

Abstract
Cholangiocarcinomas (CCAs) are tumors that develop along the biliary tract. Depending on their site of origin, they have different features and require specific treatments. Classification of CCAs into intrahepatic, perihilar, and distal subgroups has helped standardize the registration, treatment, and study of this lethal malignancy. Physicians should remain aware that cirrhosis and viral hepatitis B and C are predisposing conditions for intrahepatic CCA. Treatment options under development include locoregional therapies and a chemotherapy regimen of gemcitabine and cisplatin. It is a challenge to diagnose perihilar CCA, but an advanced cytologic technique of fluorescence in situ hybridization for polysomy can aid in diagnosis. It is important to increase our understanding of the use of biliary stents and liver transplantation in the management of perihilar CCA, as well as to distinguish distal CCAs from pancreatic cancer, because of different outcomes from surgery. We review advances in the classification, diagnosis, and staging of CCA, along with treatment options.
Keywords: Intrahepatic Cholangiocarcinoma, Perihilar Cholangiocarcinoma, Distal Cholangiocarcinoma
Cholangiocarcinomas (CCAs) are a heterogenous group of tumors that arise from the cholangiocytes that line the biliary tree. CCAs are classified based on their anatomic location, as follows: (1) intrahepatic CCA (iCCA), (2) perihilar CCA (pCCA), or (3) distal CCA (dCCA) (Figure 1).1 iCCA is the second-most common primary liver cancer; its incidence has increased by 22% between 1979 and 2004.2 The increased incidence, which only partially can be explained by more frequent diagnosis and greater awareness, has been accompanied by a 39% increase in mortality.2 A recent re-analysis of age-standardized incidence rates in the United States showed an increase in iCCA from 1990 to 2001, and then a decrease by 2007.
Figure 1.

Anatomic classification of CCA describing iCCA, pCCA, and dCCA.
The incidence of extrahepatic CCA increased from 2001 to 2007, coinciding with the 2001 implementation of a new version of the International Classification of Diseases for Oncology. As the reanalysis indicated, pCCA often was misclassified as iCCA in earlier registries—the increase in iCCA reported by many registries could result from an increase in pCCA.3 Age-adjusted rates for CCA are highest among Hispanics and Asians (2.8–3.3/100,000) and lowest among non-Hispanic whites and blacks (2.1/100,000). Men appear to have a slightly greater mortality from the disease than women (1.9 and 1.5 per 100,000, respectively).2
In a large, single-center study, iCCA accounted for less than 10% of cases of CCA, pCCA accounted for 50%, and dCCA accounted for approximately 40%.4 It makes sense that tumors that develop in the small vs large bile ducts have different symptoms and patterns of progression. Different subtypes of CCA also have been associated with different genetic factors.5
Risk Factors
Most CCAs are sporadic and have no identifiable risk factors. However, the incidence of CCA might vary among populations of different geographic origin because of differences in risk factors (Supplementary Table 1). In Southeast Asia (specifically Thailand), the incidence of CCA is as high as 113/100,000 people,6 and hepatobiliary flukes, Opisthorchis viverrini, and Clonorchis sinensis, are risk factors. Bile duct cystic disorders also are risk factors for CCA and also are more prevalent in Asia than in Europe or North America; the lifetime incidence of CCA in people with these disorders ranges from 6% to 30%,6 with an odds ratio from 10.7 to 47.1.7,8
Single extrahepatic, diverticulum-like, and multiple extrahepatic and intrahepatic cysts have especially strong associations with CCA.9 Patients with bile duct cystic disorders are diagnosed with CCA at a mean age of 32 years, which is considerably younger than the age of CCA diagnosis in the general population (70–80 y).6,10 Many patients still develop CCA after excision of choledochal cysts because tumors can develop in noncystic portions of the biliary tree.6 Caroli's disease, characterized by congenital, multifocal, segmental dilatation of the intrahepatic bile ducts, is another cystic disorder that increases risk for CCA.6 Furthermore, 7% of patients with hepatolithiasis (particularly that characterized by calcium bilirubinate stones) develop iCCA.11,12 Although hepatolithiasis is observed more frequently in Asia, it also has been associated with CCA in Western populations.8,13–15
A less-commonly recognized risk factor for CCA is biliary– enteric drainage, which can cause bile stasis, inflammation, stone formation, and infection to promote epithelial cell trans-formation.16 Thorotrast, which was used as a radiocontrast agent in medical radiography in the 1930s and 1940s, was reported to increase the risk for CCA 300-fold, compared with the general population.6 Diabetes, obesity, alcohol consumption, and smoking exposure also might be risk factors for CCA, but data are limited and have not been verified.6 A large number of genetic polymorphisms also have been reported to increase risk of CCA, but require further analysis.6
Primary Sclerosing Cholangitis
Primary sclerosing cholangitis (PSC) is one of the best-known risk factors for CCA; the lifetime prevalence for CCA among patients with PSC ranges from 5% to 10%,17–21 with an overall risk of 0.5% to 1.5%/y.17,22 Many factors that modify risk for CCA in patients with PSC are under investigation, but these have only modest odds ratios and/or have not been confirmed in prospective studies. These factors include older age at diagnosis of PSC, smoking, alcohol use, a longer duration of associated inflammatory bowel disease, colorectal cancer or dysplasia in patients with ulcerative colitis, variceal bleeding, increased levels of bilirubin, proctocolectomy, the presence of biliary stones, and certain polymorphisms in the gene NKG2D.21 As many as 50% of patients diagnosed with PSC are diagnosed with CCA within 1 year,21 but little is known about risk factors for this cancer in these patients. Patients with PSC therefore should be monitored carefully for strictures, changes in biochemical parameters, or serum markers or symptoms of cancer.
Intrahepatic Cholangiocarcinoma
Risk Factors
Cirrhosis and viral hepatitis C and B are risk factors for CCA,6 although the association varies with tumor type (they have the strongest association with iCCA), and it is stronger for hepatitis C than B.23 It has been proposed that cholangiocarcinogenesis in patients with cirrhosis involves hepatic progenitor cells (HPCs).11
Classification of Intrahepatic Cholangiocarcinomas and Mixed Hepatocellular-Cholangiocellular Carcinomas
iCCA is a CCA subtype that arises from the intrahepatic biliary tract. Patients with iCCA often present with an intrahepatic mass lesion. Based on macroscopic growth patterns, iCCA can be divided into mass-forming, periductal-infiltrating, intraductal, and undefined subtypes.1,24 The mass-forming subtype is the most common and spreads via venous and lymphatic vessels. Patients with periductal-infiltrating tumors with mass-forming features have the worst prognosis of all patients with iCCA.24 A superficial spreading type is a rare type of iCCA that does not usually invade the liver parenchyma, but instead spreads along the biliary duct lumen11 and has better outcomes than other iCCAs.
iCCA are usually adenocarcinomas that are well, moderately, or poorly differentiated. Some researchers have proposed that rare types of iCCA, based on histologic analyses (Supplementary Table 2), actually might be different tumors—many are not adenocarcinomas.11 An increasingly recognized subtype of iCCA could arise from biliary ductules that contain bipotential HPCs; these tumors have similar gene expression patterns and clinicopathologic and molecular profiles to mixed hepatocellular carcinoma (HCC)–CCAs.25 These tumors might arise via transformation of HPCs in a stem cell niche that have the potential to mature into hepatocytes and bile duct cells.11,26 However, hepatocytes recently were reported to transdifferentiate directly into CCAs.27 All types of CCAs are associated with rapid proliferation of tumor-associated stroma cells, which contributes to desmoplasia.
Diagnosis
Patients with iCCA often present with nonspecific symptoms such as cachexia, abdominal pain, night sweats, and fatigue.1 Patients with cirrhosis often are asymptomatic, with a mass identified in cross-sectional imaging studies (Figure 2A). If a mass is identified in the liver of a patient without cirrhosis, risk factors, or history of nonhepatic primary cancer, then iCCA should be considered. Computed tomography (CT) and magnetic resonance imaging (MRI) greatly assist in the diagnosis of CCA. iCCA takes up contrast agent progressively during the arterial and venous phases of studies— especially if the lesion is larger than 20 mm.28 iCCAs with extensive desmoplasia take up the contrast agent more slowly than tumors without desmoplasia, whereas active inflammation at the tumor parenchyma interface causes a rim pattern of peripheral enhancement. In contrast, HCCs typically take up the contrast agent rapidly during the arterial phase, and quickly wash out the contrast during the venous phase of the study. If surgery is considered for a patient with a small intrahepatic lesion without cirrhosis then a diagnostic biopsy might not be required because its results will not change the management strategy. In all other cases, a biopsy specimen should be obtained to confirm the diagnosis of iCCA (Figure 2B).
Figure 2.

Algorithm for diagnosis and management of patients with iCCA (A) and cirrhosis or (B) without cirrhosis.
Either CT or MRI is appropriate for evaluation of tumor size, the presence of satellite lesions, the status of vascular structures, and for volumetric assessment of potential liver remnants; findings can be used to plan further treatment. Multidetector CT might be more accurate than MRI in predicting resectability, with an accuracy of 85% to 100%29; CT also might be better for identifying extrahepatic metastases. The use of positron emission tomography (PET) in the diagnosis of CCA is limited; PET detects iCCA with sensitivity values ranging from 18% (for infiltrating types) to more than 80% (for mass-forming types).30 PET sensitivity for the detection of extrahepatic CCA is 55%. The specificity values range from 33% to 80%, depending on the tumor's location.31 In general, PET scanning is more helpful in detecting larger iCCAs and metastases32; with advances in CT and MRI technologies, PET usually adds little to the diagnostic algorithm and should not be used routinely.
Carbohydrate antigen 19-9 (CA19-9) is a serum marker that can be measured to identify patients with iCCA, with 62% sensitivity and 63% specificity.1 When the cut-off value is set at 100 U/mL CA 19-9, and patients do not have PSC, tests for this marker identify patients with resectable disease with 33% sensitivity, and patients with unresectable disease with 72% sensitivity.33 Cut-off values of CA19-9 of 129 U/mL or greater detect iCCA in patients with PSC with sensitivity and specificity values of 79% and 98%, respectively.34 Levels of CA19-9 of 1000 U/mL or greater have been associated strongly with unresectable disease.33 Seven percent of the general population is negative for Lewis antigen and have undetectable levels of CA19-9. This makes interpreting a serum CA19-9 test useless in the Lewis antigen–negative population.35 Although tests for serum levels of CA19-9 are not very useful in identifying individuals with iCCA, they can be used in conjunction with other diagnostic tools.
In mixed tumors, cholangiocellular elements can be identified by immunohistochemical analysis for cytokeratins (CKs) 19 and 7. Tumors positive for CK19 and CK7 can be considered mixed HCC–CCA.36,37 Identification of a mixed tumor has clinical implications because even if only a small number of its cells (5%) are positive for CK19, it has a high risk for recurrence after surgery compared with HCCs without cholangiocellular features.38
Treatment
Treatment options for iCCA are limited and associated with high rates of tumor recurrence and short survival times. When surgical resection can be offered, the median survival time is 36 months, with a recurrence rate of 62.2% after a median of 26 months of follow-up evaluation.39 Sixty percent of patients who undergo curative resection (defined as negative tumor margins, R0) survive for 5 years, but less than 30% of all patients receive curative resections—even in centers with expertise.4 The 5-year survival rate has been reported to be better for patients with iCCA (63%) than patients with pCCA (30%) or dCCA (27%).4 Factors associated with reduced survival time after resection include positive tumor margins and lymph node metastases.4 Cirrhosis also reduces patient survival times after surgery for iCCA.40 Child–Pugh scores B and C, model for end-stage liver disease scores of 9 or greater, and portal hypertension are relative contraindications for resection of iCCA in patients with cirrhosis. Multifocal tumors have high rates of recurrence (>90%) and usually preclude curative resection.39
Although liver transplants might seem to be a good option for patients with iCCA, the 5-year rate of tumor recurrence after transplantation is higher than 70%, and the median time of disease-free survival is only 8 months, which is unacceptably high.37 One center treats patients with neoadjuvant therapy and advocates liver transplantation for those who respond.41 Liver explants often contain both CCA and HCC, and some tumors thought to be small HCCs are, in fact, iCCA or mixed HCC-CCA,37,42 so it is reasonable to collect biopsy samples from any lesion with imaging features atypical for HCC before liver transplantation.
The Advanced Biliary Cancer (ABC)-02 study showed that systemic chemotherapy with a combination of gemcitabine and cisplatin prolonged survival times of patients with inoperable CCA, making it a treatment standard.43 This treatment approach appeared to be most effective for patients with iCCA or gallbladder cancer, increasing survival times by approximately 3 months, compared with gemcitabine alone. Patients with cirrhosis were not included in the study—further trials are needed to determine if this therapeutic strategy also is effective in these patients. A recent meta-analysis supports adjuvant therapy for patients with lymph node–positive disease.44 Targeted therapies also are being investigated.45
Other viable therapeutic options for inoperable iCCA without extrahepatic metastases include transarterial chemoembolization, radiofrequency ablation, and transarterial radioembolization. Patients who receive transarterial chemoembolization or transarterial radioembolization had median survival times of 20 and 43.7 months, respectively, after diagnosis.46,47 However, this was an uncontrolled study. Patients with tumors smaller than 3 cm who were treated with radiofrequency ablation had an overall median survival time of 38.5 months.48 No studies have compared transarterial radioembolization with transarterial chemoembolization or systemic chemotherapy with locoregional therapy. However, for patients amenable to locoregional therapy, this approach might be a palliative treatment option— especially for patients whose performance status (a major prognostic factor) precludes more aggressive approaches.
Perihilar Cholangiocarcinoma
Classification
pCCAs develop anywhere from the second-order biliary ducts to above the site of cystic duct origin; they can have exophytic (mass-forming) and intraductal growth patterns. Intraductal pCCAs can be nodular or periductal infiltrating (also referred to as a sclerosing subtype). Periductal-infiltrating tumors can form an associated mass and are the most common subtype of pCCA. Intraductal papillary neoplasias include a range of lesions, from preneoplastic to invasive carcinomas, and often are well differentiated. They can be divided further into papilloma type, intraductal growing type, mucin-producing type, and cystic type.11,25 pCCA spread by perineural invasion and lymphatic metastasis.1
Diagnosis
pCCAs of the major bile ducts that cause cholestasis present earlier than iCCA; 90% of patients with pCCAs have painless jaundice, 10% have cholangitis, and 56% have systemic symptoms such as malaise, abdominal discomfort, nausea, anorexia, or weight loss.49,50 Weight loss with jaundice is common; patients with jaundice often regain their weight after biliary tract stenting. Depending on the presence or absence of other liver diseases (cirrhosis, PSC, and so forth), patients can have the cutaneous stigmata associated with chronic liver disease, or a history of inflammatory bowel disease. Tests for CA19-9 are helpful in diagnosis when used in combination with other diagnostic tests. However, levels of CA19-9 can increase with other hepatobiliary conditions (such as cholangitis or largeduct obstruction), so the test is less specific for pCCA than iCCA.
A new surgical staging system for pCCA has been introduced to improve and standardize determination of prognosis and tumor reporting.51 This new system keeps the Bismuth–Corlette classification for assessment of biliary tree involvement (common bile duct, confluence, right and/or left hepatic ducts, and both ducts involvement),52 but also considers tumor size (>1 cm, 1–3 cm, or ≥3 cm), tumor morphology (sclerosing, mass-forming, mixed, or polypoid), degree and specific location of hepatic artery and portal vein encasement (vessel involvement >180° indicates encasement), volume of the potential liver remnant, other liver diseases (fibrosis, nonalcoholic steatohepatitis, or PSC), status of lymph node groups (hilar and along the hepatic artery vs celiac and periaortic), and presence of distant metastases.
MRI, CT, endoscopic retrograde cholangiography (ERC), and, perhaps, endoscopic ultrasound (EUS) are used most frequently to diagnose and stage pCCA (Figure 3). Cross-sectional imaging studies can reveal a biliary stricture with proximal bile duct dilatation, periductal thickening with or without associated mass, and vascular encasement; determine whether lymph nodes are involved; and identify distant metastases. When segmental or lobar atrophy is present, ipsilateral encasement of the portal vein often is detected. MRI with magnetic resonance cholangiopancreatography (MRCP) is a valuable diagnostic tool (Figure 4), identifying pCCA with 89% sensitivity and 76% accuracy.34 MRCP with 3-dimensional liver acquisition with volume acceleration sequences allows for detailed evaluation of the hilar and distal extrahepatic bile ducts and should be used when available. CT has a high negative predictive value for advanced disease (85%–100%),29 but detects small intrahepatic and distant metastases with limited sensitivity. Abdominal ultrasound with vascular Doppler studies detect CCA with 57% sensitivity and 94% specificity21,34; these values vary between medical centers and with the size of the lesion, and the involvement of the hepatic artery and biliary tree can be underesti-mated.29 EUS is becoming an important tool for staging pCCA—it is particularly useful in assessing lymph node and omental metastases, which can be identified by EUS-directed fine-needle aspiration (FNA). However, transperitoneal FNA of the primary tumor during EUS can result in tumor seeding.53 Therefore, EUS with FNA is valuable in tumor staging, but primary tumor FNA should be used only in cases in which needle tract seeding would not change patient management.54
Figure 3.

Algorithm for diagnosis and management of pCCA. LN, lymph node.
Figure 4.
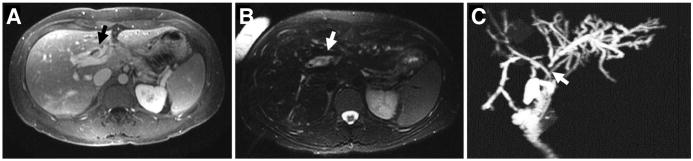
(A and B) MRI and (C) MRCP images of pCCA (indicated by arrows) superimposed on PSC.
ERC is virtually required for evaluation and assessment of pCCA. ERC allows acquisition of samples via brush cytology and endoscopic biopsy, and can be used in therapy, with dilatation and stent placement for relief of biliary obstruction. ERC identification of biliary stenosis with a dominant stricture, polypoid duct lesion, or marked proximal bile duct dilatation is a strong indicator of pCCA. The challenges to diagnosis of CCA relate to its highly desmoplastic nature and potential sampling errors. Cytologic specimens can be classified as negative for malignancy, atypical, suspicious for malignancy, and diagnostic of cancer. Specimens obtained by brush cytology have a sensitivity of 15% when only diagnostic of cancer results are used for the diagnosis of CCA and 48% when diagnostic of cancer plus suspicious for malignancy cytologic interpretations are combined for cancer diagnosis.55 Because of the low level of sensitivity of cytology analysis, many centers use fluorescence in situ hybridization, which identifies chromosome abnormalities, such as amplifications or aneusomy (a marker for aneuploidy), that are present in most cancer cells.55 The addition of fluorescence in situ hybridization to conventional cytology analysis increases sensitivity values to 38% to 58% and accuracy values to 77% to 83%.56–60 Tests to detect deletion of chromosome 9p21 have increased the diagnostic yield of fluorescence in situ hybridization analysis to 93%.56
Percutaneous transhepatic cholangiography is used as an alternative to ERC, especially when endoscopy is unsuccessful or technically unfeasible. Immunoglobulin (Ig)G4-related diseases can mimic CCA, so serum levels of IgG4 should be measured in all patients with perihilar or distal bile-duct strictures.61,62 If IgG4-related disease is suspected, immunohistochemical confirmation of significant IgG4 immunostaining in biopsy or cytologic specimens is desirable (Figures 3 and 5). Even when IgG4-related diseases are excluded, approximately 10% of patients who present with what appears to be pCCA are found to have benign disease during surgery.63
Figure 5.

Algorithm for diagnosis and management of dCCA. LN, lymph node.
Treatment
Until recently, the only curative treatment for pCCA was surgery with lobar (or extended lobar) hepatic and bile duct resection, with regional lymphadenectomy and Roux-en-Y hepaticojejunostomy; 20% to 30% of patients survived until 5 years after surgery.4,64 However, it is a challenge to stage pCCA accurately, which is required to select the best treatment plan. A recently proposed staging system properly incorporates different tumor characteristics to guide management.51 Contralateral or bilateral vascular encasement often precludes surgery or obligates vascular reconstruction. Bilateral extension of pCCA to the level of the secondary biliary branches also precludes surgical resection. The presence of only regional (cystic, pericholedochal, hepatic arterial, portal, and posterior pancreaticoduodenal) lymph node metastases is not an absolute contraindication to resection, although patient outcomes are not as good as for patients without lymph node metastases.65
Patients with PSC often require liver transplantation, rather than resection, because of parenchymal liver disease, skip lesions, and an oncogenic field effect. Sufficient volume of the liver remnant is required for patient recovery. If the tumor is potentially resectable, but the remnant lobe is of limited volume, perioperative preparation with relief of biliary obstruction with ERC or percutaneous transhepatic cholangiography and portal vein embolization of the affected lobe for induction of hypertrophy of the contralateral nondiseased liver lobe can be considered.65 The role of preoperative biliary drainage is controversial. Clearly, it is not required for dCCA.66 However, many surgeons and hepatologists advocate preoperative biliary drainage for pCCAs that are to be resected because of the heavy metabolic and regenerative demands on the liver remnant. Having the liver remnant drainage accomplished before these demands makes physiological sense.
A subset of patients with early stage pCCA respond well to neoadjuvant chemoradiation therapy followed by liver transplantation; Murad et al67 reported 5-year rates of recurrence-free survival of 68% in a multicenter study—a rate similar to that for liver transplantation for other indications. Criteria for liver transplantation for patients without PSC include a tumor less than 3 cm in radial diameter, no intrahepatic or extrahepatic metastases, and unresectability. In patients with PSC, the criteria are a tumor less than 3 cm and no evidence for metastases; pCCA is therefore no longer a contraindication to liver transplantation.68 The presence of substantial residual tumor on the explant is associated with disease recurrence,69 so more studies are needed to determine the effects of adjuvant therapy for patients who have received liver transplants.
Patients with pCCA who are not candidates for resection or liver transplantation might consider systemic chemotherapy with gemcitabine and cisplatin. It is important to provide biliary drainage, in case of obstructive jaundice, to improve patients' comfort and ability to tolerate chemotherapy. An optimal level of bilirubin of 2 mg/dL or less should be achieved within 6 weeks if the level was 10 mg/dL or higher before stenting, and in 3 weeks if the level was less than 10 mg/dL.70 The jury is still out with regard to safety and effectiveness of unilateral vs bilateral biliary stenting. Some would argue that drainage of 50% of liver volume is necessary to prolong survival,71,72 whereas others have associated bilateral stenting with a higher rate of complications, including bacterial cholangitis, when bilateral contrast is injected.73
Selection of the proper stent to relieve biliary obstruction is becoming increasingly complex, given the growing number of options. Plastic stents should be used until diagnostic analyses and management decisions have been finalized. If patients are not candidates for surgery or liver transplantation, metal stents provide longer periods of patency than plastic stents, and are cost effective, with reasonable performance status and life expectancy of 6 months or longer.74–76 Covered metal stents might provide longer periods of patency than uncovered stents because they preclude tumor ingrowth.77–80 The ultimate decision should be individualized and based on local expertise.
Distal Cholangiocarcinoma
Classification and Diagnosis
dCCA develops anywhere between the cystic duct origin and the ampulla of Vater (without its involvement). dCCA arises from the precursor lesions intraductal papillary neoplasm or biliary intraepithelial neoplasia.11 Well-to-moderately differentiated adenocarcinoma is the most common histologic subtype.1 It is difficult to distinguish dCCA from early cancer of the head of the pancreas. However, dCCA is less aggressive than pancreatic cancer and merits more aggressive surgical intervention.
Similar to pCCA, most patients present with painless jaundice, which leads to further evaluation. Blood tests typically show an increase in cholestatic parameters; cross-sectional studies show thickening and/or stricture of the extrahepatic bile duct with proximal bile duct dilatation, and, more rarely than for pancreatic cancer, an associated mass (Figure 5). MRI with MRCP and CT can help to delineate the tumor burden—especially hepatic artery and portal vein involvement and the extension into the pancreas. ERC is an important diagnostic and therapeutic tool, but intraductal ultrasonography also is helpful in diagnosis.81 EUS with FNA can aid in evaluation of lymph node metastases and status of the vascular structures. The role of direct intraductal visualization with cholangioscopy for diagnosis of dCCA is under investigation.81 The data from studies dedicated specifically to dCCA are limited because of drawbacks of the prior CCA classification.
Treatment
Surgery for dCCA usually requires a Whipple procedure. When surgery is performed, positive lymph nodes are identified in 68% of patients with distal common bile duct tumors, compared with 28% and 29% for perihilar and intrahepatic CCAs, respectively.4 The overall 5-year survival rate of patients with dCCA after R0 resection is 27%, with a median survival time of 25 months.4 Perioperative chemotherapy or radiation therapy do not change the outcomes for patients with any subtype of CCA. Radiation therapy, based on our experience, can even precipitate the development of a difficult-to-manage cholangiopathy. Negative tumor margins are the most important predictor of patient survival. When R0 resection is not achievable, patients are given a combination of chemotherapy and relief of biliary obstruction.
Summary
CCAs arise from different topographic regions of the biliary tree; each subtype is characterized by its unique behavior (Figure 6). Cross-sectional imaging studies and ERC with brush cytology analysis are mainstays of evaluation that can be enhanced by EUS and assays for serum levels of CA19-9. Treatment options for CCA are limited and should be tailored for each tumor subtype with respect to its extent, other liver diseases, level of vascular involvement, and presence of metastases. The only effective therapies are resection with negative tumor margins, for all CCA subtypes, and liver transplantation, for a subset of early stage pCCAs. Systemic chemotherapy with gemcitabine and cisplatin is a pragmatic practice standard for patients with inoperable tumors, although more effective therapies need to be developed. There is insufficient evidence for the efficacy of chemoradiation as a neoadjuvant, aside from liver transplantation neoadjuvant protocols. Increasing our understanding of the pathogenesis of CCA could lead to more effective therapies.
Figure 6.

Key points in approach to CCA. FISH, fluorescent in situ hybridization.
Supplementary Material
Supplementary Table 1. Risk Factors for CCA
Supplementary Table 2. Histologic Types of iCCA
NOTE. Adapted with permission from Nakanuma et al.11
Acknowledgments
The authors would like to thank Dr David Nagorney for kindly contributing to the discussion of surgical management of cholangiocarcinoma, Dr Joachim Mertens for manuscript proofreading, and Ms Courtney Hoover for outstanding secretarial support.
Funding: Supported by National Institutes of Health grants DK59427 (G.J.G.) and T32 DK007198 (N.R.), and the Mayo Foundation.
Abbreviations used in this paper
- CA19-9
carbohydrate antigen 19-9
- CCA
cholangiocarcinoma
- CK
cytokeratin
- CT
computed tomography
- dCCA
distal cholangiocarcinoma
- ERC
endoscopic retrograde cholangiography
- EUS
endoscopic ultrasound
- FNA
fine-needle aspiration
- HCC
hepatocellular carcinoma
- HPC
hepatic progenitor cells
- iCCA
intrahepatic cholangiocarcinoma
- MRCP
magnetic resonance cholangiopancreatography
- MRI
magnetic resonance imaging
- pCCA
perihilar cholangiocarcinoma
- PET
positron emission tomography
- PSC
primary sclerosing cholangitis
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Supplementary Material: Note: To access the supplementary material accompanying this article, visit the online version of Clinical Gastroenterology and Hepatology at www.cghjournal.org, and at http://dx.doi.org/10.1016/j.cgh.2012.09.009.
References
- 1.Blechacz B, Komuta M, Roskams T, et al. Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev Gastroenterol Hepatol. 2011;8:512–522. doi: 10.1038/nrgastro.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Everhart JE, Ruhl CE. Burden of digestive diseases in the United States part III: liver, biliary tract, and pancreas. Gastroenterology. 2009;136:1134–1144. doi: 10.1053/j.gastro.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 3.Khan SA, Emadossadaty S, Ladep NG, et al. Rising trends in cholangiocarcinoma: is the lCD classification system misleading us? J Hepatol. 2012;56:848–854. doi: 10.1016/j.jhep.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 4.DeOliveira ML, Cunningham SC, Cameron JL, et al. Cholangiocarcinoma: thirty-one-year experience with 564 patients at a single institution. Ann Surg. 2007;245:755–762. doi: 10.1097/01.sla.0000251366.62632.d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kipp BR, Voss JS, Kerr SE, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol. 2012;43:1552–1558. doi: 10.1016/j.humpath.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Tyson GL, El-Serag HB. Risk factors for cholangiocarcinoma. Hepatology. 2011;54:173–184. doi: 10.1002/hep.24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee TY, Lee SS, Jung SW, et al. Hepatitis B virus infection and intrahepatic cholangiocarcinoma in Korea: a case-control study. Am J Gastroenterol. 2008;103:1716–1720. doi: 10.1111/j.1572-0241.2008.01796.x. [DOI] [PubMed] [Google Scholar]
- 8.Welzel TM, Graubard BI, El-Serag HB, et al. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma in the United States: a population-based case-control study. Clin Gastroenterol Hepatol. 2007;5:1221–1228. doi: 10.1016/j.cgh.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Söreide K, Körner H, Havnen J, et al. Bile duct cysts in adults. Br J Surg. 2004;91:1538–1548. doi: 10.1002/bjs.4815. [DOI] [PubMed] [Google Scholar]
- 10.Shaib Y, El-Serag HB. The epidemiology of cholangiocarcinoma. Semin Liver Dis. 2004;24:115–125. doi: 10.1055/s-2004-828889. [DOI] [PubMed] [Google Scholar]
- 11.Nakanuma Y, Sato Y, Harada K, et al. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J Hepatol. 2010;2:419–427. doi: 10.4254/wjh.v2.i12.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang MH, Chen CH, Yen CM, et al. Relation of hepatolithiasis to helminthic infestation. J Gastroenterol Hepatol. 2005;20:141–146. doi: 10.1111/j.1440-1746.2004.03523.x. [DOI] [PubMed] [Google Scholar]
- 13.Donato F, Gelatti U, Tagger A, et al. Intrahepatic cholangiocarcinoma and hepatitis C and B virus infection, alcohol intake, and hepatolithiasis: a case-control study in Italy. Cancer Causes Control. 2001;12:959–964. doi: 10.1023/a:1013747228572. [DOI] [PubMed] [Google Scholar]
- 14.Shaib YH, El-Serag HB, Davila JA, et al. Risk factors of intrahepatic cholangiocarcinoma in the United States: a case-control study. Gastroenterology. 2005;128:620–626. doi: 10.1053/j.gastro.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 15.Welzel TM, Mellemkjaer L, Gloria G, et al. Risk factors for intrahepatic cholangiocarcinoma in a low-risk population: a nationwide case-control study. Int J Cancer. 2007;120:638–641. doi: 10.1002/ijc.22283. [DOI] [PubMed] [Google Scholar]
- 16.Tocchi A, Mazzoni G, Liotta G, et al. Late development of bile duct cancer in patients who had biliary-enteric drainage for benign disease: a follow-up study of more than 1,000 patients. Ann Surg. 2001;234:210–214. doi: 10.1097/00000658-200108000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burak K, Angulo P, Pasha TM, et al. Incidence and risk factors for cholangiocarcinoma in primary sclerosing cholangitis. Am J Gastroenterol. 2004;99:523–526. doi: 10.1111/j.1572-0241.2004.04067.x. [DOI] [PubMed] [Google Scholar]
- 18.Farrant JM, Hayllar KM, Wilkinson ML, et al. Natural history and prognostic variables in primary sclerosing cholangitis. Gastroenterology. 1991;100:1710–1717. doi: 10.1016/0016-5085(91)90673-9. [DOI] [PubMed] [Google Scholar]
- 19.Helzberg JH, Peterson JM, Boyer JL. Improved survival with primary sclerosing cholangitis. A review of clinicopathologic features and comparison of symptomatic and asymptomatic patients. Gastroenterology. 1887;92:1869–1875. doi: 10.1016/0016-5085(87)90618-4. [DOI] [PubMed] [Google Scholar]
- 20.Kornfeld D, Ekbom A, Ihre T. Survival and risk of cholangiocarcinoma in patients with primary sclerosing cholangitis. A population-based study. Scand J Gastroenterol. 1997;32:1042–1045. doi: 10.3109/00365529709011222. [DOI] [PubMed] [Google Scholar]
- 21.Razumilava N, Gores GJ, Lindor KD. Cancer surveillance in patients with primary sclerosing cholangitis. Hepatology. 2011;54:1842–1852. doi: 10.1002/hep.24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergquist A, Ekbom A, Olsson R, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002;36:321–327. doi: 10.1016/s0168-8278(01)00288-4. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Takasaki K, Nakano M, et al. Minute nodular intrahepatic cholangiocarcinoma. Cancer. 1998;82:2145–2149. [PubMed] [Google Scholar]
- 24.Yamasaki S. Intrahepatic cholangiocarcinoma: macroscopic type and stage classification. J Hepatobiliary Pancreat Surg. 2003;10:288–291. doi: 10.1007/s00534-002-0732-8. [DOI] [PubMed] [Google Scholar]
- 25.Komuta M, Govaere O, Vandecaveye V, et al. Histological diversity in cholangiocellular carcinoma reflects the different cholangiocyte phenotypes. Hepatology. 2012;55:1876–1888. doi: 10.1002/hep.25595. [DOI] [PubMed] [Google Scholar]
- 26.Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. doi: 10.1038/sj.onc.1209558. [DOI] [PubMed] [Google Scholar]
- 27.Fan B, Malato Y, Calvisi DF, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911–2915. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rimola J, Forner A, Reig M, et al. Cholangiocarcinoma in cirrhosis: absence of contrast washout in delayed phases by magnetic resonance imaging avoids misdiagnosis of hepatocellular carcinoma. Hepatology. 2009;50:791–798. doi: 10.1002/hep.23071. [DOI] [PubMed] [Google Scholar]
- 29.Vilgrain V. Staging cholangiocarcinoma by imaging studies. HPB (Oxford) 2008;10:106–109. doi: 10.1080/13651820801992617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anderson CD, Rice MH, Pinson CW, et al. Fluorodeoxyglucose PET imaging in the evaluation of gallbladder carcinoma and cholangiocarcinoma. J Gastrointest Surg. 2004;8:90–97. doi: 10.1016/j.gassur.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Petrowsky H, Wildbrett P, Husarik DB, et al. Impact of integrated positron emission tomography and computed tomography on staging and management of gallbladder cancer and cholangiocarcinoma. J Hepatol. 2006;45:43–50. doi: 10.1016/j.jhep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Lan BY, Kwee SA, Wong LL. Positron emission tomography in hepatobiliary and pancreatic malignancies: a review. Am J Surg. 2012;204:232–241. doi: 10.1016/j.amjsurg.2011.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel AH, Harnois DM, Klee GG, et al. The utility of CA 19–9 in the diagnoses of cholangiocarcinoma in patients without primary sclerosing cholangitis. Am J Gastroenterol. 2000;95:204–207. doi: 10.1111/j.1572-0241.2000.01685.x. [DOI] [PubMed] [Google Scholar]
- 34.Charatcharoenwitthaya P, Enders FB, Halling KC, et al. Utility of serum tumor markers, imaging, and biliary cytology for detecting cholangiocarcinoma in primary sclerosing cholangitis. Hepatology. 2008;48:1106–1117. doi: 10.1002/hep.22441. [DOI] [PubMed] [Google Scholar]
- 35.Nehls O, Gregor M, Klump B. Serum and bile markers for cholangiocarcinoma. Semin Liver Dis. 2004;24:139–154. doi: 10.1055/s-2004-828891. [DOI] [PubMed] [Google Scholar]
- 36.Durnez A, Verslype C, Nevens F, et al. The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology. 2006;49:138–151. doi: 10.1111/j.1365-2559.2006.02468.x. [DOI] [PubMed] [Google Scholar]
- 37.Sapisochin G, Fidelman N, Roberts JP, et al. Mixed hepatocellular cholangiocarcinoma and intrahepatic cholangiocarcinoma in patients undergoing transplantation for hepatocellular carcinoma. Liver Transpl. 2011;17:934–942. doi: 10.1002/lt.22307. [DOI] [PubMed] [Google Scholar]
- 38.Uenishi T, Kubo S, Yamamoto T, et al. Cytokeratin 19 expression in hepatocellular carcinoma predicts early postoperative recurrence. Cancer Sci. 2003;94:851–857. doi: 10.1111/j.1349-7006.2003.tb01366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Endo I, Gonen M, Yopp AC, et al. Intrahepatic cholangiocarcinoma: rising frequency, improved survival, and determinants of outcome after resection. Ann Surg. 2008;248:84–96. doi: 10.1097/SLA.0b013e318176c4d3. [DOI] [PubMed] [Google Scholar]
- 40.Li YY, Li H, Lv P, et al. Prognostic value of cirrhosis for intrahepatic cholangiocarcinoma after surgical treatment. J Gastrointest Surg. 2011;15:608–613. doi: 10.1007/s11605-011-1419-8. [DOI] [PubMed] [Google Scholar]
- 41.Rana A, Hong JC. Orthotopic liver transplantation in combination with neoadjuvant therapy: a new paradigm in the treatment of unresectable intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2012;28:258–265. doi: 10.1097/MOG.0b013e32835168db. [DOI] [PubMed] [Google Scholar]
- 42.Goodman ZD, Ishak KG, Langloss JM, et al. Combined hepatocellular-cholangiocarcinoma. A histologic and immunohistochemical study. Cancer. 1985;55:124–135. doi: 10.1002/1097-0142(19850101)55:1<124::aid-cncr2820550120>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 43.Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 44.Horgan AM, Amir E, Walter T, et al. Adjuvant therapy in the treatment of biliary tract cancer: a systematic review and meta-analysis. J Clin Oncol. 2012;30:1934–1940. doi: 10.1200/JCO.2011.40.5381. [DOI] [PubMed] [Google Scholar]
- 45.Andersen JB, Spee B, Blechacz BR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031. e1015. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kiefer MV, Albert M, McNally M, et al. Chemoembolization of intrahepatic cholangiocarcinoma with cisplatinum, doxorubicin, mitomycin C, ethiodol, and polyvinyl alcohol: a 2-center study. Cancer. 2011;117:1498–1505. doi: 10.1002/cncr.25625. [DOI] [PubMed] [Google Scholar]
- 47.Hoffmann RT, Paprottka PM, Schön A, et al. Transarterial hepatic yttrium-90 radioembolization in patients with unresectable intrahepatic cholangiocarcinoma: factors associated with prolonged survival. Cardiovasc Intervent Radiol. 2012;35:105–116. doi: 10.1007/s00270-011-0142-x. [DOI] [PubMed] [Google Scholar]
- 48.Kim JH, Won HJ, Shin YM, et al. Radiofrequency ablation for the treatment of primary intrahepatic cholangiocarcinoma. AJR Am J Roentgenol. 2011;196:W205–W209. doi: 10.2214/AJR.10.4937. [DOI] [PubMed] [Google Scholar]
- 49.Blechacz B, Gores GJ. Cholangiocarcinoma: advances in pathogenesis, diagnosis, and treatment. Hepatology. 2008;48:308–321. doi: 10.1002/hep.22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagorney DM, Donohue JH, Farnell MB, et al. Outcomes after curative resections of cholangiocarcinoma. Arch Surg. 1993;128:871–877. doi: 10.1001/archsurg.1993.01420200045008. discussion, 877-879. [DOI] [PubMed] [Google Scholar]
- 51.Deoliveira ML, Schulick RD, Nimura Y, et al. New staging system and a registry for perihilar cholangiocarcinoma. Hepatology. 2011;53:1363–1371. doi: 10.1002/hep.24227. [DOI] [PubMed] [Google Scholar]
- 52.Bismuth H, Corlette MB. Intrahepatic cholangioenteric anastomosis in carcinoma of the hilus of the liver. Surg Gynecol Obstet. 1975;140:170–178. [PubMed] [Google Scholar]
- 53.Heimbach JK, Sanchez W, Rosen CB, et al. 2011 Trans-peritoneal fine needle aspiration biopsy of hilar cholangiocarcinoma is associated with disease dissemination. HPB (Oxford) 2011;13:356–360. doi: 10.1111/j.1477-2574.2011.00298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levy MJ, Heimbach JK, Gores GJ. Endoscopic ultrasound staging of cholangiocarcinoma. Curr Opin Gastroenterol. 2012;28:244–252. doi: 10.1097/MOG.0b013e32835005bc. [DOI] [PubMed] [Google Scholar]
- 55.Barr Fritcher EG, Kipp BR, Slezak JM, et al. Correlating routine cytology, quantitative nuclear morphometry by digital image analysis, and genetic alterations by fluorescence in situ hybridization to assess the sensitivity of cytology for detecting pancreatobiliary tract malignancy. Am J Clin Pathol. 2007;128:272–279. doi: 10.1309/BC6DY755Q3T5W9EE. [DOI] [PubMed] [Google Scholar]
- 56.Gonda TA, Glick MP, Sethi A, et al. Polysomy and p16 deletion by fluorescence in situ hybridization in the diagnosis of indeterminate biliary strictures. Gastrointest Endosc. 2012;75:74–79. doi: 10.1016/j.gie.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 57.Fritcher EG, Kipp BR, Halling KC, et al. A multivariable model using advanced cytologic methods for the evaluation of indeterminate pancreatobiliary strictures. Gastroenterology. 2009;136:2180–2186. doi: 10.1053/j.gastro.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 58.Kipp BR, Stadheim LM, Halling SA, et al. A comparison of routine cytology and fluorescence in situ hybridization for the detection of malignant bile duct strictures. Am J Gastroenterol. 2004;99:1675–1681. doi: 10.1111/j.1572-0241.2004.30281.x. [DOI] [PubMed] [Google Scholar]
- 59.Moreno Luna LE, Kipp B, Halling KC, et al. Advanced cytologic techniques for the detection of malignant pancreatobiliary strictures. Gastroenterology. 2006;131:1064–1072. doi: 10.1053/j.gastro.2006.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bangarulingam SY, Bjornsson E, Enders F, et al. Long-term outcomes of positive fluorescence in situ hybridization tests in primary sclerosing cholangitis. Hepatology. 2010;51:174–180. doi: 10.1002/hep.23277. [DOI] [PubMed] [Google Scholar]
- 61.Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–715. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 62.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 63.Baskin-Bey ES, Devarbhavi HC, Nagorney DM, et al. Idiopathic benign biliary strictures in surgically resected patients with presumed cholangiocarcinoma. HPB (Oxford) 2005;7:283–288. doi: 10.1080/13651820500292954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosen CB, Heimbach JK, Gores GJ. Surgery for cholangiocarcinoma: the role of liver transplantation. HPB (Oxford) 2008;10:186–189. doi: 10.1080/13651820801992542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagorney DM, Kendrick ML. Hepatic resection in the treatment of hilar cholangiocarcinoma. Adv Surg. 2006;40:159–171. doi: 10.1016/j.yasu.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 66.van der Gaag NA, Rauws EA, van Eijck CH, et al. Preoperative biliary drainage for cancer of the head of the pancreas. N Engl J Med. 2010;362:129–137. doi: 10.1056/NEJMoa0903230. [DOI] [PubMed] [Google Scholar]
- 67.Murad SD, Kim WR, Harnois DM, et al. Efficacy of neoadjuvant chemoradiation, followed by liver transplantation, for perihilar cholangiocarcinoma at 12 US centers. Gastroenterology. 2012;143:88–98. doi: 10.1053/j.gastro.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hong JC, Jones CM, Duffy JP, et al. Comparative analysis of resection and liver transplantation for intrahepatic and hilar cholangiocarcinoma: a 24-year experience in a single center. Arch Surg. 2011;146:683–689. doi: 10.1001/archsurg.2011.116. [DOI] [PubMed] [Google Scholar]
- 69.Darwish Murad S, Kim WR, Therneau T, et al. Predictors of pretransplant dropout and posttransplant recurrence in patients with perihilar cholangiocarcinoma. Hepatology. 2012;56:972–981. doi: 10.1002/hep.25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weston BR, Ross WA, Wolff RA, et al. Rate of bilirubin regression after stenting in malignant biliary obstruction for the initiation of chemotherapy: how soon should we repeat endoscopic retrograde cholangiopancreatography? Cancer. 2008;112:2417–2423. doi: 10.1002/cncr.23454. [DOI] [PubMed] [Google Scholar]
- 71.Vienne A, Hobeika E, Gouya H, et al. Prediction of drainage effectiveness during endoscopic stenting of malignant hilar strictures: the role of liver volume assessment. Gastrointest Endosc. 2010;72:728–735. doi: 10.1016/j.gie.2010.06.040. [DOI] [PubMed] [Google Scholar]
- 72.Deviere J, Baize M, de Toeuf J, et al. Long-term follow-up of patients with hilar malignant stricture treated by endoscopic internal biliary drainage. Gastrointest Endosc. 1988;34:95–101. doi: 10.1016/s0016-5107(88)71271-7. [DOI] [PubMed] [Google Scholar]
- 73.Chang WH, Kortan P, Haber GB. Outcome in patients with bifurcation tumors who undergo unilateral versus bilateral hepatic duct drainage. Gastrointest Endosc. 1998;47:354–362. doi: 10.1016/s0016-5107(98)70218-4. [DOI] [PubMed] [Google Scholar]
- 74.Soderlund C, Linder S. Covered metal versus plastic stents for malignant common bile duct stenosis: a prospective, randomized, controlled trial. Gastrointest Endosc. 2006;63:986–995. doi: 10.1016/j.gie.2005.11.052. [DOI] [PubMed] [Google Scholar]
- 75.Raju RP, Jaganmohan SR, Ross WA, et al. Optimum palliation of inoperable hilar cholangiocarcinoma: comparative assessment of the efficacy of plastic and self-expanding metal stents. Dig Dis Sci. 2011;56:1557–1564. doi: 10.1007/s10620-010-1550-5. [DOI] [PubMed] [Google Scholar]
- 76.Yeoh KG, Zimmerman MJ, Cunningham JT, et al. Comparative costs of metal versus plastic biliary stent strategies for malignant obstructive jaundice by decision analysis. Gastrointest Endosc. 1999;49:466–471. doi: 10.1016/s0016-5107(99)70044-1. [DOI] [PubMed] [Google Scholar]
- 77.Kullman E, Frozanpor F, Söderlund C, et al. Covered versus uncovered self-expandable nitinol stents in the palliative treatment of malignant distal biliary obstruction: results from a randomized, multicenter study. Gastrointest Endosc. 2010;72:915–923. doi: 10.1016/j.gie.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 78.Krokidis M, Fanelli F, Orgera G, et al. Percutaneous treatment of malignant jaundice due to extrahepatic cholangiocarcinoma: covered Viabil stent versus uncovered Wallstents. Cardiovasc Intervent Radiol. 2010;33:97–106. doi: 10.1007/s00270-009-9604-9. [DOI] [PubMed] [Google Scholar]
- 79.Isayama H, Komatsu Y, Tsujino T, et al. A prospective randomised study of “covered” versus “uncovered” diamond stents for the management of distal malignant biliary obstruction. Gut. 2004;53:729–734. doi: 10.1136/gut.2003.018945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kahaleh M, Tokar J, Conaway MR, et al. Efficacy and complications of covered Wallstents in malignant distal biliary obstruction. Gastrointest Endosc. 2005;61:528–533. doi: 10.1016/s0016-5107(04)02593-3. [DOI] [PubMed] [Google Scholar]
- 81.Siddiqui AA, Mehendiratta V, Jackson W, et al. Identification of cholangiocarcinoma by using the spyglass spyscope system for peroral cholangioscopy and biopsy collection. Clin Gastroenterol Hepatol. 2012;10:466–471. doi: 10.1016/j.cgh.2011.12.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Risk Factors for CCA
Supplementary Table 2. Histologic Types of iCCA
NOTE. Adapted with permission from Nakanuma et al.11