

Abstract
This review introduces the pathology of aminoglycoside antibiotic and the cisplatin chemotherapy classes of drugs, discusses oxidative stress in the inner ear as a primary trigger for cell damage, and delineates the ensuing cell death pathways. Among potentially ototoxic (damaging the inner ear) therapeutics, the platinum-based anti-cancer drugs and the aminoglycoside antibiotics are of critical clinical importance. Both drugs cause sensorineural hearing loss in patients, a side effect that can be reproduced in experimental animals. Hearing loss is reflected primarily in damage to outer hair cells, beginning in the basal turn of the cochlea. In addition, aminoglycosides might affect the vestibular system while cisplatin seems to have a much lower likelihood to do so. Finally, based on an understanding the mechanisms of ototoxicity pharmaceutical ways of protection of the cochlea are presented
Keywords: aminoglycosides, cisplatin, ototoxicity, oxidative stress, otoprotection
Introduction: Ototoxic drugs
Many therapeutics can be classified as “ototoxic”, i.e. having side effects on the inner ear. More specifically, they can be “cochleotoxic” (affecting the hearing organ) or “vestibulotoxic” (affecting balance). Despite their potential ototoxicity, such drugs remain in current use, either because the risk of damage to the ear is small, or because there simply are no comparable alternatives. Historically, cochlear and vestibular complications have been experienced by patients and recorded by clinicians for centuries, but it was not until the 1940s that ototoxicity caught widespread awareness. At that time, the first aminoglycoside, streptomycin, was discovered by Selman Waksman and his graduate student, Albert Schatz (Schatz et al., 1944) and introduced as a highly successful drug against tuberculosis. Within a short time it became obvious that many patients suffered vestibular and hearing deficits, as well as kidney problems (Hinshaw and Feldman, 1945). The ensuing years saw the introduction of further natural and semi-synthetic compounds in search of improved aminoglycosides, but side effects remained associated with all of them. Today, aminoglycosides remain in widespread use as broad-spectrum antibiotics, including specific applications against life-threatening sepsis, Mycobacterium tuberculosis and opportunistic pulmonary Pseudomonas infections in cystic fibrosis patients. Arguably, however, the largest market for aminoglycosides is in developing countries. The drugs offer the benefits of being extremely inexpensive to produce, having broad-spectrum efficacy against most infections, causing a relatively small incidence of allergic reactions, and being easily accessible, in many areas even without a prescription. The advent of aminoglycoside antibiotics was considered a crowning achievement in medicine and earned Waksman the Nobel Prize in 1952.
Unlike the immediate recognition of aminoglycoside antibiotics as efficacious antibacterial drugs, the discovery of cisplatin as an effective anticancer agent was an accidental one. Cisplatin was already synthesized in 1845 by Peyrone, earning it the common name of Peyrone’s salt. Over 70 years later, Barnett Rosenberg discovered the biological application of cisplatin while studying the effects of electric fields on bacterial growth, deducing that the platinum compounds from his electrodes were inhibiting cell division (Rosenberg et al., 1965). He and his colleagues then demonstrated cisplatin as a successful antitumor agent in rats and mice, efficacious against advanced tumors and tumors resistant to other drugs (Rosenberg et al., 1969). Today, cisplatin is used singly or in combination therapy, especially in the treatment of head, neck, lung, bladder, cervical, ovarian, testicular, and gastrointestinal cancers, as well as malignant gliomas and metastatic cancers like metastatic melanoma, mesothelioma, and those of the prostate and breast (Boulikas and Vougiouka, 2004). The later synthesized platinum-based antineoplastic agents carboplatin and oxaliplatin are indicated in limited applications: carboplatin against specific types of breast cancer and in combination therapy against lung cancer, oxaliplatin in combination therapy against colorectal cancer. Cisplatin is considered the most ototoxic in this class, but is up to 45 times more effective against certain neoplasms than carboplatin and oxaliplatin.
This review is dedicated to the detailed discussion of cisplatin and aminoglycosides but we should not neglect to mention other ototoxins. Salicylate (aspirin) has long been associated with elevated hearing threshold and tinnitus (ringing in the ear), two effects that disappear with cessation of drug intake. Loop diuretics like furosemide, ethacrynic acid, and bumetanide have transient side effects on the inner ear given singly, but create disastrous repercussions in combination with aminoglycoside antibiotics. Recently, observational evidence has earned some phosphodiesterase inhibitors (such as Cialis, Viagra, Revatio, and Levitra) a safety warning that administration may be associated with sudden hearing loss and/or vestibular disturbances (Maddox et al., 2009). Other potentially ototoxic substances include organometals like organotins and mercury preparations, as well as solvents like toluene and styrene used in industrial settings, putting workers at risk for auditory side effects.
Mechanisms of action as therapeutic agents
Cisplatin forms stable covalent bonds by an alkylation-like reaction with nuclear DNA. Guanine is an especially susceptible target at the 7-nitrogen atom where cisplatin forms inter-and intra-strand cross-links (Rozencweig et al., 1977); the intra-strand crosslinks appear to determine the cytotoxicity of cisplatin for tumors (Wang and Lippard, 2005). High mobility group (HMG) proteins recognize and bind to DNA at the 1,2-d(GpG) crosslinks, and a ternary complex of cisplatin-DNA-HMGB1 can block transcription factors, thereby preventing both transcription and replication. This action, in turn, may send out DNA damage signals that trigger apoptosis. Contravening this therapeutic action, intra-strand crosslinks can be removed by cellular repair mechanisms, for example, nucleotide excision repair (McHugh et al., 2001), which appears to be important in controlling cisplatin sensitivity of tumors. Testicular tumors, which are highly sensitive to cisplatin, exhibit low levels of mRNA for the DNA repair enzymes XPA and ERCC1-XPF (Rabik and Dolan, 2007). The fate of a particular cell toward cell survival or apoptosis following cisplatin-induced DNA lesions therefore depends upon its tolerance of the insult and its ability to repair (Liedert et al., 2006).
In contrast, aminoglycoside antibiotics therapeutically target prokaryotic cells rather specifically and exert their antibacterial effects mainly through an inhibition of protein synthesis. The drugs bind to the A-translational site of the 16S rRNA of the 30S ribosomal subunit and cause an increased misreading of the mRNA. The resultant accumulation of dysfunctional proteins leads the cell into stress response, and eventually into death. Aminoglycosides are rendered inactive by resistant bacteria through enzymatic reactions of acetylation, phosphorylation or adenylation. Different aminoglycosides have different susceptibility to these inactivating enzymes, which allows for effectiveness of combination therapy against resistant strains of bacteria.
Although prokaryotic and eukaryotic ribosomes are structurally distinct, aminoglycosides do interact with mammalian ribosomes, albeit to a lesser degree, which is exploited in stop codon suppression therapy and constitutes a risk factor in the case of the sensitizing mitochondrial “1555 mutation”, discussed later. “Stop codon suppression” is an emerging application for aminoglycoside antibiotics in the therapy of genetic disorders. Close to 20% of human genetic disorders are caused by premature stop codons created by point mutations in the DNA sequence, leading to some forms of cystic fibrosis, Duchenne muscular dystrophy, Hurler syndrome, hemophilia A, and others (Kaufman, 1999). Aminoglycosides may cause a slight “misreading” of such disease-causing incorrect codons, thereby overriding the “stop” message and restoring the synthesis of functional proteins, generally to a small extent but sufficient to alleviate symptoms. Clinical trials involving patients with Duchenne muscular dystrophy have confirmed the utility of this approach (Wagner et al., 2001; Malik et al., 2010). The dilemma is to balance the therapeutic efficacy with the potential ototoxicity; novel “designer aminoglycosides” have been introduced attempting to maximize stop codon suppression and minimize side effects (Nudelman et al., 2009).
Side effects
Clinical experience to date has found the most severe effects of cisplatin administration to be on the kidney, affecting as much as one third of patients (Taguchi et al., 2005) while aminoglycosides cause nephrotoxicity in about one fifth of patients (Swan, 1997). Chemotherapy can be limited by these side effects. Other adverse actions of cisplatin may include neurotoxicity, thrombocytopenia, leukopenia, anorexia, nausea, and vomiting. Of those, neurotoxicity tends to be problematic, causing tingling sensations and numbness in the extremities, and possibly progressing even after treatment has ceased (Grunberg et al., 1989).
Both cisplatin and aminoglycosides actively accumulate in the kidneys, aminoglycosides localizing to the proximal tubules. The mechanism of accumulation is unknown, but is thought to be by way of carrier-mediated transport in the case of cisplatin, and likely by way of the glycoprotein transporter megalin in the case of aminoglycosides (Moestrup et al., 1995, Kawai et al., 2005). Cell death is evident in the proximal and distal tubules and in the loop of Henle after administration of either drug, resulting in risk of acute renal failure. Organ failure appears to be owed to decreased kidney perfusion and, in the case of aminoglycosides, tubular obstruction and reduced glomerular filtration (Lopez-Novoa et al., 2011).
The severity of ototoxicity varies among the aminoglycosides: neomycin is considered highly toxic, gentamicin, kanamycin and tobramycin somewhat less, and amikacin and netilmicin are regarded as the least toxic. Their adverse actions manifest as either vestibulotoxicity, cochleotoxicity, or both, while cisplatin appears to be exclusively cochleotoxic. Predisposition to harm the cochlea or the vestibular system also varies with the type of aminoglycoside and their preference is not related to any site-specific uptake mechanism or to drug levels in the cochlear and vestibular tissues (Dulon et al., 1986). Amikacin and neomycin tend to primarily affect the cochlea, while gentamicin tends to be quite vestibulotoxic. Indeed, the severity of gentamicin-induced vestibulotoxicity is the basis of vestibular ablation for treatment of Menière’s disease (Blakley, 1997).
Hearing loss induced by cisplatin or aminoglycosides is generally bilateral and begins at high frequencies, extending to lower frequencies with prolonged treatment. Vestibulotoxicity of aminoglycosides can manifest as loss of balance with or without vertigo. Symptoms are usually delayed days or weeks after commencement of therapy and can continue to progress after treatment has been discontinued. An exception to this temporal progression may be seen in in patients with specific risk factors. For this group of patients, hearing loss can occur very rapidly, sometimes as a result of only one dose of the drug, as discussed later.
Incidence of adverse effects
The incidence of drug-induced hearing-loss is often underreported due to the fact that high-frequency hearing loss would not commonly interfere with communication and therefore might not even be noticed by patients. Estimates of the incidence of hearing and balance deficits from aminoglycosides vary widely between clinical studies. This is owed to the varying treatment durations and doses, patient populations, and hearing assessment techniques employed, as well as the definition of “hearing loss”. By and large, however, aminoglycosides incur about a 20% incidence of auditory and a 15% incidence of vestibular disturbances in short-term treatment (Fee, 1980, Moore et al.,1984, Lerner et al., 1986). Testing at higher frequencies that exceed those utilized in routine audiograms (>8 kHz) reveals greater proportions of losses (47% of patients treated; Fausti et al., 1992). Furthermore, audiologic follow-up with patients in many studies may not be protracted enough to identify additional damage that occurs long after treatment (Einarsson et al., 2010, Einarsson et al., 2011). For patients with cystic fibrosis that receive repeated prophylactic treatments with aminoglycosides against pulmonary infections, reports of cochleotoxicity cover a wide range, but may be near 17% (Mulheran et al., 2001). The incidence of ototoxicity in tuberculosis patients is time- and dose-dependent. It may be low during the initial phase of therapy, but prolonged treatment with aminoglycosides for 6 to 12 months will lead to a measurable hearing loss in all patients (Duggal and Sarkar, 2007). Cisplatin therapy appears to be associated with a consistently higher occurrence of hearing deficits. Some studies estimate the incidence as high as 90 to 100% (Benedetti Panici et al., 1993).
Risk factors
While ototoxic side effects are largely determined by dose and duration of treatment, several risk factors influence individual susceptibility. Diet and nutritional status can confer risk for both cisplatin and aminoglycoside therapy (Garetz et al., 1994) and patients with hypoalbuminemia and anemia may develop more profound hearing loss than those with normal levels of albumin and hemoglobin (Blakley, 1997). This is particularly relevant for cisplatin treatment as hypoalbuminemia is very common in cancer patients, one study finding 82% of cancer patients in the ICU to have albumin levels below the normal range (Namendys-Silva et al., 2011). Anemia can also be caused by particular cancers that interfere with hematopoiesis, as well as by certain cancer treatments.
Other factors that enhance the risk of cisplatin-induced hearing loss include renal insufficiency, pre-existing hearing losses, noise insults, cranial irradiation, as well as co-treatment with other drugs like vincristine (Bokemeyer et al., 1998). In the case of aminoglycosides, concomitant treatment with ethacrynic acid can cause a precipitous hearing loss (Mathog and Klein, 1969) and exposure to intense noise may also aggravate ototoxicity. Finally, owing to changes in metabolism and organ function, young children and the elderly are more susceptible to ototoxicity from cisplatin (Laurell and Jungnelius, 1990, Li et al., 2004). Such a connection has not been confirmed for aminoglycosides, but animal studies suggest an enhanced sensitivity around the onset of cochlear functional differentiation in utero (Raphael et al., 1983).
Genetics and hearing loss
The pharmacological efficacy as well as the side effects of drugs is influenced by uptake and excretion, as well as drug metabolism and detoxification mechanisms. Variations in these parameters are controlled by the genetic make-up of the individual. It is therefore not surprising that genetic factors modulate the ototoxicity of both cisplatin and aminoglycoside antibiotics. The pharmacogenomics of cisplatin-induced ototoxicity has been recently reviewed (Mukherjea and Rybak, 2011) so that a brief account may suffice here.
Glutathione S-transferases (GST) are crucial enzymes in detoxification, catalyzing the conjugation of potentially damaging electrophiles, like cisplatin, with glutathione in an innate protective mechanism. GST proteins are expressed in the inner ear (el Barbary et al., 1993) and polymorphisms of genes coding for these proteins, primarily GSTM1, GSTP1 and GSTT1, have been implicated in modulating the response to cisplatin ototoxicity. In a study of 173 testicular cancer survivors treated with cisplatin, GSTP1 polymorphisms AA or AG were found in 145 patients, of whom 34 (23.4%) reported severe hearing loss. Of 28 patients with the protective GSTP1 GG only 2 patients (7%) had hearing loss. In contrast, the presence of GSTM1 was associated with heightened susceptibility to cisplatin-induced hearing impairment (Oldenburg et al., 2007).
Associations between genetic variants and cisplatin ototoxicity were also found for thiopurine S-methyl transferase (TMPT) and catechol-O-methyl transferase (COMT). An increasing number of TMPT and COMT risk alleles correlated with earlier onset and greater severity of cisplatin-induced hearing loss (Ross et al., 2009). Both of these enzymes are methyl-transfereases that utilize S-adenosylmethionine as a methyl donor in methionine synthesis. Ototoxicity may be linked to increased S-adenosylmethionine levels caused by reduced enzyme activity of TMPT and COMT but an exact mechanism remains to be established. A novel gene, COMT2, has been reported to code for the enzyme COMT2, a unique isoform of COMT. COMT2 is expressed in the inner and outer hair cells of the cochlea (Du et al., 2008) and a concomitant loss of COMT function may promote cisplatin ototoxicity (Pussegoda, 2010).
Mutations in the mitochondrial genome are also well-defined risk factors in cisplatin-and aminoglycoside-induced cochlear damage. In a group of twenty cancer survivors with cisplatin-induced ototoxicity, five were found to cluster in a rare European mitochondrial single nucleotide polymorphism associated with Leber’s Hereditary Optic Atrophy (Peters et al., 2003). Quite striking is the effect on aminoglycoside ototoxicity of a A1555G mutation in the 12S ribosomal RNA (Prezant et al., 1993) whose carriers may sustain profound deafness after a single injection. This mutation pervades all geographic and ethnic groups (reviewed by Fischel-Ghodsian, 2005) and may account for deafness in about 20% of patients with aminoglycoside ototoxicity. Patients with the A1555G genotype may also develop spontaneous non-syndromic deafness. Several other mitochondrial mutations identified contribute an additional, however relatively small, risk of hearing deficits. It is interesting to note that mutations that increase susceptibility to aminoglycoside-induced hearing loss appear to alter the morphology of the mitochondrial 12S ribosomal subunit such that it is structurally more similar to the bacterial ribosomal RNA and thus becomes a better target for aminoglycoside-induced protein misreading (Matt et al., 2012). Evidence, however, whether protein synthesis is indeed affected in the cochlea in vivo remains indirect. Intriguingly, the vestibular system is not involved in the enhanced response to aminoglycosides (Tono et al., 2001), leaving the question unresolved on how the mutation interacts with proposed mechanisms of toxicity.
Pathology of inner-ear damage
The cochlear and vestibular pathology of aminoglycoside-induced ototoxicity has been long established through the study of human temporal bones. In both organs of the inner ear, the hair cells are accepted as the primary targets of damage underlying the loss of function (Causse et al., 1949, Ruedi, 1952, review of early studies by Hawkins, 1976). In the organ of Corti, the pathologies of cisplatin and aminoglycoside ototoxicity are remarkably similar. Outer hair cells are generally the most susceptible to damage and are affected gradually from the base of the cochlea to its apex (Hawkins, 1976, Schuknecht, 1993). This pattern of pathology leads to an initial hearing loss of high frequencies, which are processed in the base, followed by a progressive loss into lower speech frequencies. In addition, the drugs induce a lateral gradient of damage as well, with hair cell death occurring in the first row (innermost) of outer hair cells first, followed by the second and third rows as the lesion progresses (as seen in fig. 1). Inner hair cells are more resistant and they usually disappear only after outer hair cells in the vicinity have been completely ablated.
Figure 1. Typical morphological damage by aminoglycoside antibiotics.
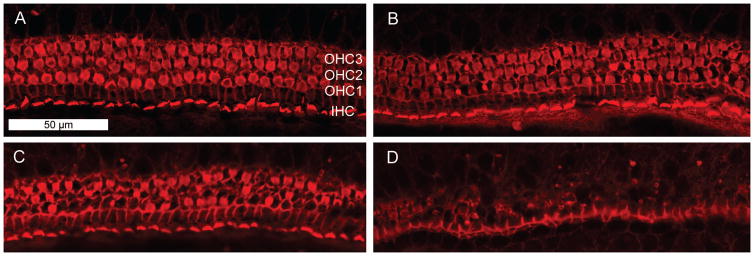
Explants of the postnatal mouse organ of Corti were cultured for 72 h with or without 3.5 μM gentamicin, then fixed and stained for actin with rhodamine phalloidin (Chen et al., 2009).
A: Incubation without drug. The red stain outlines the orderly arrangement of the three rows of outer hair cells (OHC1, OHC2, OHC3) and one row of inner hair cells (IHC).
B, C, D: Incubation with gentamicin. The middle section of the organ of Corti (B) shows a few missing outer hair cells, mostly in row 1. Further towards the base (C), more damage is evident. The basal segment (D) has essentially lost all outer hair cells while inner hair cells remain.
Experiment and photo courtesy of Dr. Naoki Oishi.
Additionally, aminoglycosides induce vestibulotoxicity. Investigations of vestibulotoxicity, in fact, preceded those of cochleotoxicity (Causse et al., 1949), since the first aminoglycoside introduced into clinical practice, streptomycin, was primarily vestibulotoxic. Deficits in the vestibular organ are mostly due to loss of type I and type II hair cells. In contrast, studies of human temporal bones after cisplatin treatment agree that the cells of the vestibular system are spared (Wright and Schaefer, 1982, Strauss et al., 1983, Schuknecht, 1993, Hinojosa et al., 1995, Hoistad et al., 1998).
The hair cells are not the only drug targets. Aminoglycosides have documented effects on the stria vascularis (Ruedi, 1952), including thinning of the tissue and a decrease in the number of marginal cells (Hawkins, 1973). Degeneration of spiral ganglion cells after aminoglycoside treatment appears largely to be a sequel to the loss of hair cells that the ganglia innervate (Hawkins, 1976, Johnsson et al., 1981) and continues even long after treatment has ended (Webster and Webster, 1981, Leake and Hradek, 1988). Adding to the complexity of the pattern of damage, some studies suggest that spiral ganglia can be affected without obvious insult to the hair cells (Hinojosa and Lerner, 1987, Sone et al., 1998), while the spiral ganglia of some subjects completely deafened by aminoglycosides have been found intact (Nadol, 1997). The underlying mechanisms of such variability are not known, but we may deduce from animal experimentation that neurotrophic factors regulating development and maintenance of neurons may be involved and vary between individuals. Spiral ganglion cells can likewise be affected by cisplatin. A recent study reported a case of a child who lost benefit from his cochlear implant following cisplatin therapy, despite a decade of successful adaptation of the device prior to treatment (Harris et al., 2011). As a cochlear implant relies on a functional auditory nerve, this case implies a direct effect on the spiral ganglia.
Studies in several animal models (including mice, rats, chinchillas, gerbils, and hamsters) have confirmed and expanded the observations from human temporal bones. Systemic application of either drug leads to loss of outer hair cells, beginning in the basal region of the cochlear spiral (fig. 1). Correspondingly, auditory deficits manifest as high-frequency hearing loss that progresses to lower frequencies with increasing dose or duration of treatment. Other changes include deterioration of distortion product otoacoustic emissions corroborating dysfunction of outer hair cells (Alam et al., 2000), as well as elevation of compound action potential and cochlear microphonic output (van Ruijven et al., 2005a). With more prolonged treatment, lesions spread to other cell types, eventually replacing the entire cochlear sensory epithelium with a layer of non-specialized epithelial scar tissue (Hawkins, 1976, Estrem et al., 1981, Laurell and Bagger-Sjoback, 1991). An additional hallmark of cisplatin ototoxicity is a reduction in endocochlear potential, suggesting dysfunction stria vascularis (Ravi et al., 1995, Klis et al., 2000, Tsukasaki et al., 2000). Indeed, the stria vascularis in the basal turn may show edema, bulging, and depletion of intracellular organelle concentrations. These changes occur primarily in the marginal cells, although some effects are seen in intermediate cells as well (Meech et al., 1998, Campbell et al., 1999). Aminoglycosides do not induce changes in endocochlear potentials during early phases of treatment (Davis et al., 1958, Komune et al., 1987) and may only do so after extensive treatment leading to significant atrophy of the stria vascularis. It appears that some strial changes can also happen in the absence of hair cell damage (Forge and Fradis, 1985). As another target of cisplatin treatment, the myelin sheaths of type I spiral ganglion cells detach, again corroborating findings from clinical experience. In guinea pigs, the changes to these nerve cells occur in parallel with losses of outer hair cells (van Ruijven et al., 2005a).
Pharmacokinetics and cellular uptake
Following systemic injections, more than ninety percent of cisplatin is rendered biologically inactive through binding with serum proteins (Gormley et al., 1979). About 25% of administered cisplatin is eliminated from the body during the first 24 hours, with renal clearance accounting for more than 90% of this elimination. The remaining cisplatin collects largely in the liver, kidneys, and intestines, with minimal penetration of the central nervous system (Vermorken et al., 1984). Consistent with the ototoxic damage incurred in the cochlea, platinated DNA has been localized to the nuclei of outer hair cells, marginal cells of the stria vascularis and the cells in the spiral ligament (Van Ruijven et al., 2005b).
Cellular uptake of cisplatin seems to be tied to the active regulation of intracellular copper and platinum concentrations. Tumor cells with mutations or deletions in copper transporter genes controlling drug efflux (ATP7B and ABCC2) and drug uptake (Ctr1) are resistant to cisplatin treatment. Moreover, copper and cisplatin can prevent the uptake of each other (Ishida et al., 2002). In the mouse cochlea, Ctr1 is highly expressed and localized to the main sites of cisplatin toxicity, namely the outer hair cells, inner hair cells, stria vascularis, and spiral ganglia. Intratympanic administration of copper sulfate before intraperitoneal administration of cisplatin prevented hearing loss in mice (More et al., 2010). Organic cation transporters (OCTs) which are present in both kidney and inner ear (Ciarimboli et al., 2010) also contribute to cisplatin uptake. Co-administration of the organic cation cimetidine to mice treated with cisplatin protected against ototoxicity and nephrotoxicity. Likewise, no ototoxicity and only mild nephrotoxicity were found in OCT1/2 double-knockout mice.
Unlike cisplatin, aminoglycosides exhibit negligible binding to serum proteins. Following systemic treatment, they reach peak plasma levels by 30 to 90 minutes and their plasma half-life ranges from 2 to 6 hours. The drugs are excreted by the kidneys essentially unaltered at a rate roughly proportional to the glomerular filtration rate. Aminoglycoside antibiotics enter the inner ear within minutes after an injection via the bloodstream and may reach steady-state levels as early as after one-half to three hours (Tran Ba Huy et al., 1986). Drug concentrations in the inner ear remain about one-tenth of peak serum levels (Henley and Schacht, 1988) but are less efficiently eliminated from the inner ear than from serum. Clearance from the inner ear is biphasic and the half-life of the second phase may exceed 30 days (Tran Ba Huy et al., 1986). Even eleven months after cessation of treatment, traces of gentamicin were found in hair cells (Dulon et al., 1993). This difference in half-life between serum and cochlear tissues gives the appearance of an accumulation of aminoglycosides in the inner ear after time, which was formerly thought to account for the organ specific toxicity of these drugs. That theory, however, is rendered tenuous by the fact that inner ear concentrations reached by different their preference for vestibular or cochlear toxicity (Dulon et al., 1986). There is also no correlation between uptake and toxicity to certain cell populations. While a rapid uptake into the cochlea, primarily the sensory cells, can be observed (Dai et al., 2006), there is also a widespread distribution of aminoglycosides into other cells and tissues of the inner ear (Imamura and Adams, 2003, reviewed in Steyger, 2005). This absence of a uptake/toxicity relationship suggests that hair cells may have an intrinsic susceptibility to aminoglycoside antibiotics, as described later (Sha et al., 2001).
Studies of the mechanism of aminoglycoside uptake into cells point to the possibility of multiple modes of entry, perhaps in a cell-dependent manner. The glycoprotein megalin has been strongly implicated in renal transport (Moestrup et al., 1995), but the pattern of megalin expression in the inner ear does not correlate with the pattern of uptake and damage from aminoglycosides. In fact, megalin may be entirely absent from outer hair cells, the primary target of toxicity. (Ylikoski et al., 1997, Mizuta et al., 1999). Endocytotic uptake at the apices of hair cells was suggested by early observations of lysosome accumulation in the subcuticular portion of hair cells soon after systemic treatment of guinea pigs with kanamycin (Darrouzet and Guilhaume, 1974) and later observations confirmed aminoglycosides appearing initially at the apical ends of hair cells in different species (de Groot et al., 1990, Hiel et al., 1992, Hashino et al., 1997). Further evidence that aminoglycosides may enter the hair cells in the apical region is the ostensible involvement of myosin VIIA (Richardson et al., 1997), which regulates the turnover and trafficking at the apical plasma membrane of hair cells, but not the baso-lateral membrane. Compellingly, explants from the inner ear of myosin IIVA mutant mice do not take up aminoglycosides in vitro. Other suspected mechanisms include polyamine-like transport, due to the polyamine-like nature of the drugs (Williams et al., 1987) and vesicular receptor-mediated transport at the base of hair cells (Lim, 1986).
Specific ion channels in hair cells offer another potential pathway of entry (Marcotti et al., 2005). Several channels of the TRP (transient receptor potential) class are aminoglycoside permissive (trpp1, trpa1, trpv4; Steyger, 2005), most specifically perhaps TRPA1 channels (Stepanyan et al., 2011). A recent study provided further evidence that functional hair cell mechano-transducer channels are required for aminoglycoside ototoxicity (Alharazneh et al., 2011). However, despite a postulated causal relationship between the developmental onset of mechano-electrical transduction with onset of sensitivity to aminoglycosides, these events have been established as independent in the zebrafish lateral line (Santos et al., 2006). Furthermore, uptake via mechano-transduction channels would be hair cell-specific, leaving other uptake options open for non-sensory cells.
It is interesting to note that ototoxicity has also been attributed, at least in part, to the inhibition of potassium channels by aminoglycoside-induced depletion of phosphoinositides (Leitner et al., 2011). This suggestion is intriguing, considering the early hypotheses of an involvement of these lipids in ototoxicity based on the fact that aminoglycoside antibiotics strongly bind phosphoinositides (Schacht, 1979) and more recent observations that they deplete phosphatidylinositol trisphosphate in hair cells of mice after in-vivo treatment (Jiang et al., 2006b).
Biochemical and metabolic pathology
In addition to its therapeutic action of binding to nuclear DNA, cisplatin also interacts with variety of other molecules (Bose, 2002) and disrupts microtubule formation and the cellular cytoskeleton (Gonzalez et al., 2001, Fuertes et al., 2003). Of potential import is the binding to glutathione and other sulfhydryl-containing molecules, such as metallothionins, which can lead to an impairment of cellular detoxification mechanisms. Aminoglycosides likewise interact with a host of cellular constituents and metabolites. One of the earliest effects to be noted was that of a competitive antagonism with calcium evidenced as neuromuscular block (Vital-Brazil and Corrado, 1957). Tetany is still today a potential, albeit rarely observed, complication of aminoglycoside therapy (Thakur, 2008). However, many of the described actions may be inconsequential in the pathology of ototoxicity and the prevailing opinion to date is that the formation of reactive oxygen species (ROS) is a major cause that underlies the undesired side effects of both cisplatin and the aminoglycoside antibiotics.
Maintaining homeostasis, a controlled intracellular environment in the face of external challenges, is a crucial process in preserving cell integrity and survival. In the context of drug ototoxicity, the challenge lies in the defense against drug-induced ROS that would create a redox imbalance. ROS are normally present in low concentrations in all cell types as signaling messengers or as metabolic byproducts. Their potentially detrimental oxidative action on cell constituents is limited by intrinsic protective mechanisms such as antioxidant molecules (e.g., glutathione) or enzymes (e.g. catalase, superoxide dismutase, and peroxidases). Excessive ROS production may lead to exhaustion of such defenses and a redox imbalance which may trigger cell death pathways.
Cisplatin generates ROS, as shown in cochlear tissue explants (Clerici et al., 1996, Kopke et al., 1997). Specifically, the superoxide anion has been detected (Dehne et al., 2001), which may also be involved in nephrotoxicity (McGinness et al., 1978). Additional support for the formation of ROS is offered by the detection of 4-hydroxynonenal in the cochlea after cisplatin treatment (Lee et al., 2004), as well as malondialdehyde (Rybak et al., 2000), both indicators of lipid peroxidation. The precise details of ROS generation from cisplatin treatment are speculative, but it appears that NOX3, an isoform of NADPH oxidase that catalyzes the formation of superoxide, plays a significant role (fig. 2), at least in part, in hair cell damage (Mukherjea et al., 2006). NOX3 was up-regulated in the rat cochlea following systemic cisplatin administration and in vitro after cisplatin application to cochlear cell lines derived from the immortomouse. Linking NOX3 to ototoxicity, the local application of siRNA prior to systemic cisplatin administration prevented its up-regulation in the rat cochlea and protected against hair cell damage and hearing loss (Mukherjea et al., 2010). Another candidate for superoxide generation is xanthine oxidase. Although an increased activity of this enzyme in the cochlea has not been directly demonstrated, partial protection against cisplatin is afforded by allopurinol, a xanthine oxidase inhibitor (Lynch et al., 2005a, b). As a consequence of the increased formation of ROS, glutathione and antioxidant enzymes, such as catalase, superoxide dismutase, glutathione peroxidase and glutathione reductase, are depleted in cochlear tissues (Ravi et al., 1995, Rybak et al., 2000).
Figure 2.

Administration of NOX3 siRNA reduces immunolabeling of NOX3 and p-STAT1. Rats were injected trans-tympanically with scrambled or NOX3 siRNA 48 h prior to a cisplatin injection of 11 mg/kg i.p. Three days later cochleae were removed and processed for immunohistochemistry. NOX3 siRNA reduced both the basal and cisplatin-stimulated p-STAT1 (green) and NOX3 (red) immunoreactivity in various regions of the cochlea. NOX3 and p-STAT1 immunolabeling co-localized, demonstrated by the merged images (yellow). OHC, outer hair cells; SVA, stria vascularis; SG, spiral ganglion; SL, spiral ligament. Scale bars (lower right) represent 50 μm. The images shown are a representative of three independent experiments showing similar results. (This figure was published as supplementary figure 4 in Kaur et al, 2011).
Aminoglycosides, likewise, promote the formation of ROS, albeit not necessarily the same species and by the same reactions. ROS and their reaction products have been observed in explanted tissues as well as in vivo (Lautermann et al., 1995, Clerici et al., 1996, Hirose et al., 1997) and both non-enzymatic and enzymatic mechanisms have been suggested for their generation. Gentamicin may act as an iron-chelator, forming a redox-active complex by stabilizing iron intermediates and thereby reducing molecular oxygen to superoxide radicals using electrons provided by available polyunsaturated fatty acids (Priuska and Schacht, 1995). ROS may also be generated by enzymatic pathways, akin to the stimulation of NADPH by cisplatin. Aminoglycosides affect signaling pathways linked to Rho-GTPases (Jiang et al., 2006b), which in turn may lead to activation of the NADPH oxidase complex. Mitochondrial dysfunction had also been speculated as an element in aminoglycoside ototoxicity, based on the A1555G mitochondrial mutation that conveys hypersusceptibility. A recent study now demonstrates that apramycin, a structurally unique aminoglycoside used in veterinary medicine, shows little activity toward eukaryotic ribosomes, even toward hybrid ribosomes genetically engineered to carry the mitochondrial A1555G allele. The drug, despite being a potent antibacterial, causes only little ROS formation and hair cell damage in cochlear explants and much less hearing loss in guinea pigs in vivo than other aminoglycosides (Matt et al., 2012). These data strongly support the idea that mitochondrial dysfunction might be linked to excessive formation of ROS and ototoxicity. Furthermore, the dissociation of antibacterial activity and ototoxicity may provide a novel framework for the development of less toxic aminoglycosides.
Pathways of cell death
A redox imbalance sets in motion an intricate network of cellular pathways designed to rescue the cell or ensure its orderly demise. Compensatory protective pathways operate in the early stages of drug-induced hearing loss (Jiang et al., 2005; Chen et al., 2008) but most research has focused on the modes of cell death. Both necrosis and apoptosis have been documented for aminoglycosides (Nakagawa et al., 1998, for a review Forge and Schacht, 2000, Ylikoski et al., 2002), while cisplatin appears primarily to cause apoptosis in the affected tissues, namely the outer hair cells in the organ of Corti, the stria vascularis, spiral ligament, and the spiral ganglion cells (Alam et al., 2000, Watanabe et al., 2003, Liang et al., 2005). Multiple pathways, extrinsic and intrinsic, appear to be involved and specific observations have suggested c-jun N-terminal kinases (JNK) and caspase cascades (Ylikoski et al, 2002; Eshraghi et al., 2007), as well as the nuclear translocation of endonuclease G (Endo G) and activation of μ-calpain (Jiang et al., 2006c).
Caspase activation, and specifically that of caspase-3, is considered the hallmark of apoptosis. Caspases appear pivotal to cisplatin toxicity, and members of the intrinsic apoptosis caspase cascade are activated after cisplatin treatment. Furthermore, caspase-inhibitors and notably caspase-3 and caspase-9 inhibitors effectively prevent cell death from cisplatin exposure (Liu et al., 1998, Wang et al., 2004). Activation of caspases has also been suggested as a response to aminoglycoside treatment, but support for caspase-3-mediated pathways comes largely from in-vitro studies or high-dose treatment of the chick basilar papilla or vestibular cultures of the guinea pig (Shimizu et al., 2003, Mangiardi et al., 2004). Adding to the tenuous nature of the results, caspase-inhibitors have yielded mixed success in protecting from aminoglycoside-induced cell death (Momiyama et al., 2006, Tabuchi et al., 2007).
Another branch of caspase signaling is initiated by caspase-8, a protein closely linked to the death domain-associated receptors of the cell membrane, but evidence of caspase-8 involvement in aminoglycoside and cisplatin ototoxicity is limited. Only studies using the HEI-OC1 cell line, derived from postnatal inner ear cells of the immortomouse, found activation of caspase-8 following cisplatin treatment (Devarajan et al., 2002, Jeong et al., 2007). In other studies, activation of caspase-8 is notably absent. Outer hair cells of the guinea pig organ of Corti were devoid of caspase-8 staining after systemic cisplatin treatment and a caspase-8 inhibitor was unable to protect the OHCs from apoptosis (Wang et al., 2004), raising questions as to the validity of immortal cell lines (such as HEI-OC1) to reflect mechanisms of ototoxicity (Chen et al., 2011). Likewise, caspase-8 inhibition was unsuccessful to prevent aminoglycoside-induced cell death (Cunningham et al., 2002, Tabuchi et al., 2007) while caspase-9 and caspase-3 inhibition can protect. Taken together, evidence supports caspase-9 as a predominant upstream signal for caspase-mediated apoptosis.
Consistent with a mitochondrial involvement in cell death pathways, the Bcl-2 family of proteins has been implicated in both cisplatin and aminoglycoside injury. The expression of the anti-apoptotic Bcl-2 is reduced and that of the pro-apoptotic Bax is increased in auditory hair cells from gerbils treated with cisplatin (Alam et al., 2000) and Bax translocates from the cytosol to the mitochondria (Wang et al., 2004). In aminoglycoside toxicity, the anti-apoptotic members Bcl-2 and Bcl-XL confer protection (Cunningham et al., 2004, Liu et al., 2007, Staecker et al., 2007, Pfannenstiel et al., 2009). As a consequence of permeabilized mitochondria, cytochrome c is released and might form the “apoptosome” complex along with Apaf1 and caspase-9 complexes. Cytochrome c release is indeed observed both after aminoglycoside treatment (Mangiardi et al., 2004, Matsui et al., 2004) and in cell cultures after cisplatin treatment (Devarajan et al., 2002). Further, cyclosporin A, an inhibitor of mitochondrial permeability transition pores and, hence, of the release of cytochrome c, alleviates aminoglycoside cochleotoxicity in vitro by way of blocking the mitochondria-mediated caspase cascade (Dehne et al., 2002).
Caspase-independent apoptosis and necrosis may also be important in aminoglycoside but probably not in cisplatin ototoxicity. Both caspase-dependent and caspase-independent cell death is observed in a chronic in-vivo model where the onset of cochlear deficits is delayed more than one week after treatment begins and continues to develop after the cessation of treatment. Such a paradigm is of particular interest as it closely models the clinical dosing schedule in humans. In this model, the nuclear translocation of endonuclease G (endo G) and activation of μ-calpain (Jiang et al., 2006c) are observed as well as the release of cathepsins into the cytoplasm, inferring lysosomal rupture. Lysosomal destabilization may be the result of μ-calpain activation or of an overload of sequestered iron-bound molecules in the lysosome (Jiang et al., 2006a). Cathepsin-dependent cleavage of the DNA repair enzyme PARP1 ensues, resulting in DNA fragments associated with necrotic cell death. Calpain involvement is supported by the ability of leupeptin, a calpain inhibitor, to reduce hair cell death in utricle explants (Ding et al., 2002).
Another potentially apoptotic route, the JNK/MAPK pathway, is comprised of stress response kinases that participate in a phosphorylation cascade, which can lead to activation of transcription factors like c-jun and consequent changes in gene expression. Investigation of the role of JNK in cisplatin ototoxicity by way of JNK-inhibitor studies have concluded that JNK may not be involved in cell death, but rather in the repair and maintenance of cisplatin-damaged cells (Wang et al., 2004). On the other hand, the apoptotic JNK pathway has been directly or indirectly documented in various models of aminoglycoside ototoxicity. In vitro, activation of c-jun is found following aminoglycoside exposure, and is linked to activation of the JNK pathway (Maroney et al., 1998, Ylikoski et al., 2002). JNK involvement is corroborated by a modest pharmacological attenuation from ototoxicity in the guinea pig inner ear (Ylikoski et al., 2002) and similar protective effects in vitro (Pirvola et al., 2000, Bodmer et al., 2002, Wang et al., 2003, Eshraghi et al., 2007). The JNK pathway may be activated by small G-proteins (guanine-nucleotide binding proteins), Rho, and Ras. Inhibitors of Ras can protect explant cultures from gentamicin-induced damage and reduce activation of c-jun (Battaglia 2003), as can Clostridium difficile toxin B, an inhibitor of Rho, Rac, and Cdc42 (Bodmer et al., 2002b). Like the caspase pathway, which is likely downstream of JNK activity in aminoglycoside ototoxicity, the involvement of the JNK pathway is characterized primarily in cultured explants with limited studies of its involvement in vivo.
Reflecting the complexity of the cellular responses triggered by ototoxic drugs, several other pathways have been invoked. In the case of cisplatin, evidence for the involvement of the tumor suppressor p53 has been presented in vitro with studies of cochlear cell lines and organotypic cultures of the organ of Corti, which both showed increased p53 expression (Devarajan et al., 2002, Zhang et al., 2003, Cheng et al., 2005). Inhibition of p53 activity with pifithrin-alpha in organ of Corti cultures protected the hair cells from damage. (Zhang et al., 2003). In addition, deletion of the p53 gene prevents cytochrome c translocation, caspase-3 activation, and hair cell death (Cheng et al., 2005). Also of note, apoptosis in fibrocytes of the lateral wall after cisplatin treatment is accompanied by activation of potassium channels, leading to potassium efflux and reducing intracellular ionic and osmotic strength, effects which can trigger apoptosis by activating pro-apoptotic enzymes, such as caspases and pro-apoptotic nucleases. The loss of fibrocytes can then affect ion transport and endocochlear potential generation in the stria vascularis, which in turn may affect other cells within the cochlea (Liang et al., 2005).
Finally, it has been proposed that several of the metabolic changes induced by ototoxic drugs converge on the nucleus and lead to epigenetic changes, alterations of gene expression without changes to the DNA sequence. Such effects are mediated by the covalent modification of histone core proteins, for example by their acetylation, and generally can lead to disease pathogenesis (see Mohtat and Susztak, 2010). Aminoglycosides increase the levels of histone deacetylases (Chen et al., 2009) and thereby decrease the acetylation of histones in the nuclei of outer hair cells in mice in vivo and in organotypic cultures (Jiang et al., 2006c). Implicating epigenetics as part of the overall response to aminoglycoside challenge, inhibitors of histone deacetylases rescue hair cells from ototoxic insult. Histone deacetylase inhibitors likewise can protect in vivo from cisplatin ototoxicity (Drottar et al., 2006).
Protection from ototoxic effcts
Basically, interventions to attenuate ototoxic side effects take one of two approaches: the augmentation of protective pathways or the inhibition of cell death pathways. The previous sections have already discussed a variety of studies employing such strategies in order to uncover potential causal relationships between drug actions and cell death. We will not reiterate those studies here but focus on results that are translationally promising or already have been clinically tested.
Success in vitro or in animal experiments does not assure success in clinical studies. In cisplatin therapy, there are cautions against certain classes of protective agents that might be effective against ototoxicity in animals but may interfere with therapeutic efficacy. This problem has been much discussed for sulfhydryl-containing compounds which might chemically interact with cisplatin and inactivate it (Rybak and Whitworth, 2005). Amifostine is one such compound that protects against peripheral ototoxicity in the hamster (Church et al., 2004) but showed mixed results in clinical trials. No beneficial effect was found in children with germ cell tumors treated with amifostine in combination with cisplatin, etoposide, or bleomycin (Marina et al., 2005, Sastry and Kellie, 2005). A more recent study, however, established protection in a cohort of patients, specifically pediatric patients with medulloblastoma, reducing the incidence from 37.1% to 14.5% (Fouladi et al., 2008).
Another agent that shows promise for translation into the clinic is cimetidine, a histamine H2-receptor antagonist. It completely protected animals against cisplatin-induced auditory threshold shifts, although some evidence of nephrotoxicity persisted. Importantly, cimetidine did not change the therapeutic efficacy of cisplatin on a human acute lymphoblastic leukemia T cell line (Ciarimboli et al., 2010). Since it is currently an approved drug for the treatment of heartburn and peptic ulcers, it has potential as a clinical antidote against cisplatin ototoxicity. More compounds might become available if local routes of application to the inner ear are considered, eliminating the problem of interference with cancer chemotherapy (Rybak and Whitworth, 2005).
Antioxidant therapy may be the currently most practicable method of protection against aminoglycoside-induced ototoxicity. A large number of such compounds, some of which are even classified as food supplements, can be safely administered in order to detoxify stressed cells and return them to homeostasis (Rybak and Whitworth, 2005). A variety of antioxidants show promise in animal experiments and do not interfere with the antimicrobial actions of aminoglycosides, thus providing opportunities for clinical applications. However, caution is warranted here as well, as exemplified by vitamin E which was suggested to protect against aminoglycoside damage in guinea pigs but was ineffective in a clinical setting (Kharkheli et al., 2007).
On the other hand, at least two compounds have already proven clinically successful, aspirin and N-acetylcysteine. Salicylate, the active principle of aspirin (acetyl salicylate), was first established as an effective protectant in guinea pigs (Sha and Schacht, 1999), and subsequently aspirin was tested in a randomized double-blind placebo-controlled trial in patients receiving gentamicin for acute infections (Sha et al., 2006). Fourteen of 106 patients (13%) met the criterion of hearing loss in the placebo group compared to 3/89 (3%) in the aspirin group, constituting a 75% reduction in risk. Aspirin did not influence gentamicin serum levels or the outcome of therapy. The benefit of aspirin was confirmed in an independently conducted second clinical trial (Behnoud et al., 2009). N-acetylcysteine significantly reduced the incidence of hearing loss in a small trial involving patients receiving hemodialysis and gentamicin for bacteremia (Feldman et al., 2007). An alternative to aspirin is welcome because its use carries a small risk of gastrointestinal problems and is contraindicated in individuals at risk of bleeding disorders and in children, including the bulk of the population with cystic fibrosis.
The sum of these results clearly indicates that therapeutic protection from drug-induced ototoxicity is feasible and that it is possible, albeit not always, to extrapolate from animal experimentation on ototoxicity to the clinic.
Conclusion
The platinum-based anti-cancer agents and the aminoglycoside antibiotics are two classes of drugs with critical clinical importance. Unfortunately, cisplatin as well as aminoglycosides have the potential to cause sensorineural hearing loss. This is due primarily by damage to outer hair cells, initially in the basal turn of the cochlea. Some aminoglycosides, such as streptomycin and gentamicin, have a proclivity also to damage the vestibular system; cisplatin seems to have a much lower likelihood to do so. Despite the fact that both categories of ototoxic drugs produce oxidative stress in the inner ear as a primary trigger for cell injury, the ensuing cell death pathways appear to differ. Cisplatin mostly initiates apoptosis while aminoglycosides seem to trigger multiple apoptotic and necrotic pathways. Understanding the mechanisms of ototoxicity has already helped to develop novel ways of protection of the cochlea and antioxidants have emerged as promising therapeutic agents. In addition, novel methods of delivery of protective agents should allow selective protection of the inner ear without interference against the desired therapeutic effects of cisplatin and aminoglycoside antibiotics.
Figure 3. Cisplatin-induced hearing loss is related to activation of STAT1.

(a) Bar graphs depict ABR threshold measured in rats 72 h after treatment with cisplatin (11 mg/kg i.p.) which was begun 48 h after transtympanic injection of scrambled RNA or STAT1 siRNA (0.9 ug). (b) Scanning electron microscopy of the basal turn of the cochlea of these rats. The representative figure demonstrates significant damage to outer hair cells (white arrows) by cisplatin which is prevented by pretreatment with STAT1 siRNA. (c) Quantitation of the scanning electron microscopy data. Bar graphs depict mean +/− S.E.M. The asterisks indicate statistically significant difference between the STAT1 siRNA + cisplatin group (**) and scramble + cisplatin treatment group and the scramble-treated versus the scramble + cisplatin group (p <0.05, n = 6). (This figure was published as Figure 4 in Kaur et al, (2011).
Acknowledgments
The authors’ research on drug-induced hearing loss and its prevention is supported by research grants DC 003685 (JS) and DC 02396 (LPR) from the National Institute for Deafness and Communication Disorders, National Institutes of Health.
Contributor Information
Jochen Schacht, Email: schacht@umich.edu.
Andra E. Talaska, Email: atalaska@umich.edu.
Leonard P. Rybak, Email: lrybak@siumed.edu.
Literature Cited
- Alam SA, Ikeda K, Oshima T, Suzuki M, Kawase T, Kikuchi T, Takasaka T. Cisplatin-induced apoptotic cell death in Mongolian gerbil cochlea. Hear Res. 2000;141:28–38. doi: 10.1016/s0378-5955(99)00211-7. [DOI] [PubMed] [Google Scholar]
- Alharazneh A, Luk L, Huth M, Monfared A, Steyger PS, Cheng AG, Ricci AJ. Functional hair cell mechanotransducer channels are required for aminoglycoside ototoxicity. PLoS One. 2011;6(7):e22347. doi: 10.1371/journal.pone.0022347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia A, Pak K, Brors D, Bodmer D, Frangos JA, Ryan AF. Involvement of ras activation in toxic hair cell damage of the mammalian cochlea. Neuroscience. 2003;122(4):1025–1035. doi: 10.1016/j.neuroscience.2003.08.041. [DOI] [PubMed] [Google Scholar]
- Behnoud F, Davoudpur K, Goodarzi MT. Can aspirin protect or at least attenuate gentamicin ototoxicity in humans? Saudi Med J. 2009;30(9):1165–1169. [PubMed] [Google Scholar]
- Benedetti Panici P, Greggi S, Scambia G, Baiocchi G, Lomonaco M, Conti G, Mancuso S. Efficacy and toxicity of very high-dose cisplatin in advanced ovarian carcinoma: 4-year survival analysis and neurological follow-up. Int J Gynecol Cancer. 1993;3:44–53. doi: 10.1046/j.1525-1438.1993.03010044.x. [DOI] [PubMed] [Google Scholar]
- Blakley BW. Clinical forum: a review of intratympanic therapy. Am J Otol. 1997;18:520–526. discussion 527–531. [PubMed] [Google Scholar]
- Bodmer D, Brors D, Bodmer M, Ryan AF. Rescue of auditory hair cells from ototoxicity by CEP-11 004, an inhibitor of the JNK signaling pathway. Laryngorhinootologie. 2002;81:853–856. doi: 10.1055/s-2002-36100. [DOI] [PubMed] [Google Scholar]
- Bodmer D, Brors D, Pak K, Gloddek B, Ryan A. Rescue of auditory hair cells from aminoglycoside toxicity by Clostridium difficile toxin B, an inhibitor of the small GTPases Rho/Rac/Cdc42. Hear Res. 2002b;172:81–82. doi: 10.1016/s0378-5955(02)00514-2. [DOI] [PubMed] [Google Scholar]
- Bokemeyer C, Berger CC, Hartmann JT, Kollmannsberger C, Schmoll HJ, Kuczyk MA, Kanz L. Analysis of risk factors for cisplatin-induced ototoxicity in patients with testicular cancer. Br J Cancer. 1998;77:1355–1362. doi: 10.1038/bjc.1998.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose RN. Biomolecular targets for platinum antitumor drugs. Mini Rev Med Chem. 2002;2:103–111. doi: 10.2174/1389557024605500. [DOI] [PubMed] [Google Scholar]
- Boulikas T, Vougiouka M. Recent clinical trials using cisplatin, carboplatin and their combination chemotherapy drugs (review) Oncol Rep. 2004;11:559–595. [PubMed] [Google Scholar]
- Campbell KC, Meech RP, Rybak LP, Hughes LF. D-Methionine protects against cisplatin damage to the stria vascularis. Hear Res. 1999;138:13–28. doi: 10.1016/s0378-5955(99)00142-2. [DOI] [PubMed] [Google Scholar]
- Causse R, Gondet I, Vallancien B. Action de la streptomycine sur les cellules cilees des organes vestibulaires de la souris. Compt Rend Soc Biol. 1949;143:619–620. [Google Scholar]
- Chen F, Sha SH, Schacht J. Stress-induced changes in mitrochondrial peroxiredoxin in mouse cochlear hair cells. Abs Assoc Res Otolaryngol. 2008;31:50. [Google Scholar]
- Chen FQ, Schacht J, Sha SH. Aminoglycoside-induced histone deacetylation and hair cell death in the mouse cochlea. J Neurochem. 2009;108:1226–1236. doi: 10.1111/j.1471-4159.2009.05871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F-Q, Hill K, Guan Y-J, Schacht J, Sha S-H. Activation of apoptotic pathways in the absence of cell death in an inner-ear immortomouse cell line. Hear Res. 2011;284:33–41. doi: 10.1016/j.heares.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AG, Cunningham LL, Rubel EW. Mechanisms of hair cell death and protection. Curr Opin Otolaryngol Head Neck Surg. 2005;13:343–348. doi: 10.1097/01.moo.0000186799.45377.63. [DOI] [PubMed] [Google Scholar]
- Church MW, Blakley BW, Burgio DL, Gupta AK. WR-2721 (Amifostine) ameliorates cisplatin-induced hearing loss but causes neurotoxicity in hamsters: dose-dependent effects. J Assoc Res Otolaryngol. 2004;5:227–237. doi: 10.1007/s10162-004-4011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarimboli G, Duester D, Knief A, Sperling M, Holtkamp M, Edemir B, Pavenstädt H, Lanvers-Kaminsky C, am Zehnhoff-Dinnesen A, Schinkel AH, Koepsell H, Jürgens H, Schlatter E. Organic cation transporter 2 mediates cisplatin-induced oto-and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici WJ, Hensley K, DiMartino DL, Butterfield DA. Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear Res. 1996;98:116–124. doi: 10.1016/0378-5955(96)00075-5. [DOI] [PubMed] [Google Scholar]
- Cunningham LL, Cheng AG, Rubel EW. Caspase activation in hair cells of the mouse utricle exposed to neomycin. J Neurosci. 2002;22:8532–8540. doi: 10.1523/JNEUROSCI.22-19-08532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham LL, Matsui JI, Warchol ME, Rubel EW. Overexpression of Bcl-2 prevents neomycin-induced hair cell death and caspase-9 activation in the adult mouse utricle in vitro. J Neurobiol. 2004;60:89–100. doi: 10.1002/neu.20006. [DOI] [PubMed] [Google Scholar]
- Dai CF, Mangiardi D, Cotanche DA, Steyger PS. Uptake of fluorescent gentamicin by vertebrate sensory cells in vivo. Hear Res. 2006;213:64–78. doi: 10.1016/j.heares.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrouzet J, Guilhaume A. Ototoxicity of kanamycin studied day by day. Experimental electron microscopic study. Rev Laryngol Otol Rhinol (Bord) 1974;95:601–621. [PubMed] [Google Scholar]
- Davis H, Deatherage BH, Rosenblut B, Fernandez C, Kimura R, Smith CA. Modification of cochlear potentials produced by streptomycin poisoning and by extensive venous obstruction. Laryngoscope. 1958;68:596–627. doi: 10.1002/lary.5540680341. [DOI] [PubMed] [Google Scholar]
- de Groot JC, Meeuwsen F, Ruizendaal WE, Veldman JE. Ultrastructural localization of gentamicin in the cochlea. Hear Res. 1990;50:35–42. doi: 10.1016/0378-5955(90)90031-j. [DOI] [PubMed] [Google Scholar]
- Dehne N, Lautermann J, Petrat F, Rauen U, de Groot H. Cisplatin ototoxicity: involvement of iron and enhanced formation of superoxide anion radicals. Toxicol Appl Pharmacol. 2001;174:27–34. doi: 10.1006/taap.2001.9171. [DOI] [PubMed] [Google Scholar]
- Dehne N, Rauen U, de Groot H, Lautermann J. Involvement of the mitochondrial permeability transition in gentamicin ototoxicity. Hear Res. 2002;169:47–55. doi: 10.1016/s0378-5955(02)00338-6. [DOI] [PubMed] [Google Scholar]
- Devarajan P, Savoca M, Castaneda MP, Park MS, Esteban-Cruciani N, Kalinec G, Kalinec F. Cisplatin-induced apoptosis in auditory cells: role of death receptor and mitochondrial pathways. Hear Res. 2002;174:45–54. doi: 10.1016/s0378-5955(02)00634-2. [DOI] [PubMed] [Google Scholar]
- Ding D, Stracher A, Salvi RJ. Leupeptin protects cochlear and vestibular hair cells from gentamicin ototoxicity. Hear Res. 2002;164:115–126. doi: 10.1016/s0378-5955(01)00417-8. [DOI] [PubMed] [Google Scholar]
- Drottar M, Liberman MC, Ratan RR, Roberson DW. The histone deacetylase inhibitor sodium butyrate protects against cisplatin-induced hearing loss in guinea pigs. Laryngoscope. 2006;116:292–296. doi: 10.1097/01.mlg.0000197630.85208.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Schwander M, Moresco EM, Viviani P, Haller C, Hildebrand MS, Pak K, Tarantino L, Roberts A, Richardson H, Koob G, Najmabadi H, Ryan AF, Smith RJ, Müller U, Beutler B. A catechol-O-methyltransferase that is essential for auditory function in mice and humans. Proc Natl Acad Sci USA. 2008;105:14609–14614. doi: 10.1073/pnas.0807219105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggal P, Sarkar M. Audiologic monitoring of multi-drug resistant tuberculosis patients on aminoglycoside treatment with long term follow-up. BMC Ear Nose Throat Disord. 2007;7:5. doi: 10.1186/1472-6815-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon D, Aran JM, Zajic G, Schacht J. Comparative uptake of gentamicin, netilmicin, and amikacin in the guinea pig cochlea and vestibule. Antimicrob Agents Chemother. 1986;30:96–100. doi: 10.1128/aac.30.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulon D, Hiel H, Aurousseau C, Erre JP, Aran JM. Pharmacokinetics of gentamicin in the sensory hair cells of the organ of Corti: rapid uptake and long term persistence. C R Acad Sci III. 1993;316:682–687. [PubMed] [Google Scholar]
- Einarsson EJ, Oskarsson T, Haraldsson A, Moell C, Wiebe T, Magnusson M, Kristinsson J, Petersen H. Hearing impairment after platinum-based chemotherapy in childhood. Pediatr Blood Cancer. 2011;56:631–637. doi: 10.1002/pbc.22876. [DOI] [PubMed] [Google Scholar]
- Einarsson EJ, Petersen H, Wiebe T, Fransson PA, Grenner J, Magnusson M, Moell C. Long term hearing degeneration after platinum-based chemotherapy in childhood. Int J Audiol. 2010;49:765–771. doi: 10.3109/14992027.2010.485595. [DOI] [PubMed] [Google Scholar]
- el-Barbary A, Altschuler RA, Schacht J. Glutathione-S-transferases in the organ of Corti of the rat: enzymatic activity, subunit composition and immunohistochemical localization. Hear Res. 1993;71:80–90. doi: 10.1016/0378-5955(93)90023-t. [DOI] [PubMed] [Google Scholar]
- Eshraghi AA, Wang J, Adil E, He J, Zine A, Bublik M, Bonny C, Puel JL, Balkany TJ, Van De Water TR. Blocking c-Jun-N-terminal kinase signaling can prevent hearing loss induced by both electrode insertion trauma and neomycin ototoxicity. Hear Res. 2007;226:168–177. doi: 10.1016/j.heares.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Estrem SA, Babin RW, Ryu JH, Moore KC. Cis-diamminedichloroplatinum (II) ototoxicity in the guinea pig. Otolaryngol Head Neck Surg. 1981;89:638–645. doi: 10.1177/019459988108900424. [DOI] [PubMed] [Google Scholar]
- Fausti SA, Henry JA, Schaffer HI, Olson DJ, Frey RH, McDonald WJ. High-frequency audiometric monitoring for early detection of aminoglycoside ototoxicity. J Infect Dis. 1992;165:1026–1032. doi: 10.1093/infdis/165.6.1026. [DOI] [PubMed] [Google Scholar]
- Fee WE., Jr Aminoglycoside ototoxicity in the human. Laryngoscope. 1980;90:1–19. doi: 10.1288/00005537-198010001-00001. [DOI] [PubMed] [Google Scholar]
- Feldman L, Efrati S, Eviatar E, Abramsohn R, Yarovoy I, Gersch E, Averbukh Z, Weissgarten J. Gentamicin-induced ototoxicity in hemodialysis patients is ameliorated by N-acetylcysteine. Kidney Int. 2007;72(3):359–363. doi: 10.1038/sj.ki.5002295. [DOI] [PubMed] [Google Scholar]
- Fischel-Ghodsian N. Genetic factors in aminoglycoside toxicity. Pharmacogenomics. 2005;6:27–36. doi: 10.1517/14622416.6.1.27. [DOI] [PubMed] [Google Scholar]
- Forge A, Fradis M. Structural abnormalities in the stria vascularis following chronic gentamicin treatment. Hear Res. 1985;20:233–244. doi: 10.1016/0378-5955(85)90028-0. [DOI] [PubMed] [Google Scholar]
- Forge A, Schacht J. Aminoglycoside antibiotics. Audiol Neurootol. 2000;5:3–22. doi: 10.1159/000013861. [DOI] [PubMed] [Google Scholar]
- Fouladi M, Chintagumpala M, Ashley D, Kellie S, Gururangan S, Hassall T, Gronewold L, Stewart CF, Wallace D, Broniscer A, Hale GA, Kasow KA, Merchant TE, Morris B, Krasin M, Kun LE, Boyett JM, Gajjar A. Amifostine protects against cisplatin-induced ototoxicity in children with average-risk medulloblastoma. J Clin Oncol. 2008;26:3749–3755. doi: 10.1200/JCO.2007.14.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes MA, Castilla J, Alonso C, Perez JM. Cisplatin biochemical mechanism of action: from cytotoxicity to induction of cell death through interconnections between apoptotic and necrotic pathways. Curr Med Chem. 2003;10:257–266. doi: 10.2174/0929867033368484. [DOI] [PubMed] [Google Scholar]
- Garetz SL, Altschuler RA, Schacht J. Attenuation of gentamicin ototoxicity by glutathione in the guinea pig in vivo. Hear Res. 1994;77:81–87. doi: 10.1016/0378-5955(94)90255-0. [DOI] [PubMed] [Google Scholar]
- Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657–663. doi: 10.1124/mol.59.4.657. [DOI] [PubMed] [Google Scholar]
- Gormley PE, Bull JM, LeRoy AF, Cysyk RL. Kinetics of cis-dichlorodiamineplatium. Clin Pharmacol Ther. 1979;25:351–357. doi: 10.1002/cpt1979253351. [DOI] [PubMed] [Google Scholar]
- Grunberg SM, Sonka S, Stevenson LL. Progressive parestesias aftere cessation of therapy with very high-dose cisplatin. Cancer Chemother Pharmacol. 1989;2:62–64. doi: 10.1007/BF00694340. [DOI] [PubMed] [Google Scholar]
- Harris MS, Gilbert JL, Lormore KA, Musunuru SA, Fritsch MH. Cisplatin ototoxicity affecting cochlear implant benefit. Otol Neurotol. 2011;32:969–972. doi: 10.1097/MAO.0b013e3182255893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashino E, Shero M, Salvi RJ. Lysosomal targeting and accumulation of aminoglycoside antibiotics in sensory hair cells. Brain Res. 1997;777:75–85. doi: 10.1016/s0006-8993(97)00977-3. [DOI] [PubMed] [Google Scholar]
- Hawkins E., Jr Ototoxic mechanisms. A working hypothesis. Audiology. 1973;12:383–393. doi: 10.3109/00206097309071652. [DOI] [PubMed] [Google Scholar]
- Hawkins JE. Drug ototoxicity. In: Keidel WD, Neff WD, editors. Sensory Physiology. Vol. 5. Berlin: Springer Verlag; 1976. pp. 707–748. [Google Scholar]
- Henley CM, 3rd, Schacht J. Pharmacokinetics of aminoglycoside antibiotics in blood, inner-ear fluids and tissues and their relationship to ototoxicity. Audiology. 1988;27:137–146. doi: 10.3109/00206098809081584. [DOI] [PubMed] [Google Scholar]
- Hiel H, Bennani H, Erre JP, Aurousseau C, Aran JM. Kinetics of gentamicin in cochlear hair cells after chronic treatment. Acta Otolaryngol. 1992;112:272–277. doi: 10.1080/00016489.1992.11665417. [DOI] [PubMed] [Google Scholar]
- Hinojosa R, Lerner SA. Cochlear neural degeneration without hair cell loss in two patients with aminoglycoside ototoxicity. J Infect Dis. 1987;156:449–455. doi: 10.1093/infdis/156.3.449. [DOI] [PubMed] [Google Scholar]
- Hinojosa R, Riggs LC, Strauss M, Matz GJ. Temporal bone histopathology of cisplatin ototoxicity. Am J Otol. 1995;16:731–740. [PubMed] [Google Scholar]
- Hinshaw HC, Feldman L. Streptomycin in treatment of clinical tuberculosis: a preliminary report. Proc Mayo Clinic. 1945;20:313–318. [Google Scholar]
- Hirose K, Hockenbery DM, Rubel EW. Reactive oxygen species in chick hair cells after gentamicin exposure in vitro. Hear Res. 1997;104:1–14. doi: 10.1016/s0378-5955(96)00169-4. [DOI] [PubMed] [Google Scholar]
- Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Böttger Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci USA. 2008;105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoistad DL, Ondrey FG, Mutlu C, Schachern PA, Paparella MM, Adams GL. Histopathology of human temporal bone after cis-platinum, radiation, or both. Otolaryngol Head Neck Surg. 1998;118:825–832. doi: 10.1016/S0194-5998(98)70276-1. [DOI] [PubMed] [Google Scholar]
- Imamura S, Adams JC. Distribution of gentamicin in the guinea pig inner ear after local or systemic application. J Assoc Res Otolaryngol. 2003;4:176–195. doi: 10.1007/s10162-002-2036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci U S A. 2002;99:14298–14302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HJ, Kim SJ, Moon PD, Kim NH, Kim JS, Park RK, Kim MS, Park BR, Jeong S, Um JY, Kim HM, Hong SH. Antiapoptotic mechanism of cannabinoid receptor 2 agonist on cisplatin-induced apoptosis in the HEI-OC1 auditory cell line. J Neurosci Res. 2007;85:896–905. doi: 10.1002/jnr.21168. [DOI] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. The NK-κB pathway protects cochlear hair cells from aminoglycoside-induced ototoxicity. J Neurosci Res. 2005;79:644–651. doi: 10.1002/jnr.20392. [DOI] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Forge A, Schacht J. Caspase-independent pathways of hair cell death induced by kanamycin in vivo. Cell Death Differ. 2006a;13:20–30. doi: 10.1038/sj.cdd.4401706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. Rac/Rho pathway regulates actin depolymerization induced by aminoglycoside antibiotics. J Neurosci Res. 2006b;83:1544–1551. doi: 10.1002/jnr.20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. Kanamycin alters cytoplasmic and nuclear phosphoinositide signaling in the organ of Corti in vivo. J Neurochem. 2006c;99:269–276. doi: 10.1111/j.1471-4159.2006.04117.x. [DOI] [PubMed] [Google Scholar]
- Johnsson LG, Hawkins JE, Jr, Kingsley TC, Black FO, Matz GJ. Aminoglycoside-induced cochlear pathology in man. Acta Otolaryngol Suppl. 1981;383:1–19. [PubMed] [Google Scholar]
- Kaufman RJ. Correction of genetic disease by making sense from nonsense. J Clin Invest. 1999;194:367–368. doi: 10.1172/JCI8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai Y, Kohda Y, Kodawara T, Gemba M. Protective effect of a protein kinase inhibitor on cellular injury induced by cephaloridine in the porcine kidney cell line LLC-PK(1) J Toxicol Sci. 2005;30:157–163. doi: 10.2131/jts.30.157. [DOI] [PubMed] [Google Scholar]
- Kharkheli E, Kevanishvili Z, Maglakelidze T, Davitashvili O, Schacht J. Does vitamin E prevent gentamicin-induced ototoxicity? Georgian Med News. 2007:14–17. [PubMed]
- Klis SF, O’Leary SJ, Hamers FP, De Groot JC, Smoorenburg GF. Reversible cisplatin ototoxicity in the albino guinea pig. Neuroreport. 2000;11:623–626. doi: 10.1097/00001756-200002280-00037. [DOI] [PubMed] [Google Scholar]
- Komune S, Ide M, Nakano T, Morimitsu T. Effects of kanamycin sulfate on cochlear potentials and potassium ion permeability through the cochlear partitions. ORL J Otorhinolaryngol Relat Spec. 1987;49:9–16. doi: 10.1159/000275900. [DOI] [PubMed] [Google Scholar]
- Kopke RD, Liu W, Gabaizadeh R, Jacono A, Feghali J, Spray D, Garcia P, Steinman H, Malgrange B, Ruben RJ, Rybak L, Van de Water TR. Use of organotypic cultures of Corti’s organ to study the protective effects of antioxidant molecules on cisplatin-induced damage of auditory hair cells. Am J Otol. 1997;18:559–571. [PubMed] [Google Scholar]
- Laurell G, Bagger-Sjoback D. Dose-dependent inner ear changes after i.v. administration of cisplatin. J Otolaryngol. 1991;20:158–167. [PubMed] [Google Scholar]
- Laurell G, Jungnelius U. High-dose cisplatin treatment: hearing loss and plasma concentrations. Laryngoscope. 1990;100:724–734. doi: 10.1288/00005537-199007000-00008. [DOI] [PubMed] [Google Scholar]
- Lautermann J, McLaren J, Schacht J. Glutathione protection against gentamicin ototoxicity depends on nutritional status. Hear Res. 1995;86:15–24. doi: 10.1016/0378-5955(95)00049-a. [DOI] [PubMed] [Google Scholar]
- Leake PA, Hradek GT. Cochlear pathology of long term neomycin induced deafness in cats. Hear Res. 1988;33:11–33. doi: 10.1016/0378-5955(88)90018-4. [DOI] [PubMed] [Google Scholar]
- Lee JE, Nakagawa T, Kim TS, Endo T, Shiga A, Iguchi F, Lee SH, Ito J. Role of reactive radicals in degeneration of the auditory system of mice following cisplatin treatment. Acta Otolaryngol. 2004;124:1131–1135. doi: 10.1080/00016480410017521. [DOI] [PubMed] [Google Scholar]
- Leitner MG, Halaszovich CR, Oliver D. Aminoglycosides inhibit KCNQ4 channels in cochlear outer hair cells via depletion of phosphatidylinositol (4,5) bisphosphate. Mol Pharmacol. 2011;79:51–60. doi: 10.1124/mol.110.068130. [DOI] [PubMed] [Google Scholar]
- Lerner SA, Schmitt BA, Seligsohn R, Matz GJ. Comparative study of ototoxicity and nephrotoxicity in patients randomly assigned to treatment with amikacin or gentamicin. Am J Med. 1986;80:98–104. doi: 10.1016/0002-9343(86)90486-9. [DOI] [PubMed] [Google Scholar]
- Li Y, Womer RB, Silber JH. Predicting cisplatin ototoxicity in children: the influence of age and the cumulative dose. Eur J Cancer. 2004;40:2445–2451. doi: 10.1016/j.ejca.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Liang F, Schulte BA, Qu C, Hu W, Shen Z. Inhibition of the calcium- and voltage-dependent big conductance potassium channel ameliorates cisplatin-induced apoptosis in spiral ligament fibrocytes of the cochlea. Neuroscience. 2005;135:263–271. doi: 10.1016/j.neuroscience.2005.05.055. [DOI] [PubMed] [Google Scholar]
- Liedert B, Pluim D, Schellens J, Thomale J. Adduct-specific monoclonal antibodies for the measurement of cisplatin-induced DNA lesions in individual cell nuclei. Nucleic Acids Res. 2006;34:e47. doi: 10.1093/nar/gkl051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DJ. Effects of noise and ototoxic drugs at the cellular level in the cochlea: a review. Am J Otolaryngol. 1986;7:73–99. doi: 10.1016/s0196-0709(86)80037-0. [DOI] [PubMed] [Google Scholar]
- Liu W, Staecker H, Stupak H, Malgrange B, Lefebvre P, Van De Water TR. Caspase inhibitors prevent cisplatin-induced apoptosis of auditory sensory cells. Neuroreport. 1998;9:2609–2614. doi: 10.1097/00001756-199808030-00034. [DOI] [PubMed] [Google Scholar]
- Liu YH, Ke XM, Qin Y, Gu ZP, Xiao SF. Adeno-associated virus-mediated Bcl-xL prevents aminoglycoside-induced hearing loss in mice. Chin Med J (Engl) 2007;120:1236–1240. [PubMed] [Google Scholar]
- Lopez-Novoa JM, Quiros Y, Vicente L, Morales AI, Lopez-Hernandez FJ. New insights into the mechanism of aminoglycoside nephrotoxicity: an integrative point of view. Kidney Int. 2011;79:33–45. doi: 10.1038/ki.2010.337. [DOI] [PubMed] [Google Scholar]
- Lynch ED, Gu R, Pierce C, Kil J. Combined oral delivery of ebselen and allopurinol reduces multiple cisplatin toxicities in rat breast and ovarian cancer models while enhancing anti-tumor activity. Anticancer Drugs. 2005a;16:569–579. doi: 10.1097/00001813-200506000-00013. [DOI] [PubMed] [Google Scholar]
- Lynch ED, Gu R, Pierce C, Kil J. Reduction of acute cisplatin ototoxicity and nephrotoxicity in rats by oral administration of allopurinol and ebselen. Hear Res. 2005b;201:81–89. doi: 10.1016/j.heares.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Maddox PT, Saunders J, Chandrasekhar SS. Sudden hearing loss from PDE-5 inhibitors: A possible cellular stress etiology. Laryngoscope. 2009;119:1586–1589. doi: 10.1002/lary.20511. [DOI] [PubMed] [Google Scholar]
- Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, Lewis S, Shilling CJ, Kota J, Serrano-Munuera C, Hayes J, Mahan JD, Campbell KJ, Banwell B, Dasouki M, Watts V, Sivakumar K, Bien-Willner R, Flanigan KM, Sahenk Z, Barohn RJ, Walker CM, Mendell JR. Gentamicin-induced readthrough of stop condons in Duchenne muscular dystrophy. Ann Neurol. 2010;67:771–780. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- Mangiardi DA, McLaughlin-Williamson K, May KE, Messana EP, Mountain DC, Cotanche DA. Progression of hair cell ejection and molecular markers of apoptosis in the avian cochlea following gentamicin treatment. J Comp Neurol. 2004;475:1–18. doi: 10.1002/cne.20129. [DOI] [PubMed] [Google Scholar]
- Marcotti W, van Netten SM, Kros CJ. The aminoglycoside antibiotic dihydrostreptomycin rapidly enters mouse outer hair cells through the mechano-electrical transducer channels. J Physiol. 2005;567:505–521. doi: 10.1113/jphysiol.2005.085951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Chang KW, Malogolowkin M, London WB, Frazier AL, Womer RB, Rescorla F, Billmire DF, Davis MM, Perlman EJ, Giller R, Lauer SJ, Olson TA. Amifostine does not protect against the ototoxicity of high-dose cisplatin combined with etoposide and bleomycin in pediatric germ-cell tumors: a Children’s Oncology Group study. Cancer. 2005;104:841–847. doi: 10.1002/cncr.21218. [DOI] [PubMed] [Google Scholar]
- Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathog RH, Klein WJ., Jr Ototoxicity of ethacrynic acid and aminoglycoside antibiotics in uremia. N Engl J Med. 1969;280:1223–1224. doi: 10.1056/NEJM196905292802208. [DOI] [PubMed] [Google Scholar]
- Matsui JI, Gale JE, Warchol ME. Critical signaling events during the aminoglycoside-induced death of sensory hair cells in vitro. J Neurobiol. 2004;61:250–266. doi: 10.1002/neu.20054. [DOI] [PubMed] [Google Scholar]
- Matt T, Leong NC, Lang K, Sha S-H, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Dubbaka SR, Xie J, Vasella A, Ramakrishnan V, Schacht J, Böttger EC. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc Natl Acad Sci US. 2012;109:10984–10989. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness JE, Proctor PH, Demopoulos HB, Hokanson JA, Kirkpatrick DS. Amelioration of cis-platinum nephrotoxicity by orgotein (superoxide dismutase) Physiol Chem Phys. 1978;10:267–277. [PubMed] [Google Scholar]
- McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2:483–490. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- Meech RP, Campbell KC, Hughes LP, Rybak LP. A semiquantitative analysis of the effects of cisplatin on the rat stria vascularis. Hear Res. 1998;124:44–59. doi: 10.1016/s0378-5955(98)00116-6. [DOI] [PubMed] [Google Scholar]
- Mizuta K, Saito A, Watanabe T, Nagura M, Arakawa M, Shimizu F, Hoshino T. Ultrastructural localization of megalin in the rat cochlear duct. Hear Res. 1999;129:83–91. doi: 10.1016/s0378-5955(98)00221-4. [DOI] [PubMed] [Google Scholar]
- Moestrup SK, Cui S, Vorum H, Bregengard C, Bjorn SE, Norris K, Gliemann J, Christensen EI. Evidence that epithelial glycoprotein 330/megalin mediates uptake of polybasic drugs. J Clin Invest. 1995;96:1404–1413. doi: 10.1172/JCI118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohtat D, Susztak K. Fine tuning gene expression: the epigenome. Semin Nephrol. 2010;30:468–476. doi: 10.1016/j.semnephrol.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama J, Hashimoto T, Matsubara A, Futai K, Namba A, Shinkawa H. Leupeptin, a calpain inhibitor, protects inner ear hair cells from aminoglycoside ototoxicity. Tohoku J Exp Med. 2006;209:89–97. doi: 10.1620/tjem.209.89. [DOI] [PubMed] [Google Scholar]
- Moore RD, Smith CR, Lietman PS. Risk factors for the development of auditory toxicity in patients receiving aminoglycosides. J Infect Dis. 1984;149:23–30. doi: 10.1093/infdis/149.1.23. [DOI] [PubMed] [Google Scholar]
- More SS, Akil O, Ianculescu AG, Geier EG, Lustig LR, Giacomini KM. Role of the copper transporter, CTR1, in platinum-induced ototoxicity. J Neurosci. 2010;30:9500–9509. doi: 10.1523/JNEUROSCI.1544-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjea D, Whitworth CA, Nandish S, Dunaway GA, Rybak LP, Ramkumar V. Expression of the kidney injury molecule 1 in the rat cochlea and induction by cisplatin. Neuroscience. 2006;139:733–740. doi: 10.1016/j.neuroscience.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Mukherjea D, Jajoo S, Kaur T, Sheehan KE, Ramkumar V, Rybak LP. Transtympanic administration of short interfering (si)RNA against cochlear specific NADPH oxidase isoform NOX3 abrogates cisplatin-induced hearing loss in the rat. Antioxidants & Redox Signal. 2010;13:589–598. doi: 10.1089/ars.2010.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjea D, Rybak LP. The pharmacogenomics of cisplatin-induced ototoxicity. Pharmacogenomics. 2011;12:1039–1050. doi: 10.2217/pgs.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulheran M, Degg C, Burr S, Morgan DW, Stableforth DE. Occurrence and risk of cochleotoxicity in cystic fibrosis patients receiving repeated high-dose aminoglycoside therapy. Antimicrob Agents Chemother. 2001;45:2502–2509. doi: 10.1128/AAC.45.9.2502-2509.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadol JB., Jr Patterns of neural degeneration in the human cochlea and auditory nerve: implications for cochlear implantation. Otolaryngol Head Neck Surg. 1997;117:220–228. doi: 10.1016/s0194-5998(97)70178-5. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y. Apoptosis of guinea pig cochlear hair cells following chronic aminoglycoside treatment. Eur Arch Otorhinolaryngol. 1998;255:127–131. doi: 10.1007/s004050050027. [DOI] [PubMed] [Google Scholar]
- Namendys-Silva SA, Gonzalez-Herrera MO, Texcocano-Becerra J, Herrera-Gomez A. Hypoalbuminemia in critically ill patients with cancer: incidence and mortality. Am J Hosp Palliat Care. 2011;28:253–257. doi: 10.1177/1049909110384841. [DOI] [PubMed] [Google Scholar]
- Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem. 2009;52:2836–2845. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuki K, Ohtani I, Aikawa T, Sato Y. The ototoxicity and the accumulation in the inner ear fluids of the various aminoglycoside antibiotics. Ear Res Jap. 1982;13:85–87. [Google Scholar]
- Oldenburg J, Kraggerud SM, Cvancarova M, Lothe RA, Fossa SD. Cisplatin-induced long-term hearing impairment is associated with specific glutahione-S-transferase genotypes in testicular cancer survivors. J Clin Oncol. 2007;25:708–714. doi: 10.1200/JCO.2006.08.9599. [DOI] [PubMed] [Google Scholar]
- Peters U, Preisler-Adams S, Lanvers-Kaminsky C, Jurgens H, Lamprecht-Dinnesen A. Sequence variations of mitochondrial DNA and individual sensitivity to the ototoxic effect of cisplatin. Anticancer Res. 2003;23:1249–1255. [PubMed] [Google Scholar]
- Pfannenstiel SC, Praetorius M, Plinkert PK, Brough DE, Staecker H. Bcl-2 gene therapy prevents aminoglycoside-induced degeneration of auditory and vestibular hair cells. Audiol Neurootol. 2009;14:254–266. doi: 10.1159/000192953. [DOI] [PubMed] [Google Scholar]
- Pirvola U, Xing-Qun L, Virkkala J, Saarma M, Murakata C, Camoratto AM, Walton KM, Ylikoski J. Rescue of hearing, auditory hair cells, and neurons by CEP-1347/KT7515, an inhibitor of c-Jun N-terminal kinase activation. J Neurosci. 2000;20:43–50. doi: 10.1523/JNEUROSCI.20-01-00043.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- Priuska EM, Schacht J. Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem Pharmacol. 1995;50:1749–1752. doi: 10.1016/0006-2952(95)02160-4. [DOI] [PubMed] [Google Scholar]
- Pussegoda K. Genetic variants associated with cisplatin-induced hearing loss. Clin Genet. 2010;78:33–35. doi: 10.1111/j.1399-0004.2010.01414_2.x. [DOI] [PubMed] [Google Scholar]
- Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxocity associated with platinating agents. Cancer Treat Rev. 2007;33(1):9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael Y, Fein A, Nebel L. Transplacental kanamycin ototoxicity in the guinea pig. Arch Otorhinolaryngol. 1983;238:45–51. doi: 10.1007/BF00453740. [DOI] [PubMed] [Google Scholar]
- Ravi R, Somani SM, Rybak LP. Mechanism of cisplatin ototoxicity: antioxidant system. Pharmacol Toxicol. 1995;76:386–394. doi: 10.1111/j.1600-0773.1995.tb00167.x. [DOI] [PubMed] [Google Scholar]
- Richardson GP, Forge A, Kros CJ, Fleming J, Brown SD, Steel KP. Myosin VIIA is required for aminoglycoside accumulation in cochlear hair cells. J Neurosci. 1997;17:9506–9519. doi: 10.1523/JNEUROSCI.17-24-09506.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg B, Vancamp L, Krigas T. Inhibition of Cell Division in Escherichia Coli by Electrolysis Products from a Platinum Electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Platinum compounds: a new class of potent antitumour agents. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- Ross CJD, Katzov-Eckert H, Dube MP, Brooks B, Rassekh SR, Barhdadi A, Feroz-Zada Y, Visscher H, Brown AMK, Rieder MJ, Rogers PC, Phillips MS, Carleton BC, Hayden MR CPNDS Consortium . Genetic variants in TMPT and COMT are associated with hearing loss in children receiving cisplatin chemotherapy. Nat Genet. 2009;41:1345–1349. doi: 10.1038/ng.478. [DOI] [PubMed] [Google Scholar]
- Rozencweig M, von Hoff DD, Slavik M, Muggia FM. Cis-diamminedichlorophlatinum (II). A new anticancer drug. Ann Intern Med. 1977;86:803–812. doi: 10.7326/0003-4819-86-6-803. [DOI] [PubMed] [Google Scholar]
- Ruedi L. Further observations concerning the toxic effects of streptomycin and quinine on the auditory organ of guinea pigs. Laryngoscope. 1952;62:333–357. doi: 10.1288/00005537-195204000-00001. [DOI] [PubMed] [Google Scholar]
- Rybak LP, Husain K, Morris C, Whitworth C, Somani S. Effect of protective agents against cisplatin ototoxicity. Am J Otol. 2000;21:513–520. [PubMed] [Google Scholar]
- Rybak LP, Whitworth CA. Ototoxicity: therapeutic opportunities. Drug Discov Today. 2005;10:1313–1321. doi: 10.1016/S1359-6446(05)03552-X. [DOI] [PubMed] [Google Scholar]
- Santos F, MacDonald G, Rubel EW, Raible DW. Lateral line hair cell maturation is a determinant of aminoglycoside susceptibility in zebrafish (Danio rerio) Hear Res. 2006;213:25–33. doi: 10.1016/j.heares.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Sastry J, Kellie SJ. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr Hematol Oncol. 2005;22:441–445. doi: 10.1080/08880010590964381. [DOI] [PubMed] [Google Scholar]
- Schacht J. Isolation of an aminoglycoside receptor from guinea pig inner ear tissues and kidney. Arch Otorhinolaryngol. 1979;224:129–134. doi: 10.1007/BF00455236. [DOI] [PubMed] [Google Scholar]
- Schatz A, Bugie E, Waksman SA. Streptomycin, a substance exhibiting antibiotic activity against gram-positive and gram-negative bacteria. Proc Soc Exp Biol Med. 1944;55:66–69. doi: 10.1097/01.blo.0000175887.98112.fe. [DOI] [PubMed] [Google Scholar]
- Schuknecht HF. Pathology of the Ear. Philidelphia: Lea & Febiger; 1993. [Google Scholar]
- Sha SH, Qiu JH, Schacht J. Aspirin to prevent gentamicin-induced hearing loss. N Engl J Med. 2006;354:1856–1857. doi: 10.1056/NEJMc053428. [DOI] [PubMed] [Google Scholar]
- Sha SH, Schacht J. Salicylate attenuates gentamicin-induced ototoxicity. Lab Invest. 1999;79:807–813. [PubMed] [Google Scholar]
- Sha SH, Zajic G, Epstein CJ, Schacht J. Overexpression of copper/zinc-superoxide dismutase protects from kanamycin-induced hearing loss. Audiol Neurootol. 2001;6:117–123. doi: 10.1159/000046818. [DOI] [PubMed] [Google Scholar]
- Shimizu A, Takumida M, Anniko M, Suzuki M. Calpain and caspase inhibitors protect vestibular sensory cells from gentamicin ototoxicity. Acta Otolaryngol. 2003;123:459–465. doi: 10.1080/00016480310001312. [DOI] [PubMed] [Google Scholar]
- Sone M, Schachern PA, Paparella MM. Loss of spiral ganglion cells as primary manifestation of aminoglycoside ototoxicity. Hear Res. 1998;115:217–223. doi: 10.1016/s0378-5955(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Staecker H, Liu W, Malgrange B, Lefebvre PP, Van De Water TR. Vector-mediated delivery of bcl-2 prevents degeneration of auditory hair cells and neurons after injury. ORL J Otorhinolaryngol Relat Spec. 2007;69:43–50. doi: 10.1159/000096716. [DOI] [PubMed] [Google Scholar]
- Stepanyan RS, Indzhykulian AA, Vélez-Ortega AC, Boger ET, Steyger PS, Friedman TB, Frolenkov GI. TRPA1-mediated accumulation of aminoglycosides in mouse cochlear outer hair cells. J Assoc Res Otolaryngol. 2011;12:729–740. doi: 10.1007/s10162-011-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyger PS. Cellular uptake of aminoglycosides. Volta Rev. 2005;105:299–324. [Google Scholar]
- Strauss M, Towfighi J, Lord S, Lipton A, Harvey HA, Brown B. Cis-platinum ototoxicity: clinical experience and temporal bone histopathology. Laryngoscope. 1983;93:1554–1559. doi: 10.1288/00005537-198312000-00007. [DOI] [PubMed] [Google Scholar]
- Swan SK. Aminoglycoside nephrotoxicity. Semin Nephrol. 1997;17:27–33. [PubMed] [Google Scholar]
- Tabuchi K, Pak K, Chavez E, Ryan AF. Role of inhibitor of apoptosis protein in gentamicin-induced cochlear hair cell damage. Neuroscience. 2007;149:213–222. doi: 10.1016/j.neuroscience.2007.06.061. [DOI] [PubMed] [Google Scholar]
- Taguchi T, Nazneen A, Abid MR, Razzaque MS. Cisplatin-associated nephrotoxicity and pathological events. Contrib Nephrol. 2005;148:107–121. doi: 10.1159/000086055. [DOI] [PubMed] [Google Scholar]
- Thakur CP. Tetany in kala azar patients treated with paromomycin. Indian J Med Res. 127:489–493. [PubMed] [Google Scholar]
- Tono T, Kiyomizu K, Matsuda K, Komune S, Usami S, Abe S, Shinkawa H. Different clinical characteristics of aminoglycoside-induced profound deafness with and without the 1555 A-->G mitochondrial mutation. ORL J Otorhinolaryngol Relat Spec. 2001;63:25–30. doi: 10.1159/000055702. [DOI] [PubMed] [Google Scholar]
- Tran Ba Huy P, Bernard P, Schacht J. Kinetics of gentamicin uptake and release in the rat. Comparison of inner ear tissues and fluids with other organs. J Clin Invest. 1986;77:1492–1500. doi: 10.1172/JCI112463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukasaki N, Whitworth CA, Rybak LP. Acute changes in cochlear potentials due to cisplatin. Hear Res. 2000;149:189–198. doi: 10.1016/s0378-5955(00)00182-9. [DOI] [PubMed] [Google Scholar]
- van Ruijven MW, de Groot JC, Klis SF, Smoorenburg GF. The cochlear targets of cisplatin: an electrophysiological and morphological time-sequence study. Hear Res. 2005a;205:241–248. doi: 10.1016/j.heares.2005.03.023. [DOI] [PubMed] [Google Scholar]
- van Ruijven MW, De Groot JC, Klis SF, Smoorenburg GF. Time sequence of degeneration pattern in the guinea pig cochlea during cisplatin administration. A quantitative histological study. Hear Res. 2005b;197:44–54. doi: 10.1016/j.heares.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Vermorken JB, van der Vijgh WJ, Klein I, Hart AA, Gall HE, Pinedo HM. Pharmacokinetics of free and total platinum species after short-term infusion of cisplatin. Cancer Treat Rep. 1984;68:505–513. [PubMed] [Google Scholar]
- Vital-Brazil O, Corrado AP. The curariform action of streptomycin. J Pharm Exp Ther. 1957;68:452–459. [PubMed] [Google Scholar]
- Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, Hoffman EP, Fischbeck KH. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutation. Ann Neurol. 2001;49:706–711. [PubMed] [Google Scholar]
- Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- Wang J, Ladrech S, Pujol R, Brabet P, Van De Water TR, Puel JL. Caspase inhibitors, but not c-Jun NH2-terminal kinase inhibitor treatment, prevent cisplatin-induced hearing loss. Cancer Res. 2004;64:9217–9224. doi: 10.1158/0008-5472.CAN-04-1581. [DOI] [PubMed] [Google Scholar]
- Wang J, Van De Water TR, Bonny C, de Ribaupierre F, Puel JL, Zine A. A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J Neurosci. 2003;23:8596–8607. doi: 10.1523/JNEUROSCI.23-24-08596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Inai S, Jinnouchi K, Baba S, Yagi T. Expression of caspase-activated deoxyribonuclease (CAD) and caspase 3 (CPP32) in the cochlea of cisplatin (CDDP)-treated guinea pigs. Auris Nasus Larynx. 2003;30:219–225. doi: 10.1016/s0385-8146(03)00049-x. [DOI] [PubMed] [Google Scholar]
- Webster M, Webster DB. Spiral ganglion neuron loss following organ of Corti loss: a quantitative study. Brain Res. 1981;212:17–30. doi: 10.1016/0006-8993(81)90028-7. [DOI] [PubMed] [Google Scholar]
- Williams SE, Smith DE, Schacht J. Characteristics of gentamicin uptake in the isolated crista ampullaris of the inner ear of the guinea pig. Biochem Pharmacol. 1987;36:89–95. doi: 10.1016/0006-2952(87)90385-6. [DOI] [PubMed] [Google Scholar]
- Wright CG, Schaefer SD. Inner ear histopathology in patients treated with cis-platinum. Laryngoscope. 1982;92:1408–1413. doi: 10.1288/00005537-198212000-00013. [DOI] [PubMed] [Google Scholar]
- Ylikoski J, Pirvola U, Suoqiang Z, Erikkson U, Solin M-L. Aminoglycoside ototoxicity: high affinity receptors are expressed in secretory epithelia. Aud Neurosci. 1997;3:415–424. [Google Scholar]
- Ylikoski J, Xing-Qun L, Virkkala J, Pirvola U. Blockade of c-Jun N-terminal kinase pathway attenuates gentamicin-induced cochlear and vestibular hair cell death. Hear Res. 2002;163:71–81. doi: 10.1016/s0378-5955(01)00380-x. [DOI] [PubMed] [Google Scholar]
- Zhang M, Liu W, Ding D, Salvi R. Pifithrin-alpha suppresses p53 and protects cochlear and vestibular hair cells from cisplatin-induced apoptosis. Neuroscience. 2003;120:191–205. doi: 10.1016/s0306-4522(03)00286-0. [DOI] [PubMed] [Google Scholar]