

Abstract
In this study, we report the preparation of a novel microcapsule of ~ 100 μm with a liquid (as compared to solid-like alginate hydrogel) core and an alginate-chitosan-alginate (ACA) shell for encapsulation and culture of embryonic stem (ES) cells in the miniaturized 3D space of the liquid core. Murine R1 ES cells cultured in the microcapsules were found to survive (> 90%) well and proliferate to form either a single aggregate of pluripotent cells or embryoid body (EB) of more differentiated cells in each microcapsule within 7 days, dependent on the culture medium used. This novel microcapsule technology allows massive production of the cell aggregates or EBs of uniform size and controllable pluripotency, which is important for the practical application of stem cell based therapy. Moreover, the semipermeable ACA shell was found to significantly reduce immunoglobulin G (IgG) binding to the encapsulated cells by up to 8.2 times, compared to non-encapsulated cardiac fibroblasts, mesenchymal stem cells, and ES cells. This reduction should minimize inflammatory and immune responses induced damage to the cells implanted in vivo becasue IgG binding is an important first step of the undesired host responses. Therefore, the ACA microcapsule with selective shell permeability should be of importance to advance the emerging cell-based medicine.
Introduction
Transplantation of encapsulated cells (particularly stem cells) has been proposed to be a promising cell-based treatment for a wide variety of malfunctions such as diabetes, metabolic deficiencies, liver failure, cancer, and neurodegenerative and cardiovascular diseases.1–8 Alginate has been one of the most commonly used biomaterials for cell encapsulation, probably due to its natural origin (in seaweeds) and excellent biocompatibility.8–12 A unique property of alginate is that it forms hydrogel under mild condition (in the presence of divalent cations such as Ca2+) that is not very harmful to living cells,11–13 which we and others have utilized to prepare cell-loaded plain (or pure) alginate microbeads by electrospray and microfluidic flow focusing.14–16
Plain alginate microbeads, however, might not be used to effectively block the humoral component of host immune system such as immunoglobulin G (IgG) to achieve effective immunoisolation (i.e., to protect the encapsulated cells from being attacked by the host immune system) that is desired for nonautologous cell transplantation. Therefore, poly-l-lysine (PLL, a polycation) has been used to coat the plain alginate (a polyanion) microbeads to allow for further permeability control and the PLL coated alginate microbeads were usually further coated with alginate to form the so-called alginate-PLL-alginate or APA microcapsules.17–20 However, recent studies suggest that PLL in APA microcapsules might induce inflammatory cytokine release from host immune cells, which can cause cellular necrosis and contribute to fibrotic overgrowth around encapsulated cells after implantated in vivo.20–23
Chitosan, a polysaccharide of natural origin, has attracted much attention recently as a promising alternative to PLL for coating plain alginate microbeads due to its excellent biocompatibility.24–28 Nonetheless, there is only one study so far that reported successful replacement of PLL with chitosan to prepare ACA (alginate-chitosan-alginate) microcapsules of ~ 250 μm with a solid-like core of alginate hydrogel for encapsulating bacterial (E. coli) cells.29 This study also showed that ACA microcapsules have better mechanical and chemical stability than APA microcapsules. However, no study has been reported to encapsulate any cells in ACA microencapsules with a liquid core.
In addition, although microcapsules greater than 250 μm have been used dominantly in the past studies of cell encapsulation with promising outcomes, smaller microcapsules may be desired.1–8 This is because the typical distance between two capillaries in a normal epithelial tissue is ~ 100 μm and cells in the middle zone between two capillaries would suffer deprivation of nutrients and oxygen when the distance becomes greater under diseased conditions.7,8,22,30–34 Cell death in microcapsules should be minimized as dead/dying cells can release chemokines that can induce undesired inflammatory/immune responses.35,36 Moreover, smaller microcapsules have been shown to exhibit better mechanical stability to minimize microcapsule burst and consequently better biocompatibility.37–40 Therefore, microcapsules of ~ 100 μm (whose volume is only one fifteenth of that of the 250 μm microcapsules assuming a spherical morphology) or smaller may be desired for sufficient transport of nutrients, oxygen, and metabolites for all encapsulated cells to survive and better outcome after implantation in vivo.
In this study, we report the successful preparation of ~ 100 μm ACA microcapsules with a liquid (as compared to solid-like alginate hydrogel) core that is desired for ES cell culture. Murien R1 ES cells encapsulated in the ACA microcapsules were observed to survive (> 90%) and proliferate well in the miniaturized 3D space of the liquid core. We further demonstrated that the ACA microcapsules exhibit robust selectivity of permeability to external molecules: They can block IgG while allowing efficient transport of smaller molecules necessary for the survival and pluripotency control of the encapsulated ES cells. These results suggest that the ~ 100 μm ACA microcapsule should be very useful for cell microencapsulation to advance the emerging cell-based medicine.
Materials and Methods
Materials
Purified sodium alginate (type M) was purchased from Medipol (Lausanne, Switzerland). Chitosan of pharmaceutical grade (80 kD, ~ 95% deacetylation) was obtained from Weikang Biological Products Co. Ltd (Shanghai, China). The live/dead viability/cytotoxicity kit for mammalian cells, trypsin/EDTA, and regular DMEM (high glucose) were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum, penicillin, and streptomycin were purchased from Hyclone (Logan, Utah). Konckout® DMEM and Konckout® serum, LIF (leukemia inhibitory factor) were purchased from Millipore (Billerica, MA). Fluorescein isothiocyanate-immunoglobulin G (FITC-IgG) of human origin, FITC-dextran, and all other chemicals were purchased from Sigma (St. Louis, MO) unless specifically indicated otherwise.
Cell culture
R1 murine embryonic stem (ES) cells from ATCC (Manassas, VA) were cultured in ES cell medium made of Konckout® DMEM supplemented with 15% Konckout® serum, 1000 U/ml LIF, 4 mM L-glutamine, 0.1 M 2-mercaptoethanol, 10 μg/ml gentamicin, and 100 U/ml penicillin and 100 μg/ml streptomycin using gelatin coated tissue culture flasks. C3H10T1/2 mesenchymal stem cells (MSCs) from ATCC were cultured in regular DMEM with 10% fetal bovine serum, 100 U/mL penicillin and 100 mg/L streptomycin. Primary neonatal cardiac fibroblasts were cultured in DMEM/F-10 mixture (1:1) supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. All cells were cultured at 37 °C in a humidified 5% CO2 incubator. On the day of experiments, the cells in culture were collected by detaching from culture dishes or flasks with trypsin/EDTA digestion, centrifuged for 3 minutes at 960 rpm, and resuspended in a 0.9 % (w/v) sodium chloride solution (physiological saline) for further experimental use.
Preparation of alginate-chitosan-alginate (ACA) microcapsules
The procedure for preparing ACA microcapsules is illustrated in Scheme 1. First, detached cells in physiological saline were centrifuged and re-suspended at a density of 5 × 106 cells/ml in 2% (w/v) sodium alginate solution with 0.25 M mannitol. The cell suspension was then transferred into a 3 mL syringe for preparing plain alginate microbeads encapsulated with cells using electrospray. This was done by pushing the cell suspension through a 30 gauge syringe needle with a blunt tip and spraying it under an electrostatic field into 150 mM calcium chloride solution in a gelling bath, as detailed elsewhere by us.14 The distance between the syringe needle tip and the surface of the calcium chloride solution in the gelling bath was 5.5 mm, a 0.84 kV electrostatic potential was applied on the needle tip, and a spay flow of 2.5 ml/hr was used. All solutions used for cell microencapsulation were buffered using 10 mM HEPES to maintain the solution pH between 7.2 and 7.4.
Scheme 1.
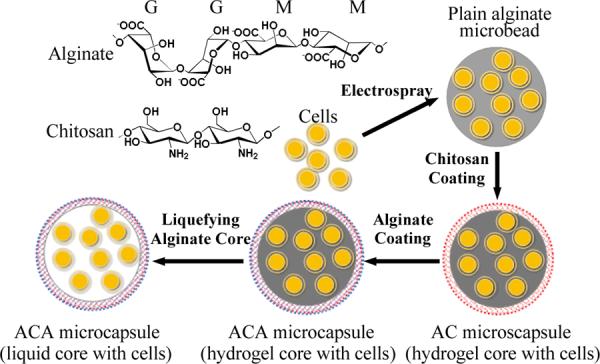
A schematic illustration of the procedure for preparing ACA microcapsules with a liquid core loaded with cells by 1, electrospraying to produce cell loaded plain alginate microbeads (a solid microsphere of alginate hydrogel with no shell); 2, coating the microbeads with chitosan to form aliginate-chitosan (AC) microcapsules (with an AC shell); 3, coating the AC microcapsules further with alginate to form alginate-chitosan-alginate (ACA) microcapsules (with an ACA shell and solid-like core of alginate hydrogel loaded with cells); and 4, liquefying the alginate hydrogel core to give rise to the ACA microcapsules with a liquid core loaded with cells: The letters G and M represent the guluronate and mannuronate blocks in alginate, respectively.
To prepare ACA microcapsules encapsulated with cells of high viability, the plain alginate microbeads generated by electrospray were washed using 0.5 M mannitol solution for 3 – 10 minutes and suspended in 0.1 – 0.5 % (wt) chitosan solution (pH 6.5 – 6.6) for 3 – 10 minutes for chitosan coating to obtain alginate-chitosan (AC) microcapsules. The chitosan solution was made by dissolving chitosan (80 kD) in saline at pH 4.83 (adjusted by adding 85 μl acetic acid in 20 ml saline) and the pH of the solution was brought back to 6.5 – 6.6 by titration using NaOH. The AC microcapsules were then suspended in saline with 0.15 % (w/v) sodium alginate to counteract the rudimental charges of the coated chitosan (i.e., further alginate coating), giving rise to the ACA microcapsules with a solid-like core of alginate hydrogel. Lastly, the solid-like alginate hydrogel core was liquefied by ion chelation to remove Ca2+ from alginate in the core using a 55 mM sodium citrate solution for 5 min to obtain alginate-chitosan-alginate (ACA) microcapsules with a liquid core and a thin alginate-chitosan-alginate (ACA) membrane or shell. The ACA microcapsules with a liquid core are of importance for encapsulating and culturing embryonic stem (ES) cells because it is desired for the encapsulated ES cells to proliferate, aggregate, and form embryoid body (EB) for further differentiation.41–43 ACA microcapsules with a liquid core without cells were also prepared in the same way for characterization of their chemistry and shell permeability.
Characterization of the ACA microcapsules
To measure the surface charge (represented by surface zeta potential), microbeads or microcapsules without cells were suspended in de-ionized (DI) water and their surface charge measured quickly using a Brookhaven (Holtsville, NY) zeta potential analyzer. The chemistry of the microcapsule synthesis was studied using a PerkinElmer Spectrum 100 ATR-FTIR, for which the samples were suspended in DI water and then freeze-dried for further analysis. To test shell permeability, the microbeads or microcapsules were added into 2 ml of 0.05% (w/v) FITC-IgG (MW: 160 kD) or FITC-dextran (MW: 4.4 kD) solution and incubated for either 1 or 12 hr at 37 °C. Afterwards, the microcapsules were studied using a Zesis LSM 510 META confocal laser-scanning microscope to examine the distribution of the fluorescence probes (FITC-IgG and FITC-dexran) inside and outside of the microparticles.
Viability, culture, and proliferation of encapsulated ES cells
Viability of the encapsulated cells was determined using the standard live/dead assay kit from Invitrogen. Green (live cells) and red (dead cells) fluorescence images were collected separately and merged using NIH ImageJ to determine cell viability as the ratio of viable cells to total cells counted. The encapsulated ES cells were cultured in cell culture inserts (BD Falcon) immersed in medium in the companion plate (BD Falcon) of the inserts (to avoid microcapsule loss during changing medium) under different conditions: Complete ES cell medium for 0–7 days followed by either ES cell medium without LIF or regular culture medium (containing regular DMEM, 10% serum, 100 U/ml penicillin and 100 μg/ml streptomycin) for 7–0 days for a total of 7 days with the culture medium being replenished daily unless specifically indicated otherwise. Proliferation of ES cells in microcapsules under the various culture conditions was monitored as the formation of either a single cell aggregate of pluripotent cells or embryoid body (EB) in each microcapsule.
Quantitative real time RT-PCR (qRT-PCR) study of gene expression
For qRT-PCR studies, RNAs were isolated from the encapsulated cells using the TRIzol®/Plant RNeasy® Mini Kit method with modification.44 Briefly, the encapsulated cells were washed twice using physiological saline. After cell lysis using TRIzol® reagent (Invitrogen), 0.2 ml of chloroform per 1 ml of TRIzol® reagent was used for phase separation of the lysate. The aqueous phase containing RNAs was gently collected. Next, 0.5 ml of isopropanol per 1 ml TRIzol® reagent was added to the aqueous phase to precipitate RNAs. Afterward, the precipitated RNAs were dissolved using the RLT buffer from the Plant RNeasy® Mini Kit (Qiagen) and RNA purification was performed following the manufacture's instructions. Next, reverse transcription was carried out to generate complementary DNAs (cDNAs) using the iScript™ cDNA synthesis kit (Bio-Rad). This was done by mixing the purified RNAs with the reverse transcriptase and mix reagent from the kit according to the manufacture's instruction. The reverse transcription reaction was performed using a GeneAmp PCR system (model 9700). Quantitative PCR studies were done with the SYBR Green mix (Bio-Rad) using a Bio-Rad CFX96 real time PCR machine. Relative gene expression was calculated using the ΔΔCt method built in the Bio-Rad software where Ct is cycle threshold defined as the number of cycles required for amplifying a gene detected as fluorescence signal (using fluorescence probe in the above-mentioned SYBR Green mix in a sample) to exceed the background fluorescence level. In this method, the difference in Ct between a specific gene of interest (GOI such as Oct4) and house keeping gene (HKG such as GAPDH here) and the difference in Ct of HKG between control and non-control conditions are calculated as ΔCt,GOI and ΔCt,HKG, respectively. The further differenece (i.e., ΔΔCt,GOI) between ΔCt, GOI and ΔCt, HKG is used to calculate the times of amplification of the GOI with respect to HKG as 2ΔΔCt,GOI, which should eliminate any potential random error in calculating the amplication induced by accidental variations (e.g., due to pipetting) in the signal of HKG between different conditions. The following genes and primers were used in this study: Oct4, 5'-GAAGCCCTCCCTACAGCAGA-3' and 5'-CAGAGCAGTGACGGGAACAG-3'; Sox2, 5'-GCATGTCCTACTCGCAGCAG-3 and 5'-GCTGATCATGTCCCGGAGGT-3'; Nanog, 5'-CCCCAC AAGCCTTGGAATTA-3' and 5'-CTCAAATCCCAGCAACCACA-3'; Klf2, 5'-CTGCTGGAGGCCAA GCCCAA -3' and 5'AGGTGGTCGGACCTGGAGAA-3'; Nestin, 5'-GGAGTGTCGCTTAGAGGTGC-3' and 5'-T CCAGAAAGCCAAGAGAAGC-3'; gooscoid (GSC), 5'-AAACGCCGAGAAGTGGAACA AG-3' and 5'-AAGGCAGGGTGTGTGCAAGTAG-3'; Brachyury (T), 5'-CTCTAATGTCCTCCCTTG TTGCC-3' and 5'-TGCAGATTGTCTTTGGCTACTTTG-3'; α-fetoprotein (AFP), 5'-GCCACCGAGG AGGAAG TG-3' and 5'-AGTCTTCTTGCGTGCCAG C-3'; and GAPDH, 5'-CTCTGGCTCAGAGGG TTTGG-3' and 5'-ACAGAAACCAGT GGGCTTTGA -3'. Oct4, Sox2, Nanog, and Klf2 are pluripotent genes while nestin is for ectoderm, GSC and T for mesoderm, and AFP for endoderm.
Flow cytometry study of IgG-cell binding
For flow cytometry studies, cells encapsulated in pure alginate microbeads or ACA microcapsules were incubated with 150 μg/ml FITC-IgG overnight. The microbeads or microcapsules were then collected and rinsed 3 times with 0.5 M mannitol. The cells encapsulated in pure alginate microbeads were released by ion chelation to liquefy the Ca2+ cross-linked alginate using 150 mM sodium citrate solution. To release cells from the ACA microcapsule, the ACA shell were chemically broken using a buffer solution containing 200 mM sodium bicarbonate and 60 mM sodium citrate. The released cells were collected by centrifuge, followed by fixation using 4% formaldehyde. Afterward, the samples were analyzed using a BD LSR II Flow Cytometer. Nonencapsulated (attached) cells either with or without incubating with FITC-IgG overnight were also studied as the positive and negative control, respectively. The flow cytometry data of encapsulated cells with two peaks were analyzed using Origin (v8.6, OriginLab, Northampton, MA) to identify the peak as a result of IgG binding to cells, based on the single flow cytometry peak of nonencapsulated cells either with or without incubating with IgG, as detailed in Fig. S1. The results of flow cytometry data are reported as the percentage of IgG bound (or positive) cells (counts) to total cells (counts) studied.
Statistical analysis
All results are reported as the mean ± standard deviation (SD) of data from at least three separate runs. Student's two-tailed t-test assuming equal variance was calculated using Microsoft® Excel to determine statistical significance (p < 0.05).
Results and discussion
Preparation and characterization of ACA microcapsules without cells
To prepare the ACA microcapsules, plain or pure alginate microbeads of ~ 100 μm were first fabricated by electrospray as we did before.14 Coating the small alginate microbeads with either chitosan or PLL, however, turns out to be extremely challenging because of microcapsule collapse (Fig. 1A). We resolved this problem by washing (before coating in chitosan/PLL solution) the microbeads in 0.5 M mannitol solution instead of physiological saline although the latter has been commonly used in the procedure for coating PLL on 250 μm or larger alginate microbeads. The concentration of chitosan solution was found to have great influence on the outcome of coating. When low (0.1%) concentration of chitosan solution was used, the microcapsules became broken/collapsed/wrinkled and were not easily visible after liquefying the alginate core to obtain ACA microcapsules with a liquid core (Fig. 1B). This is presumably because the amount of chitosan that could be coated on the alginate microbead is little during a time frame of 3–10 min and the ACA shell would be too loose to maintain the spherical morphology after liquefying the alginate core. On the other hand, the resultant ACA microcapsules tended to stick to each other when the chitosan concentration was 0.5% or higher (Fig. 1C). This is possibly because the positive charge of chitosan is not always perfectly counteracted by the negatively charged alginate in one microcapsule, which allows the interaction between alginate and chitosan in different microcapsules. Moreover, the use of 0.5% chitosan could compromise the viability of cells encapsulated in the core during chitosan coating, as shown in Fig. S2. However, a coating time between 3 to 10 min had no significant impact on the microcapsule size and morphology, which should allow permeability control of the coated shell. Fig. 1D shows liquid core-shell ACA microcapsules of ~ 100 μm with good morphology prepared by coating for 3 min in 0.4% chitosan solution, a condition used to make ACA microcapsules for all further characterization and ES cell encapsulation studies in this paper.
Fig. 1.

Typical phase images of microcapsules formed after coating (coating time: 3 min) plain alginate microbeads with chitosan either without mannitol washing before coating in 0.4 % chitosan solution (A) or with mannitol washing before coating in 0.1 % (B), 0.5 % (C), and 0.4 % (D) chitosan solution together with surface zeta potential (E) and ATR-FTIR spectra (F) showing successful preparation of the ACA (alginate-chitosan-alginate) microcapsules (liquid core): 1, sodium alginate; 2, chitosan; 3, plain alginate microbeads; 4, AC microcapsules; 5, ACA microcapsules; and scale bar (applicable to all micrographs), 100 μm
As shown in Fig. 1E, the plain alginate (a polyanion) microbeads had a negative surface zeta potential and it became positive for the alginate-chitosan (AC) microcapsules, indicating successful coating of chitosan (a polycation) over the microbeads. The ACA microcapsules showed a negative surface zeta potential, presumably due to successful coating of alginate over the chitosan layer.
The successful preparation of ACA microcapsules was further confirmed by the ATR-FTIR data (Fig. 1F). The spectra of AC and ACA microcapsules display characteristic peaks of both alginate and chitosan (see Fig. S3 for the FTIR spectra over a wider range of wavenumber and Table S1 for detailed peak assignment). The peaks in the 950–1200 cm−1 region that are a result of various vibrations of the carbohydrate ring, the building blocks of both alginate and chitosan, are clearly observable on all the spectra. The peak at 1410 cm−1 due to symmetric stretch of carboxylate ion (COO−) in alginate is present on the spetra of sodium alginate, pure alginate microbead, AC microcapsules, and ACA microcapsules. The antisymmetric strecth of carbonylate ion at 1600 cm−1 of sodium alginate has been reported to be sensitive to the presence of crosslinking agents such as the divalent cations (Ca2+) used in this study.45 This band shifted to 1592 cm−1 (right shift) for the plain (Ca2+ crosslinked) alginate microbeads compared to sodium alginate, which presumably was a result of the increase in the strength of ionic interaction after sodium ions were replaced by calcium ions in the alginate microbeads. On the contrary, this band shifted from 1587 cm−1 (for AC microcapsules) to 1595 cm−1 (for ACA microcapsules) (left shift), presumably due to the replacement of Ca2+ with Na+ during the last coating and liquefying steps using sodium alginate and citrate, respectively. Moreover, a new peak (arrows) near 1538 cm−1 appeared on both the AC and ACA microcapsule spectra, which presumably is a result of the formation of physically crosslinked alginate-chitosan complex in the AC and ACA shell after chitosan coating.
Capability of the ACA microcapsule for encapsulating and culturing ES cells to form a single aggregate of uniform size and controllable pluripotency per microcapsule
Typical phase and fluorescence images of R1 ES cells immediately after encapsulating in the ACA microcapsules with a liquid core showing high cell viability (> 90%, green stain) are given in Fig. 2A and B, respectively. Approximately 5 cells could be encapsulated in each microcapsule on average. The microbeads for preparing the ACA microcapsules were made by electrospray using 0.25 M mannitol solution (rather than 0.9% sodium chloride solution or physiological saline that was commonly used in the past) containing 2% alginate and 5 × 106 cells/ml cells. We found that using the mannitol solution in this step is crucial for maintaining high cell viability and the cell viability was usually less than 70% when physiological saline was used. This observation could be partially attributed to the anti-oxidative capability of mannitol.46–48
Fig. 2.
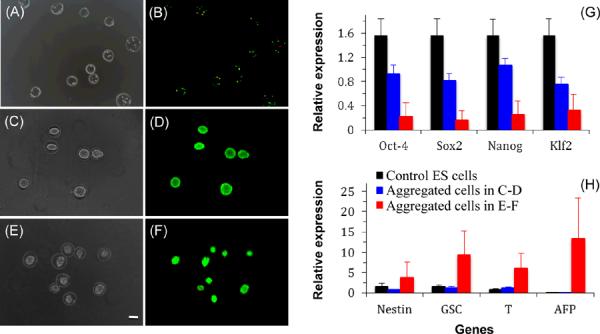
Typical phase and merged fluorescence images of the cell-loaded ACA microcapsules (liquid core) showing live (green) and dead (red) ES cells at day 0 (A–B), after 7-day cutlture in complete ES cell medium (C–D), after 2-day culture in complete ES cell medium flowed by 5-day culture in regular culture medium (E–F) showing the formation of a single cell aggregate in each microcapsule, together with quantitative real time RT-PCR (qRT-PCR) data of four pluripotent genes (G) and four more differentiated genes (H) in the aggregated cells and control ES cells: Nestin gene for ectoderm; GSC (gooscoid) and T (brachyury) genes for mesoderm; AFP (α-fetoprotein) gene for endoderm; and scale bar (applicable to all micrographs), 100 μm
Typical phase and fluorescence images showing the formation of a single cell aggregate of uniform size in each ACA microcapsule with high cell viability after 7-day culture of the encapsulated ES cells in complete ES cells medium are given in Fig. 2C and D, respectively. For ES cells encapsulated in ACA microcapsules with an alginate hydrogel core, however, the cells proliferated to form multiple aggregates of non-uniform size and irregular shape in each microcapsule (Fig. S4). This is presumably because the crosslined alginate fiber can block the continuous aggregation of ES cells in the core. Moreover, we found that culturing the ES cells during the first 2 days after encapsulation in complete ES cell medium is necessary to maintain high viability of the cells so that they can proliferate to form cell aggregates. Otherwise, the cell would die and no cell aggregate could be observable (data not shown). This might be a result of the differentiation of some ES cells in the absence of supplements (particularly LIF) in the ES cell medium when non-ES cell medium was used, the lack of extracellular matrix (ECM) in the microcapsule core for the differentiated cells to attach, and the inability of the relative small number of differentiated cells in the microcapsule to produce their own ECM timely for them to survive. Fig. 2E and F shows the phase and fluorescence image of cell aggregates formed by culturing the ES cells in complete ES cell medium during the first two days and then in regular medium (containing regular DMEM, 10% serum, 100 U/ml penicillin and 100 μg/ml streptomycin) for 5 days. The formation of cell aggregates with high cell viability is clearly observable in the figure although the cell aggregates are also smaller than that formed using complete ES cell medium for 7 days showing in Fig. 2C–D. In all cases, the ACA microcapsules (liquid core) were found to be stable and retain their morphological integrity after the 7 days in the ES cell culture medium. Since the ES cells proliferate to confluence in 7 days culture in the microcapsules, we further tested the capability of the microcapsules for cell culture of longer time using C3H10T1/2 mesenchymal stem cells (MSCs) that proliferate much slower. It was found that the microcapsules could retain their integrity and the MSCs could survive well and proliferate to form aggregates under an extended culture of at least 49 days (Fig. S5).
We further performed quantitative real time RT-PCR (qRT-PCR) study of gene expression in the aggregated cells made with either 7-day (Fig. 2C–D) or 2-day (Fig. 2E–F) culture in the complete ES cell medium together with control ES cells. The results of expression for four pluripotent and four more differentiated genes are shown in Fig. 2G and H, respectively. As expected, the expression of the four pluripotent genes (Oct-4, Sox2, Nanog, and Klf2) are the highest in the control ES cells, followed by the aggregated cells in the microcapsules with 7-day culture in ES cell medium, and it is the lowest in the aggregated cells with only 2-day culture in ES cell medium. On the contrary, the four more differentiated genes (nestin for ectoderm, GSC and T for mesoderm, and AFP for endoderm) are much more highly expressed in the aggregated cells with only 2-day culture in ES cell medium compared to both the control ES cells and the aggregated cells with 7-day culture in ES cell medium. Therefore, the aggregated cells with 7-day culture in ES cell medium are still very pluripotent while the cells in the aggregates with only 2-day culture in ES cell medium are mostly differentiated into the ectodermic, mesodermic, or endodermic lineages, which is typical for cells in embryoid bodies (EBs).
The above proliferation and qRT-PCR data demonstrate the critical role of culture time in ES cell medium in maintaining the survival and pluripotency of the encapsulated ES cells. In other words, cell aggregates at various pluripotent/differentiated stages could be easily produced by control the time of culture in ES cell medium. Considering that hundreds of microcapsules can be easily produced using electrospray in a few minutes (compared to the labor-intensive hanging drop method that has been commonly used to make ES cell aggregates or EBs), this study provides the capability of conveniently preparing a large number of ES cell aggregates or EBs with uniform size and the desired pluripotency (a crucial step for ES cell differentiation), which should make the clinical applications of ES cells more promising where availability of a large number of EBs is very much desired.41–43 Moreover, these data indicate the capability of the ACA microcapsule shell in allowing effective transport of nutrients, oxygen, and metabolic products including metabolic wastes for the encapsulated ES (and differentiated) cells to survive and proliferate.
Capability of the ACA microcapsules for immunosiolation
To encapsulate cells for transplantation, besides allowing efficient transport of nutrients, oxygen, and metabolic products (including wastes and therapeutic agents), the microcapsule should have a shell permeability capable of blocking the humoral component of immune system such as immunoglobulin G (IgG) as IgG binding to target cells could further induce undesired inflammatory and immune responses that can cause cell death and implant failure.13,22,30,49–51 Although it has been taken for granted that the host immune system would not be able to recognize embryonic stem cells because of their undifferentiated nature and the lack of expression of major histocompatibity complex (MHC) on their membrane, data from our flow cytometry studies (Fig. 3A) show that these cells can still be recognized and attacked by the host immune system (via IgG binding) for non-autologous transplantation. As shown in the figure, as with the much more differentiated mouse cardiac fibroblasts, human IgG can bind to both the R1 ES and C3H10T1/2 mesenchymal stem cells, a critical first step of immune attack to implanted cells of non-autologous origin. Moreover, we found that the ACA microcapsules are significantly more efficient in blocking the transport of IgG to bind with the encapsulated cells than the plain alginate microbeads (p = 0.016, 0.027, and 0.002 for cardiac fibroblasts, mesenchymal stem cells, and ES cells, respectively). The plain alginate microbeads are particularly ineffective in reducing IgG binding to the ES cells although it can decrease the binding to cardiac fibroblasts and mesenchymal stem cells by 2.1 and 3.6 times, respectively. The ACA microcapsules can decrease percentage of IgG binding to all the three types of cells to single digit by up to 8.2 times from 49 to 6%, 58 to 8%, and 21 to 5% compared to the nonencapsulated cardiac fibroblasts, mesenchymal stem cells, and ES cells, respectively.
Fig. 3.
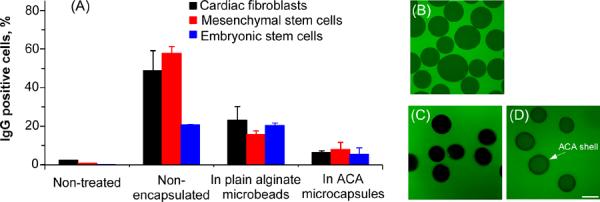
Flow cytometry data (A) showing the percentage of IgG bound (positive) cells after incubating cardiac fibroblasts (differentiated cells), C3H10T1/2 mesenchymal stem cells, and R1 ES cells with FITC-IgG (160 kD) overnight and typical confocal fluorescence images of the middle plane through plain alginate microbeads (B) and ACA microcapsules (liquid core, C) after incubating with FITC-IgG (1 hr for B and 12 hr for C) together with that of the ACA microcapsules after incubating with FITC-dextran (4.4 kD) for 1 hr (D): The ACA shell is clearly visible as the dark ring in panel (D) for the ACA microcapsules with a liquid core, but not in (B) for the plain alginate microbeads. Scale bar (applicable to all micrographs): 100 μm
We further confirmed these observations from flow cytometry studies using confocal microscopy. As shown in Fig. 3B, human IgG (labeled green) could diffuse into plain alginate microcapsules after only 1 hour incubation with them. However, IgG was not observable in the ACA microcapsules (dark appearance) even after 12 hr incubation (Fig. 3C). Considering that the diameter of IgG is ~ 20–40 nm,52–54 this result suggests that the pores of the ACA shell are possibly on nanometer scale and less than ~ 20 nm if the transport of IgG across the shell is soley dependent on size. Moreover, small molecules such as the 4.4 kD dextran (labeled green), which is larger than most nutrients for cell survival, can diffuse into the ACA microcapsules efficiently within one hour incubation (Fig. 3D). In addition, the ACA membrane or shell is clearly visible as the dark ring in Fig. 3D for the ACA microcapsules (liquid core) while no such shell structure is observable for the plain alginate microbeads in Fig. 3B, further indicating successful preparation of ACA microcapsules with a liquid core. Therefore, the ACA microcapsules that we made have robust selectivity of shell permeability that allows not only effective transport of nutrients, oxygen, and metabolic products for cell survival and pluripotency control but minimized diffusion of the humoral components of immune system such as IgG to bind and attack the encapsulated cells. Nonetheless, further in vivo studies are required to confirm the capability of the ACA microcapsules in protecting the cells from adverse inflammatory and immune responses after transplantation.
Summary and conclusions
We successfully prepared for the first time ~ 100 μm (the typical distance between two capillaries in normal epithelial tissue) alginate-chitosan-alginate (ACA) microcapsules with a liquid core for encapsulating embryonic stem (ES) cells with high (> 90%) viability. R1 ES cells encapsulated in the microcapsules were found to survive well and proliferate to form a single cell aggregate of controllable pluripotency in each microcapsule within 7 days. The ACA shell of the microcapsules was found to be selectively permeable to external molecules: It can effectively block IgG (160 kD) while allowing efficient transport of nutrients, oxygen, and metabolic products for cell survival and therapy. Therefore, the ACA microcapsule should be a promising system for encapsulating living cells (particularly those of non-autologous origin) to advance the emerging cell-based medicine.
Supplementary Material
Acknowledgements
This work was partially supported by grants from NSF (CBET-1154965) and NIH (R01EB012108).
Footnotes
Electronic Supplementary Information Available (Figures S1–S5 and Table S1)
References
- 1.Chang TM. Science. 1964;146:524–525. doi: 10.1126/science.146.3643.524. [DOI] [PubMed] [Google Scholar]
- 2.Lim F, Sun AM. Science. 1980;210:908–910. doi: 10.1126/science.6776628. [DOI] [PubMed] [Google Scholar]
- 3.Ding HF, Liu R, Li BG, Lou JR, Dai KR, Tang TT. Biochem Biophys Res Commun. 2007;362:923–927. doi: 10.1016/j.bbrc.2007.08.094. [DOI] [PubMed] [Google Scholar]
- 4.Stensvaag V, Furmanek T, Lonning K, Terzis AJ, Bjerkvig R, Visted T. Cell Transplant. 2004;13:35–44. doi: 10.3727/000000004772664879. [DOI] [PubMed] [Google Scholar]
- 5.Tobias CA, Han SS, Shumsky JS, Kim D, Tumolo M, Dhoot NO, Wheatley MA, Fischer I, Tessler A, Murray M. J Neurotrauma. 2005;22:138–156. doi: 10.1089/neu.2005.22.138. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Zhu SJ, Wang W, Wei YJ, Hu SS. Gene Ther. 2008;15:40–48. doi: 10.1038/sj.gt.3303049. [DOI] [PubMed] [Google Scholar]
- 7.Visted T, Bjerkvig R, Enger PO. Neuro Oncol. 2001;3:201–210. doi: 10.1093/neuonc/3.3.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orive G, Hernandez RM, Rodriguez Gascon A, Calafiore R, Chang TM, de Vos P, Hortelano G, Hunkeler D, Lacik I, Pedraz JL. Trends Biotechnol. 2004;22:87–92. doi: 10.1016/j.tibtech.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Murua A, Portero A, Orive G, Hernandez RM, de Castro M, Pedraz JL. J Control Release. 2008;132:76–83. doi: 10.1016/j.jconrel.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 10.Orive G, Hernandez RM, Gascon AR, Calafiore R, Chang TM, De Vos P, Hortelano G, Hunkeler D, Lacik I, Shapiro AM, Pedraz JL. Nat Med. 2003;9:104–107. doi: 10.1038/nm0103-104. [DOI] [PubMed] [Google Scholar]
- 11.Lee KY, Mooney DJ. Prog Polym Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Augst AD, Kong HJ, Mooney DJ. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 13.de Vos P, Faas MM, Strand B, Calafiore R. Biomaterials. 2006;27:5603–5617. doi: 10.1016/j.biomaterials.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 14.Zhang W, He X. J Biomech Eng. 2009;131:074515. doi: 10.1115/1.3153326. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Tumarkin E, Peerani R, Nie Z, Sullan RM, Walker GC, Kumacheva E. J Am Chem Soc. 2006;128:12205–12210. doi: 10.1021/ja0635682. [DOI] [PubMed] [Google Scholar]
- 16.Kim C, Chung S, Kim YE, Lee KS, Lee SH, Oh KW, Kang JY. Lab Chip. 2011;11:246–252. doi: 10.1039/c0lc00036a. [DOI] [PubMed] [Google Scholar]
- 17.Orive G, Tam SK, Pedraz JL, Halle JP. Biomaterials. 2006;27:3691–3700. doi: 10.1016/j.biomaterials.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 18.Uludag H, De Vos P, Tresco PA. Adv Drug Deliv Rev. 2000;42:29–64. doi: 10.1016/s0169-409x(00)00053-3. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Wang W, Ma J, Guo X, Yu X, Ma X. Biotechnol Prog. 2006;22:791–800. doi: 10.1021/bp050386n. [DOI] [PubMed] [Google Scholar]
- 20.Tam SK, Bilodeau S, Dusseault J, Langlois G, Halle JP, Yahia LH. Acta Biomater. 2011;7:1683–1692. doi: 10.1016/j.actbio.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Zhang WJ, Li BG, Zhang C, Xie XH, Tang TT. Cytotherapy. 2008;10:90–97. doi: 10.1080/14653240701762372. [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, He X. J Healthc Eng. 2011;2:427–446. doi: 10.1260/2040-2295.2.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haque T, Chen H, Ouyang W, Martoni C, Lawuyi B, Urbanska A, Prakash S. Int J Artif Organs. 2005;28:631–637. doi: 10.1177/039139880502800612. [DOI] [PubMed] [Google Scholar]
- 24.Chung HJ, Go DH, Bae JW, Jung IK, Lee JW, Park KD. Current Applied Physics. 2005;5:485–488. [Google Scholar]
- 25.De S, Robinson D. J Control Release. 2003;89:101–112. doi: 10.1016/s0168-3659(03)00098-1. [DOI] [PubMed] [Google Scholar]
- 26.Santos E, Zarate J, Orive G, Hernandez RM, Pedraz JL. Adv Exp Med Biol. 2011;670:5–21. doi: 10.1007/978-1-4419-5786-3_2. [DOI] [PubMed] [Google Scholar]
- 27.De Castro M, Orive G, Hernandez RM, Bartkowiak A, Brylak W, Pedraz JL. J Biomed Mater Res A. 2009;91:1119–1130. doi: 10.1002/jbm.a.32270. [DOI] [PubMed] [Google Scholar]
- 28.Wang L, Stegemann JP. Acta Biomater. 2011;7:2410–2417. doi: 10.1016/j.actbio.2011.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin J, Yu W, Liu X, Xie H, Wang W, Ma X. J Biosci Bioeng. 2008;105:660–665. doi: 10.1263/jbb.105.660. [DOI] [PubMed] [Google Scholar]
- 30.Wilson JT, Chaikof EL. Adv Drug Deliv Rev. 2008;60:124–145. doi: 10.1016/j.addr.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakai S, Kawabata K, Ono T, Ijima H, Kawakami K. Biotechnol Bioeng. 2004;86:168–173. doi: 10.1002/bit.20006. [DOI] [PubMed] [Google Scholar]
- 32.Sakai S, Kawakami K. Adv Exp Med Biol. 2010;670:22–30. doi: 10.1007/978-1-4419-5786-3_3. [DOI] [PubMed] [Google Scholar]
- 33.Leach RM, Treacher DF. Thorax. 2002;57:170–177. doi: 10.1136/thorax.57.2.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambert R, Rey JF, Sankaranarayanan R. Endoscopy. 2003;35:437–445. doi: 10.1055/s-2003-38766. [DOI] [PubMed] [Google Scholar]
- 35.Chen K, Huang J, Gong W, Iribarren P, Dunlop NM, Wang JM. Int Immunopharmacol. 2007;7:1271–1285. doi: 10.1016/j.intimp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 36.Marshak-Rothstein A, Rifkin IR. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 37.Ross CJ, Chang PL. J Biomater Sci Polym Ed. 2002;13:953–962. doi: 10.1163/156856202320401988. [DOI] [PubMed] [Google Scholar]
- 38.Strand BL, Gaserod O, Kulseng B, Espevik T, Skjak-Baek G. J Microencapsul. 2002;19:615–630. doi: 10.1080/02652040210144243. [DOI] [PubMed] [Google Scholar]
- 39.Sugiura S, Oda T, Aoyagi Y, Matsuo R, Enomoto T, Matsumoto K, Nakamura T, Satake M, Ochiai A, Ohkohchi N, Nakajima M. Biomed Microdevices. 2007;9:91–99. doi: 10.1007/s10544-006-9011-9. [DOI] [PubMed] [Google Scholar]
- 40.Robitaille R, Pariseau JF, Leblond FA, Lamoureux M, Lepage Y, Halle JP. J Biomed Mater Res. 1999;44:116–120. doi: 10.1002/(sici)1097-4636(199901)44:1<116::aid-jbm13>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 41.Kurosawa H. J Biosci Bioeng. 2007;103:389–398. doi: 10.1263/jbb.103.389. [DOI] [PubMed] [Google Scholar]
- 42.Bratt-Leal AM, Carpenedo RL, McDevitt TC. Biotechnol Prog. 2009;25:43–51. doi: 10.1002/btpr.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rungarunlert S, Techakumphu M, Pirity MK, Dinnyes A. World J Stem Cells. 2009;1:11–21. doi: 10.4252/wjsc.v1.i1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang C, Li X, Yao Y, Wang DA. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:3762–3766. doi: 10.1016/j.jchromb.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 45.Lawrie G, Keen I, Drew B, Chandler-Temple A, Rintoul L, Fredericks P, Grondahl L. Biomacromolecules. 2007;8:2533–2541. doi: 10.1021/bm070014y. [DOI] [PubMed] [Google Scholar]
- 46.Kostopanagiotou G, Pandazi A, Matiatou S, Kontogiannopoulou S, Matsota P, Niokou D, Kitsou M, Crepi E, Christodoulaki K, Grigoropoulou I. Eur J Anaesthesiol. 2006;23:418–421. doi: 10.1017/S0265021505001912. [DOI] [PubMed] [Google Scholar]
- 47.Sava IG, Battaglia V, Rossi CA, Salvi M, Toninello A. Free Radic Biol Med. 2006;41:1272–1281. doi: 10.1016/j.freeradbiomed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 48.Ness GC, Pendleton LC, McCreery MJ. Exp Biol Med (Maywood) 2005;230:455–463. doi: 10.1177/153537020523000703. [DOI] [PubMed] [Google Scholar]
- 49.Nagao T, Suzuki K, Utsunomiya K, Matsumura M, Saiga K, Wang PC, Minamitani H, Aratani Y, Nakayama T. Nephrol Dial Transplant. 2011;26:2752–2760. doi: 10.1093/ndt/gfr032. [DOI] [PubMed] [Google Scholar]
- 50.Jackson AM, Connolly JE, Matsumoto S, Noguchi H, Onaca N, Levy MF, Naziruddin B. Transplantation. 2009;87:500–506. doi: 10.1097/TP.0b013e318195fc33. [DOI] [PubMed] [Google Scholar]
- 51.Xi J, Zhang GP, Qiao SL, Guo JQ, Wang XN, Yang YY, Zhang LN, Miao XW, Zhao D, Zhi YB, Cai SJ, Luo J, Deng RG. Immunology. 2012;136:46–53. doi: 10.1111/j.1365-2567.2012.03553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang PC, Bai C, Ho PKH, Dai Y, Wu YS. Ieee Eng Med Biol. 1997;16:42–46. doi: 10.1109/51.582175. [DOI] [PubMed] [Google Scholar]
- 53.Desai TA, Hansford D, Ferrari M. J Membrane Sci. 1999;159:221–231. [Google Scholar]
- 54.Chen Y, Cai J, Xu Q, Chen ZW. Mol Immunol. 2004;41:1247–1252. doi: 10.1016/j.molimm.2004.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.