell Responses in Children to Internal Influenza Antigens, 1 Year after Immunization with Pandemic H1N1 Influenza Vaccine, and Response to Revaccination with Seasonal Trivalent Inactivated Influenza Vaccine. The Pediatric Infectious Disease Journal (2012). [DOI] [PubMed] [Google Scholar]
- Bodewes R. et al. Vaccination against human influenza A/H3N2 virus prevents the induction of heterosubtypic immunity against lethal infection with avian influenza A/H5N1 virus. PLoS One 4, e5538 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodewes R. et al. Vaccination against seasonal influenza A/H3N2 virus reduces the induction of heterosubtypic immunity against influenza A/H5N1 virus infection in ferrets. J Virol 85, 2695–2702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronski D. M. et al. Association between the 2008-09 seasonal influenza vaccine and pandemic H1N1 illness during Spring-Summer 2009: four observational studies from Canada. PLoS medicine 7, e1000258 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjua N. Z. et al. Seasonal influenza vaccine and increased risk of pandemic A/H1N1-related illness: first detection of the association in British Columbia, Canada. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America 51, 1017–1027 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud T. K. et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin Infect Dis 52, 1–7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillie P. J. et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin Infect Dis 55, 19–25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicks M. D. et al. A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. PLoS One 7, e40385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J. et al. Enhanced immunogenicity for CD8+ T cell induction and complete protective efficacy of malaria DNA vaccination by boosting with modified vaccinia virus Ankara. Nat. Med. 4, 397–402 (1998). [DOI] [PubMed] [Google Scholar]
- Dunachie S. J. et al. A DNA prime-modified vaccinia virus ankara boost vaccine encoding thrombospondin-related adhesion protein but not circumsporozoite protein partially protects healthy malaria-naive adults against Plasmodium falciparum sporozoite challenge. Infect Immun 74, 5933–5942 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McShane H. et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat Med 10, 1240–1244 (2004). [DOI] [PubMed] [Google Scholar]
- Pavot V., Rochereau N., Genin C., Verrier B. & Paul S. New insights in mucosal vaccine development. Vaccine 30, 142–154 (2012). [DOI] [PubMed] [Google Scholar]
- Takamura S. et al. The route of priming influences the ability of respiratory virus-specific memory CD8+ T cells to be activated by residual antigen. J Exp Med 207, 1153–1160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Wong S. B., Buck C. B., Zhang J. & Siliciano R. F. Direct priming and cross-priming contribute differentially to the induction of CD8+ CTL following exposure to vaccinia virus via different routes. J Immunol 169, 4222–4229 (2002). [DOI] [PubMed] [Google Scholar]
- Belkaid Y. & Oldenhove G. Tuning microenvironments: induction of regulatory T cells by dendritic cells. Immunity 29, 362–371 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend A. R. & Skehel J. J. The influenza A virus nucleoprotein gene controls the induction of both subtype specific and cross-reactive cytotoxic T cells. J Exp Med 160, 552–563 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]