

β-Catenin and p120-catenin bind to cadherin cytoplasmic tails and are believed to modulate cadherin function and adhesion. This study shows that p120-catenin and β-catenin function in a distinct but complementary manner to regulate the size and strength of cadherin adhesive contacts.
Abstract
Vascular endothelial (VE)-cadherin, the major adherens junction adhesion molecule in endothelial cells, interacts with p120-catenin and β-catenin through its cytoplasmic tail. However, the specific functional contributions of the catenins to the establishment of strong adhesion are not fully understood. Here we use bioengineering approaches to identify the roles of cadherin–catenin interactions in promoting strong cellular adhesion and the ability of the cells to spread on an adhesive surface. Our results demonstrate that the domain of VE-cadherin that binds to β-catenin is required for the establishment of strong steady-state adhesion strength. Surprisingly, p120 binding to the cadherin tail had no effect on the strength of adhesion when the available adhesive area was limited. Instead, the binding of VE-cadherin to p120 regulates adhesive contact area in a Rac1-dependent manner. These findings reveal that p120 and β-catenin have distinct but complementary roles in strengthening cadherin-mediated adhesion.
INTRODUCTION
Cell adhesion enables tissues to maintain their structural integrity and withstand mechanical stress (Niessen et al., 2011). Cadherins are a family of transmembrane adhesion receptors that enable cells to form cell contacts that mature into adherens junctions as cytoplasmic binding partners are recruited to facilitate cytoskeletal linkages (Pokutta and Weis, 2007). The vascular endothelium, which forms a thin layer lining the interior of blood vessels, must sustain strong intercellular adhesion in order to maintain vascular barrier function and prevent hemorrhage and edema. However, the adhesion between endothelial cells must be dynamically regulated to enable angiogenesis during growth and development and to allow the passage of leukocytes from the vascular lumen to surrounding tissue at sites of inflammation. The major cadherin family member found in endothelial cells is vascular endothelial (VE)-cadherin, which mediates homophilic, calcium-dependent adhesion through its extracellular domain and binds to Armadillo-family proteins p120-catenin (p120) and β-catenin inside the cytoplasm through its juxtamembrane and catenin-binding domains, respectively. β-Catenin provides linkage between adherens junctions and the actin cytoskeleton through interactions involving α-catenin (Nelson, 2008), whereas p120 regulates cadherin stability at the plasma membrane by masking an endocytic signal on the cadherin cytoplasmic tail to prevent cadherin internalization (Davis et al., 2003; Xiao et al., 2003, 2005; Chiasson et al., 2009; Nanes et al., 2012). Studies using mouse models have demonstrated a requirement for both p120 and β-catenin to maintain vascular barrier function, and the conditional endothelial knockout of either catenin results in hemorrhages, particularly in areas subject to increased vascular flow during development (Cattelino et al., 2003; Oas et al., 2010).
In previous studies involving C-cadherin and E-cadherin, the cadherin juxtamembrane domain and its specific interaction with p120 were implicated in the strengthening of cell adhesion (Yap et al., 1998; Goodwin et al., 2003). However, the mechanisms by which strong cadherin-based adhesion is achieved in endothelial cells, and the contribution of catenins to this process, are not fully understood. The loss of endothelial p120 in vivo results in a reduction in VE-cadherin levels (Oas et al., 2010), consistent with a role for p120 in regulating cadherin turnover. Studies using cultured endothelial cells demonstrated that the interaction between p120 and VE-cadherin at the plasma membrane is required for the maintenance of endothelial barrier function (Iyer et al., 2004; Herron et al., 2011). However, the knockout of VE-cadherin is recessive embryonic lethal (Carmeliet et al., 1999; Gory-Faure et al., 1999), and the reduction of VE-cadherin levels by 50% in the heterozygotes did not lead to hemorrhaging or other vascular defects. This raises the question of whether p120 could be acting to strengthen VE-cadherin–dependent adhesion independently of cadherin levels alone. In addition, p120 is a potent regulator of the Rho family of small GTPases, which regulate actin cytoskeletal dynamics and play important roles in the establishment of cell–cell contacts and vascular barrier function (Anastasiadis, 2007; Beckers et al., 2010). Specifically, p120 activates Rac1 and inhibits RhoA (Anastasiadis et al., 2000; Noren et al., 2000). Moreover, the adhesion defects introduced by blocking p120 binding to the E-cadherin tail can be rescued by expression of constitutively active Rac1 (Goodwin et al., 2003). Therefore it is likely that the contribution of p120 to strong adhesion through VE-cadherin involves not only the stabilization of the cadherin at the cell surface, but also the localization of p120 near the membrane to locally regulate Rho family GTPases.
Adhesive strength is modulated by a number of factors, including contact area and cytoskeletal linkages (Gallant et al., 2005). Previous studies examining cadherin adhesion strengthening used model systems in which cells both adhere and spread onto surfaces (Yap et al., 1997, 1998; Ehrlich et al., 2002; Martinez-Rico et al., 2010). Thus the contributions of cadherin tail domains and catenins to adhesion strengthening independent of contact area and cytoskeletal coupling have not been resolved. We used a combination of two approaches to overcome these limitations. First, to examine the role of p120 and β-catenin in endothelial cell adhesion, we expressed chimeric proteins in which the interleukin-2 receptor (IL-2R) extracellular domain was fused to the cytoplasmic domain of VE-cadherin. We then introduced mutations that selectively uncoupled the cadherin tail to either p120 or β-catenin. Second, we used micropatterned coverslips that limited cell–substrate contact area, thereby controlling cell geometry independent of cytoplasmic linkages. Using a hydrodynamic assay to measure the strength of cell adhesion, we found that the interaction between p120 and the cadherin tail did not alter adhesion strength. In contrast, the β-catenin–binding domain was crucial to strengthening adhesion. Furthermore, we found that the interaction between p120 and VE-cadherin was necessary to promote Rac1-dependent cell spreading. These findings support a model in which p120 and β-catenin modulate cadherin-based adhesion through complementary and experimentally distinguishable mechanisms to independently regulate adhesive contact area and adhesion strength.
RESULTS
Chimeric adhesion receptors enable functional separation of cadherin intracellular domains
To examine the functional significance of the cytoplasmic domains and interactions of the VE-cadherin cytoplasmic tail in endothelial adhesion strengthening, we used adenoviral vectors to express a series of chimeric receptor proteins in primary human microvascular endothelial cells (MECs; Figure 1A). The cytoplasmic tail of VE-cadherin was fused with the extracellular and transmembrane domains of IL-2R to generate the IL-2R–VE-cadcyto construct. Similar chimeric receptors have been used in studies of cell adhesion mediated by cadherins as well as integrins (O'Toole et al., 1994; Yap et al., 1997; Bodeau et al., 2001; Cheung et al., 2010). To examine the contribution of specific domains of the VE-cadherin tail in cell adhesion, we generated two additional variants by mutating the VE-cadherin cytoplasmic domain: a triple-alanine mutation at amino acids 562–564 in the juxtamembrane domain, which blocks binding to p120 (IL-2R–VE-cadEMD→AAA) and a deletion of the catenin-binding domain (amino acids 620–702), which eliminates β-catenin interactions (IL-2R–VE-cadΔCBD; Xiao et al., 2003). IL-2R alone without a cytoplasmic tail was used as a control. Expression of the receptors in MECs was verified by immunofluorescence (Figure 1B) and Western blot (Figure 1C), which confirmed that the constructs were expressed at comparable levels. Flow cytometry was performed to further verify that cell surface expression levels of each IL-2R construct were comparable among groups (Figure 1D).
FIGURE 1:
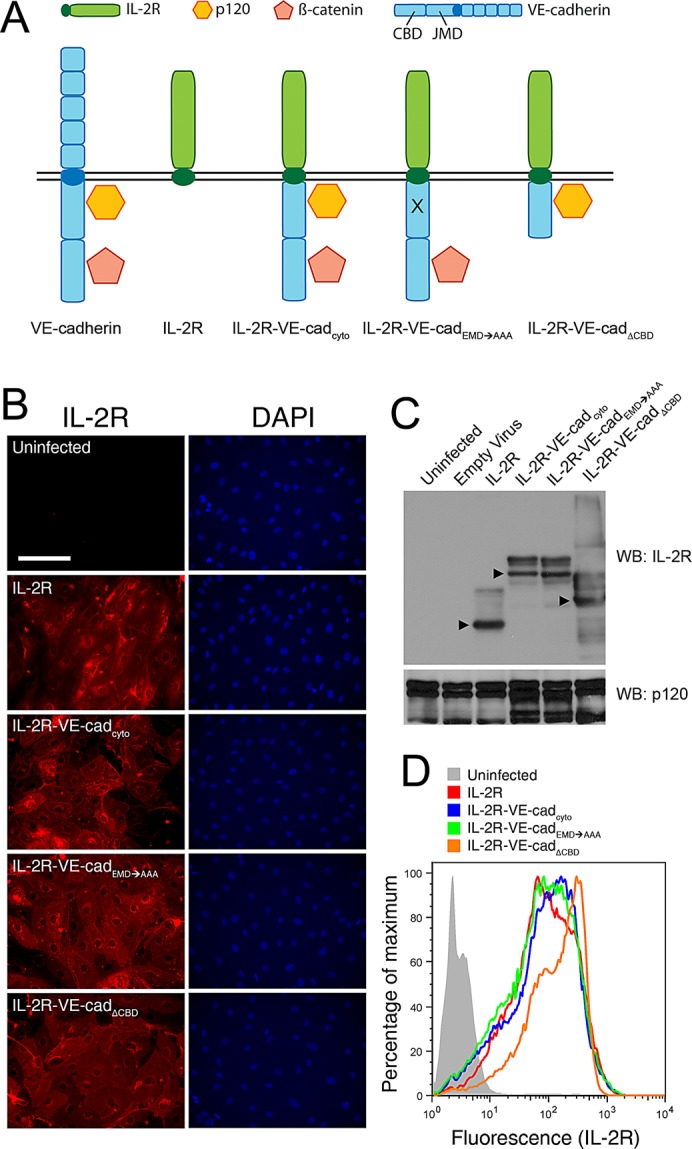
Chimeric receptors are expressed at comparable levels at the plasma membrane. (A) The four chimeric constructs used in this work as adhesive receptors are depicted, along with wild-type VE-cadherin. The extracellular domain of VE-cadherin is replaced with IL-2R to generate IL-2R–VE-cadcyto. A triple-alanine mutation in the p120-binding site that uncouples the cadherin tail from p120 is introduced to generate IL-2R–VE-cadEMD→AAA. The catenin-binding domain is deleted to generate IL-2R–VE-cadΔCBD. The four constructs are introduced into cells by way of adenoviral vectors. (B–D) Human microvascular endothelial cells were transduced with chimeric constructs containing the IL-2R extracellular domain fused to the cytoplasmic domain of VE-cadherin. (B) Expression of the constructs was verified by immunofluorescence microscopy. Cells were stained for IL-2R to detect the receptors and DAPI to show the cell nuclei and enable evaluation of infection rates. IL-2R staining was absent in uninfected cells, and for all four constructs we observed infection rates of 80% or higher. Scale bar, 100 μm. (C) Expression levels were also assessed by Western blot in which whole-cell lysates were probed for IL-2R and with p120 as a loading control. Arrowheads indicate main bands. Higher–molecular weight bands result from IL-2R glycosylation. (D) The levels of expression of the chimeric receptors at the cell surface were assessed using flow cytometry. Unpermeabilized cells were fixed and stained for IL-2R and tested for their peak fluorescence values as compared with those of uninfected control cells. Peak values for each construct occurred within a similar range, indicating that their surface expression was comparable.
To verify that the chimeric receptors were able to interact with p120 and β-catenin despite the absence of the cadherin extracellular domain, we performed immunoprecipitations using antibodies against IL-2R. The catenins did not associate with IL-2R, whereas p120 formed complexes with IL-2R–VE-cadcyto and IL-2R–VE-cadΔCBD. In contrast, β-catenin formed complexes with IL-2R–VE-cadcyto and IL-2R–VE-cadEMD→AAA. These results demonstrate that the chimeric cadherins associate in the predicted manner with p120 and β-catenin (Figure 2A).
FIGURE 2:

Cytoplasmic domains of chimeric receptors recruit p120 and β-catenin to the site of adhesion. (A) Immunoprecipitations were performed to verify that the chimeric receptors were able to complex with the appropriate catenins. Magnetic beads conjugated to antibodies against IL-2R were used to pull down the chimeric receptors, which were subsequently probed for p120 and β-catenin. p120 coprecipitated with IL-2R–VE-cadcyto and IL- 2R–VE-cadΔCBD, and β-catenin was pulled down by IL-2R–VE-cadcyto and IL-2R–VE-cadEMD→AAA. (B, C) To ensure that catenins were recruited to the sites of adhesion mediated by the chimeric constructs, cells expressing the constructs were seeded on micropatterned coverslips (see Figure 3) using IL-2R antibodies as an adhesive ligand. The cells were then extracted using Triton X-100 in a cytoskeleton stabilization buffer and stained for p120, β-catenin, and IL-2R. p120 colocalized with IL-2R–VE-cadcyto and IL-2R–VE-cadΔCBD (B), whereas β-catenin colocalized with IL-2R–VE-cadcyto and IL-2R–VE-cadEMD→AAA (C). Bars, 20 μm. (D, E) Colocalization of IL-2R chimeras with p120 (D) and β-catenin (E) was quantified as Pearson's r. Thick line, median (n = 5–6 cells per group); box, interquartile range; whiskers, full range.
We next sought to determine whether the chimeric cadherins were able to recruit β-catenin and p120 to sites of adhesion mediated by the IL-2R extracellular domain. Cells expressing the chimeric constructs or IL-2R were seeded on micropatterned coverslips, which presented an array of 20-μm adhesive islands prepared as previously described (Dumbauld et al., 2010). The adhesive areas were coated with antibodies directed against the IL-2 receptor, and cells expressing the chimeric constructs were seeded onto the micropatterned surfaces. Because the area of the islands was smaller than the fully spread area of the cells, the cells were maintained in a uniform geometry and spaced regularly across the surface of the coverslip (Figure 3B). Uninfected cells and cells transduced with an empty adenoviral vector failed to adhere to either patterned (unpublished results) or unpatterned surfaces (see discussion of Figure 5A later in the paper). Using immunofluorescence microscopy, we verified that cells expressing IL-2R–VE-cadcyto and IL-2R–VE-cadΔCBD exhibited colocalization between IL-2R and p120 (Figure 2, B and D), whereas samples expressing IL-2R–VE-cadcyto and IL-2R–VE-cadEMD→AAA displayed colocalization between IL-2R and β-catenin (Figure 2, C and E). Cells expressing IL-2R alone did not yield demonstrable colocalization with either catenin. Although both catenins can also be observed in other regions, presumably representing interactions with endogenous cadherin or other binding partners, these results show that the chimeric receptors expressing cytoplasmic VE-cadherin domains are able to selectively recruit catenins and specifically mediate adhesion to patterned surfaces.
FIGURE 3:

Micropatterned coverslips feature adhesive islands that allow for the generation of regular arrays of evenly spaced cells with well-defined morphologies. Micropatterning occurs through a stepwise process. (A) Glass coverslips are coated with titanium and gold using an electron beam evaporator, and then a PDMS stamp is used to print islands of the self-assembling monolayer across the surface of the coverslip (A, 1). This generates adhesive islands that can passively adsorb ligands that will interact specifically with adhesive receptors on the surface of cells (A, 2). Spaces between the adhesive islands are backfilled with polyethylene glycol to create nonadhesive surfaces around sites of cell adhesion (A, 3). Cells are seeded on the micropatterned coverslips and adhere individually to adhesive islands (A, 4). (B) Cells expressing IL-2R and chimeric constructs adhere to adhesive islands. Micropatterned coverslips are treated with immunoglobulin G directed against the IL-2 receptor, which can be detected with fluorescently labeled secondary antibodies. Cells expressing IL-2R seeded on the micropatterned surfaces form a regular array of single cells attached to adhesive islands.
FIGURE 5:

The interaction between p120 and the cadherin juxtamembrane domain is required to promote cell spreading. (A, B) Cells expressing the chimeric constructs were seeded sparsely on unpatterned coverslips coated with antibodies against IL-2R. The cells were allowed to adhere for 30 min before fixation. (A) Representative images of adherent cells expressing the IL-2R constructs. Uninfected cells and those infected with an empty adenoviral vector were not able to attach to the IL-2R–antibody-coated coverslips. Bar, 20 μm. (B) The spread areas of 100 cells per condition, chosen at random, were measured and plotted by quantifying the number of cells per condition whose spread areas were larger than a given area in microns. On these inverse cumulative distribution plots, each data point indicates the percentage of cells (y-axis) that have spread areas greater than a given value (x-axis). Thus a population of cells exhibiting comparatively larger spread areas will generate data points that fall further to the right on the graph than the other populations being compared. The median values of the different groups were found to be statistically different (Kruskal–Wallis test; p < 0.001). IL-2R and IL-2R–VE-cadEMD→AAA were not statistically different from each other, and IL-2R–VE-cadcyto and IL-2R–VE-cadΔCBD were not statistically different from each other. However, both members of the former pair were statistically different from both members of the latter pair (Tukey test; p < 0.05). (C, D) IL-2R, IL-2R–VE-cadcyto, and IL-2R–VE-cadEMD→AAA were expressed in mouse endothelial cells that were either control or p120 null, and their spreading ability was measured as in the previous experiment. (C) In control cells, IL-2R–VE-cadcyto exhibited significantly increased spreading over IL-2R and IL-2R–VE-cadEMD→AAA (Kruskal–Wallis test; p < 0.001; Tukey test; p < 0.05). (D) In p120-null cells, the increased spreading observed in IL-2R–VE-cadcyto was lost (Kruskal–Wallis test; p = 0.230). (D, inset) Surface expression levels of the various chimeras were similar, as measured by immunofluorescence. Thick line, median (n = 28–38 cells per group); box, interquartile range; whiskers, 90% range.
The catenin-binding domain of VE-cadherin is necessary for strong adhesion
To define the contributions of the VE-cadherin cytoplasmic domains to cell adhesion strengthening, we tested cells expressing the chimeric receptors for their ability to remain attached to a surface when subjected to a range of shear forces. Measurements of adhesion strength were made using a spinning-disk apparatus that applies hydrodynamic shear force to a large population of cells attached to patterned coverslips (Figure 3B) by rotating them in a fluid-filled chamber, generating a well-characterized range of shear forces that increase with radial position along the surface of the coverslip (Figure 4A). Cells expressing the IL-2R constructs were seeded on patterned coverslips and exposed to shear forces using the spinning-disk system. This approach, which has been extensively discussed in previous reports (García et al., 1998a; García and Gallant, 2003; Gallant et al., 2005), yields detachment profiles such as the representative data shown in Figure 4B. The chart shows the fraction of adherent cells in regions of the coverslip subject to increasing shear force relative to the fraction of adherent cells near the center, where there is no shear force. A sigmoidal curve is then fitted to the resulting data points to obtain the shear stress needed for 50% detachment (τ50), which is used as a measure of cell adhesion strength. Cells expressing IL-2R–VE-cadcyto exhibited 50% higher adhesion strength values (τ50) than those cells expressing the IL-2R alone, demonstrating the importance of the cadherin cytoplasmic tail in mediating strong attachment (Figure 4, B and C). Unexpectedly, the cells expressing the IL-2R–VE-cadEMD→AAA construct produced adhesion strength values statistically indistinguishable from those obtained with the wild-type cadherin tail. In contrast, cells expressing the IL-2R–VE-cadΔCBD construct yielded adhesion comparable to the IL-2R. Taken together, these results indicate that the p120 binding site in VE-cadherin does not contribute to adhesion strength but that the β-catenin-binding domain is required.
FIGURE 4:

Linkage between cadherins and the actin cytoskeleton is necessary to strengthen steady-state adhesion. (A) A hydrodynamic spinning-disk device was used to measure the adhesive strength of populations of cells. Cells were seeded onto micropatterned coverslips and allowed to adhere for 16 h. A coverslip with adherent cells was mounted onto the spinning-disk apparatus, and a vacuum pump was used to hold the sample in place. The sample was then submerged into the spin chamber filled with PBS+ with 2 mM dextrose. The chamber was equipped with baffles at the edges, which prevented the spinning motion of the sample from creating a vortex. The sample was then spun for 5 min at a controlled speed (ω), resulting in a gradient of shear force (τ) proportional to the distance from the coverslip center (r). Samples were then fixed, permeabilized, and stained for microscopy and quantification of adherent cells remaining on the coverslip. (B) The cells remaining attached to the coverslip were counted at various positions across the coverslip, and the values were plotted and sigmoid curves were fitted to the combined count totals. (C) Comparisons of adhesion strength values (τ50) among the chimeric constructs. IL-2R was significantly less adhesive than IL-2R–VE-cadcyto and IL-2R–VE-cadEMD→AAA (Tukey test; p < 0.050) but not significantly different from IL-2R–VE-cadΔCBD.
To verify that p120 levels in the cells were not limiting, we coexpressed exogenous p120 with IL-2R–VE-cadcyto and IL-2R–VE-cadEMD→AAA. The overexpression of p120 did not increase the adhesion strength of cells expressing IL-2R–VE-cadcyto compared with those expressing IL-2R–VE-cadEMD→AAA (Supplemental Figure S1), indicating that p120 was not limiting. This result suggests a requirement for linkage to the actin cytoskeleton through the catenin-binding domain and its association to β-catenin in order to produce strong steady-state adhesion but that p120 appears to be dispensable. Taken together, these results indicate that the loss of p120 binding to the VE-cadherin juxtamembrane domain does not significantly reduce cell adhesion strength, whereas the β-catenin–binding domain is essential for strong adhesion.
p120 binding to the VE-cadherin tail is necessary to promote cell spreading
Because we were unable to demonstrate a requirement for the interaction between p120 and VE-cadherin in establishing strong adhesion to patterned surfaces, we next tested the ability of cells expressing the chimeric constructs to spread on unpatterned substrates that did not constrain the adhesive area. Primary microvascular endothelial cells expressing IL-2R and the three chimeric receptors were able to adhere to the surface, whereas control uninfected cells or cells expressing empty adenoviral vector were unable to form attachments and were easily removed from the surface. Cells expressing the IL-2R–VE-cadcyto spread substantially within 30 min, whereas those expressing IL-2R without the cadherin tail remained rounded (Figure 5, A and B). The typical diameter of fully spread IL-2R–VE-cadΔCBD- and IL-2R–VE-cadcyto–expressing cells was approximately 30 μm, whereas the typical diameter of fully spread IL-2R– and IL-2R–VE-cadEMD→AAA–expressing cells was approximately 20 μm. Of interest, whereas IL-2R–VE-cadΔCBD–expressing cells did not exhibit a significant spreading defect compared with those expressing IL-2R–VE-cadcyto, the p120-uncoupled chimera was statistically indistinguishable from IL-2R, suggesting a key role for p120 in modulating cell spreading (Figure 5B).
To verify that these differences in cell spreading were specifically the result of disrupting the VE-cadherin–p120 interaction, we next expressed IL-2R, IL-2R–VE-cadcyto, and IL-2R–VE-cadEMD→AAA in a p120-null endothelial cell line and in a control cell line that retained p120 expression (Oas et al., 2010; Figure 5, C and D). In the p120-expressing cells, we again observed a significant increase in the spread surface area of cells expressing IL-2R–VE-cadcyto compared with those expressing IL-2R or IL-2R–VE-cadEMD→AAA (Figure 5C). However, in a p120-null background, the increased spreading seen previously in the IL-2R–VE-cadcyto–expressing cells was eliminated, and there were no differences among the three groups (Figure 5D). These results indicate that cell spreading requires both the expression of p120 and its binding to the VE-cadherin juxtamembrane domain.
Rac1 activity regulates cell spreading but not adhesion strength
Binding of p120 to the juxtamembrane domain of cadherins is believed to locally activate the small GTPase Rac1 at the plasma membrane, which in turn induces membrane ruffling and allows for the extension of lamellipodia through the localized regulation of actin dynamics (Ridley et al., 1992, 2001; Tan et al., 2008). A prediction based on these previous findings and our results presented thus far is that p120-dependent cell spreading would require Rac1 activity but the acquisition of strong adhesion, which is mediated by β-catenin, would be Rac1 independent. To test this model, we determined adhesive strength mediated by the IL-2R–VE-cadcyto chimera in the presence or absence of the Rac1 inhibitor NSC23766, using the hydrodynamic spinning-disk assay. Inhibition of Rac1 had no discernible effect on adhesion strength mediated by the IL-2R–VE-cadcyto chimera (Figure 6A). In contrast, inhibition of Rac1 dramatically reduced spreading mediated by the IL-2R–VE-cadcyto chimeras (Figure 6C). Furthermore, when cells expressing IL-2R constructs were cotransduced with constitutively active Rac1, the spread areas of all four groups were dramatically increased, regardless of the ability of p120 to bind the cadherin tail (Figure 6D). Of interest, constitutively active Rac1 expression caused cells expressing the β-catenin–uncoupled constructs to spread more than those expressing β-catenin–coupled chimeras, suggesting that actin associations limit spreading in this context. These findings indicate that p120-mediated cell spreading occurs through a Rac1-dependent pathway but that the acquisition of adhesive strength mediated by the β-catenin–binding domain of the cadherin tail occurs through a Rac1-independent process.
FIGURE 6:

Rac1 inhibition phenocopies the p120-dependent spreading defect. (A) The adhesion strength of primary microvascular endothelial cells expressing IL-2R and IL-2R–VE-cadcyto with and without Rac1 inhibitor NSC23766 treatment was assayed using the hydrodynamic spinning-disk device. The addition of NSC23766 did not significantly affect adhesion strength mediated by the VE-cadherin cytoplasmic tail (Tukey test; p < 0.050). (B–D) Cell spreading was measured in untreated endothelial cells (B), NSC23766-treated endothelial cells (C), and endothelial cells expressing a constitutively active Rac1 mutant (D). Rac1 inhibition prevented cell spreading and eliminated differences between chimeras, whereas constitutively active Rac1 increased spreading.
DISCUSSION
The results presented here indicate that the p120- and β-catenin–binding domains of the cadherin tail function differentially to regulate adhesion. Whereas p120 modulates the ability of cells to spread and increases the area of the adhesive contact, β-catenin binding to the cadherin tail modulates the strength of cadherin-mediated adhesion independent of contact area. Thus p120 and β-catenin both contribute to the overall adhesive potential of cadherin-based cell–cell contact but in mechanistically distinct manners.
Several studies have examined the contributions of cadherin tail domains and signaling pathways to cadherin-mediated adhesion strength (Nanes and Kowalczyk, 2012). Yap et al. (1998) demonstrated that the cadherin juxtamembrane domain and p120 are important in strengthening cell adhesion. Using a laminar flow assay, they allowed cells expressing C-cadherin constructs to adhere to a tube coated with the C-cadherin extracellular domain and subjected to fluid shear force. Cells expressing wild-type C-cadherin showed an increase in adhesion strength. However, this adhesion-strengthening effect was lost in those cells in which the p120 binding site was deleted or mutated (Goodwin et al., 2003). An important distinction between these previous studies and our present analysis is that we controlled the geometry of cells exposed to shear forces using micropatterned surfaces, thus allowing us to discriminate between cell spreading and adhesion strength. Indeed, Yap et al. (1998) reported reduced spreading in cells expressing C-cadherin constructs lacking the cadherin juxtamembrane domain. Similarly, we observed defects in cell spreading when binding of p120 to the cadherin tail was abrogated either by mutation of the juxtamembrane domain or the loss of endogenous p120 (Figure 4, B and C). As shown previously, contact area is a key factor in controlling adhesive strength (Gallant et al., 2005). Furthermore, it is likely that in a shear flow–based adhesion assay, cells that flatten and spread reduce their relative exposure to shear stress. Cells expressing cadherins that are uncoupled from p120 do not flatten and are thus subjected to higher shear forces. Consistent with this interpretation, when we controlled cell shape using micropatterned surfaces to constrain cell spreading and thus control cell geometry, we found that p120 had no significant role in modulating the strengthening of cadherin-based adhesion. We conclude that p120 does not directly regulate cadherin strengthening but instead regulates the area of the cadherin contact zone.
As mentioned previously, p120 binding stabilizes cadherins at the cell surface. Because surface expression levels of the different chimeras were similar (Figure 1D), disrupting p120 binding did not affect adhesion strength by reducing the amount of cadherin available to form adhesive contacts. However, the localization of p120 near the plasma membrane also influences actin dynamics through the Rho-family GTPases. p120 has been well characterized as a potent regulator of Rho-family GTPases RhoA, Rac1, and Cdc42 (Anastasiadis, 2007). In particular, the initiation of adhesion by cadherins was found to stimulate Rac1 activity (Noren et al., 2001; Kovacs et al., 2002), and this activation is dependent on binding of p120 to the cadherin tail (Goodwin et al., 2003). Rac1 activity at the plasma membrane causes actin reorganization and membrane ruffling (Ridley et al., 1992) and is known to be important in the formation of lamellipodia (Ridley, 2001; Tan et al., 2008), particularly at newly formed adhesive contacts (Ehrlich et al., 2002). The adhesive defects reported by Goodwin et al. (2003) when p120 binding to the E-cadherin tail was blocked were rescued when constitutively active Rac1 was expressed. Consistent with these results, we found that inhibition of Rac1 impaired cell spreading even in cells in which p120 was able to bind to the cadherin tail (Figure 6C). Likewise, constitutively active Rac1 rescued the spreading defect in cells expressing p120-uncoupled cadherin (Figure 6D). However, in the hydrodynamic spinning-disk assay, in which spreading area is limited, inhibition of Rac1 did not decrease adhesion strength (Figure 6A). Collectively, these findings indicate that the cadherin–p120 complex regulates the size of the adhesive contact area in a Rac1-dependent manner, although we cannot rule out the possibility that, in some circumstances, Rac1 might influence cadherin-based adhesion independently of cell spreading.
In contrast to the role of p120 in modulating adhesive contact area, β-catenin binding is dispensable for cell spreading but required for cadherin-based adhesive strength (Figure 5). β-Catenin associates with α-catenin and is believed to participate in coupling the cadherin–catenin complex to the actin cytoskeleton, although the precise mechanism by which cadherins associate with actin is not fully understood (Drees et al., 2005; Yamada et al., 2005). On the basis of our findings, we propose a model (Figure 7) in which p120 binding to the cadherin tail drives expansion of the cadherin contact in a Rac1-dependent manner. β-Catenin serves to strengthen the adhesive contact by recruiting actin-binding proteins that couple the cadherin–catenin complex to the actin cytoskeleton, thus stiffening the adhesive contact zone. Localized cytoskeletal stiffening enhances adhesion strength by increasing the shear force needed to peel off the leading edge of an adherent cell (Gallant et al., 2005; Gallant and Garcia, 2007). Thus p120 and β-catenin both contribute to the overall adhesive potential of cadherins at the cell surface, but they do so through distinct and complementary mechanisms. These findings have important implications for understanding the mechanistic basis for loss-of-function mutations in the cadherin tail domain and catenins in various model systems. Further in vivo analysis of p120-uncoupled cadherins, in parallel with the use of Rac1-uncoupled p120 gene replacement studies, will be critical for understanding how the cadherin–pl20 complex regulates cell–cell contact size and adhesion strength during development and other complex biological processes.
FIGURE 7:

Interaction between p120 and cadherins strengthens cell adhesion by promoting cell spreading. A proposed model demonstrating the distinct contributions of p120 and β-catenin to strengthening adhesion.
MATERIALS AND METHODS
Cell culture
Primary cultures of dermal microvascular endothelial cells (MECs) from human neonatal foreskin were isolated and cultured in Microvascular Endothelial Cell Growth Media-2 (EGM-2MV; Lonza, Basel, Switzerland) supplemented with cAMP (Sigma-Aldrich, St. Louis, MO). Heart endothelial cells were isolated from mice harboring a floxed allele of p120 as previously described (Oas et al., 2010) and immortalized by transduction with SV40 DNA according to a previously published method (Ades et al., 1992). Clonal cell populations were expanded, and a cell line was selected on the basis of morphology and the expression of endothelial markers VE-cadherin and PECAM-1. To induce p120 knockout, the cells were infected with an adenovirus expressing Cre (gift from L. Yang, Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA) so that parallel wild-type and p120-null lines were generated. These cells were cultured in high-glucose DMEM (Mediatech, Herndon, VA) with 20% fetal bovine serum (Sigma-Aldrich), antibiotic/antimycotic solution (Mediatech), 100 μg/ml heparin (Sigma-Aldrich), 100 μg/ml endothelial cell growth supplement (ECGS; Biomedical Technologies, Stoughton, MA), 1 mM nonessential amino acids (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 2 mM l-glutamine (Mediatech), and 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Mediatech). All endothelial cells were grown on 0.1% gelatin-coated plates to ∼80% confluency for experiments. Rac1 inhibitor NCS23766 was obtained from Tocris Bioscience (Bristol, United Kingdom) and used at a concentration of 200 μM at 30 min before assays were performed.
Adenoviruses
The IL-2R–VE-cadcyto, IL-2R–VE-cadΔCBD, and IL-2R–VE-cadJMD-AAA (here designated as IL-2R–VE-cadEMD→AAA) constructs were generated as described previously (Xiao et al., 2003). These were subcloned into the pAd-Track vector that coexpresses green fluorescent protein (GFP), and they and the IL-2R and wild-type p120 constructs were added to cells 16–20 h before seeding for experiments, and infection rates of ∼80% were used as monitored by GFP expression. Constitutively active RhoA adenovirus (Kalman et al., 1999) was provided by D. Kalman (Emory University, Atlanta, GA).
Micropatterned surfaces
Micropatterned coverslips with adhesive islands surrounded by a nonadhesive background were prepared as previously described (Gallant et al., 2005). Briefly, to generate a regular array of adhesive islands 20 μm in diameter and 75 μm from center to center, a polydimethylsiloxane (PDMS) stamp was prepared from a template (Dumbauld et al., 2010) and used for microcontact printing of self-assembled monolayers of alkanethiols on gold-coated coverslips. Glass coverslips 25 mm in diameter were cleaned and then coated with titanium (100 Å) and then gold (2000 Å) using an electron beam evaporator (Thermionics, Hayward, CA). Before microcontact printing, the stamp was sonicated in 70% ethanol for 15 min and allowed to dry. Using a cotton swab, the patterned surface of the PDMS stamp was coated with 1.0 mM hexadecanethiol in ethanol, dried using a nitrogen stream, and laid on the gold-coated coverslip for 30 s under 50–100 g of weight to ensure uniform contact. This process generates a surface with regularly spaced adhesive islands that adsorb ligands such as extracellular matrix proteins or cell membrane–associated adhesive receptors. To prevent adhesion to the areas between islands, patterned coverslips were incubated for 2 h in tri(ethylene glycol)-terminated alkanethiol to create a nonadhesive and nonfouling background. The coverslips were then washed three times with absolute ethanol, once with sterile double-distilled H2O, and once with phosphate-buffered saline plus calcium and magnesium (PBS+/+) before coating with IgG2a directed against the IL-2 receptor (purified from American Type Culture Collection [Manassas, VA] hybridoma HB8784) at a concentration of 20 μg/ml for 1 h. After ligand adsorption, the patterned coverslips were blocked in heat-inactivated bovine serum albumin (1% wt/vol) for 30 min and incubated in PBS until seeding. Cells expressing IL-2R–containing constructs were removed from culture plates using trypsin/EDTA and seeded onto the micropatterned coverslips at a density of 225 cells/mm2. The coverslips were returned to the 37°C incubator for 16 h.
Hydrodynamic spinning-disk assay
Cell adhesion strength was measured as previously described (García et al., 1998b; Gallant et al., 2005). With the use of a spinning disk, a micropatterned coverslip with adherent cells was mounted on the spinning platform, stabilized by vacuum pressure, submerged in a solution of 2 mM dextrose in PBS+/+, and spun for 5 min (Figure 4A). The hydrodynamic forces present on the surface of the coverslip are described by the equation
 |
where τ is the applied shear stress (force/area), r is the radial position relative to the center of the coverslip, ρ is the density of the solution, μ is the viscosity of the solution, and ω is the speed of rotation. After being spun, the samples were fixed in 3.7% formaldehyde, permeabilized in 0.1% Triton X-100, and stained with ethidium homodimer-1 (E1169; Life Technologies, Carlsbad, CA). The remaining adherent cells were counted on a fluorescence microscope with a motorized stage, ImagePro image analysis software (Media Cybernetics, Silver Spring, MD), and an algorithm that analyzed 61 fields of view per sample ranging from the center of the coverslip to the outer edges. The fraction of adherent cells (f) was calculated by comparing the number of cells present at each field with the number present at the center, where the shear forces are close to zero. Detachment profiles (f vs. τ) were fitted to a sigmoidal curve,
 |
where τ50 is the value of shear stress at which 50% of the cells remain adherent. This value was used as a measure of mean adhesion strength. For comparisons between groups, analysis of variance was used, and if significant differences were detected, the Tukey test was used to perform pairwise comparisons, in which p < 0.05 was considered significant.
Cell-spreading assay
Adhesive substrates were generated using the same method as for the micropatterned samples described earlier, except that instead of stamping, the entire coverslip was coated with 1 mM hexadecanethiol in ethanol before incubation in the IL-2R IgG2a ligand. Cells expressing the constructs containing IL-2R were seeded sparsely on these surfaces and allowed to attach at 37°C for 30 min. The samples were then gently washed in PBS, fixed with paraformaldehyde, and mounted on microscope slides. Light microscopy was used to photograph fields at random, and for each condition, the spread areas of a total of 100 individual cells (not bordering any other cell) were measured. To determine whether the difference in median values between groups was statistically significant, we performed the Kruskal–Wallis test (with p < 0.001 indicating significance), followed by pairwise comparisons between groups using the Tukey test (with p < 0.05 indicating significance).
Immunofluorescence staining
Cells were fixed using methanol (Acros Organics, Geel, Belgium) or 3.7% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in phosphate-buffered saline with calcium and magnesium (PBS+/+) containing 2% bovine serum albumin (Fisher Scientific), followed by permeabilization with 0.1% Triton (Roche Diagnostics Corporation, Indianapolis, IN) in PBS+/+, and then stained using antibodies against IL-2R (MAB223 clone 22722; R&D Systems, Minneapolis, MN) and 4′,6-diamidino-2-phenylindole (DAPI). Cells attached to micropatterned coverslips were washed with PBS+/+ and washed once in cytoskeleton buffer (CSK) containing 10 mM 1,4-piperazinediethanesulfonic acid buffer, 50 mM NaCl, and 3 mM MgCl2. Protease inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, and 1 μg/ml pepstatin) were added immediately before use. The cells were then washed twice in CSK containing 0.5% (vol/vol) Triton X-100 and fixed in 4% paraformaldehyde. Cells were subsequently blocked in 5% goat serum with 0.01% NaN3 and stained using antibodies against IL-2R (R&D), p120 (610135; BD Biosciences, San Diego, CA), and β-catenin (A5441; Sigma-Aldrich).
Immunoprecipitation
Immunoprecipitations were carried out as described previously (Chiasson et al., 2009), without cross-linking. Briefly, HeLa cells were grown to confluency and infected with IL-2R–containing chimeric constructs. On the day of the experiment, cells were placed on ice, rinsed with PBS, and lysed with buffer A (150 mM NaCl, 10 mM HEPES, 1 mM ethylene glycol tetraacetic acid, and 0.1 mM MgCl2, pH 7.4) plus 0.5% TX-100, scraped from the dish, and incubated on ice for 30 min. Cell lysates were centrifuged at 16,100 × g for 10 min, and supernatants were diluted to 1 mg/ml in 0.5 ml of buffer A plus 0.5% Triton X-100. The supernatants were incubated overnight at 4°C with sheep anti-mouse Dynal magnetic beads (Invitrogen) conjugated to monoclonal antibodies against IL-2R (N-19; Santa Cruz Biotechnology, Santa Cruz, CA). The beads were then washed with buffer A plus 0.1% Triton X-100 and eluted with SDS–PAGE sample buffer at 75°C for 5 min before performing Western blots, which were probed with antibodies against p120 (SC-1101; Santa Cruz Biotechnology) and β-catenin (A5441; Sigma-Aldrich).
Western blotting
Cells were cultured in complete growth medium and infected with adenoviruses for 16–24 h before being harvested in Laemmli sample buffer (Bio-Rad Laboratories, Hercules, CA), and samples were boiled for 5 min before loading on 7.5% SDS–PAGE gel for protein separation. Proteins were transferred to nitrocellulose membrane for immunoblotting and probed with antibodies against IL-2R (N19; Santa Cruz Biotechnology), and p120 (SC-1101; Santa Cruz Biotechnology). Horseradish peroxidase–conjugated secondary antibodies (Bio-Rad Laboratories) were used at 1:3000 dilution, and blots were developed with Amersham ECL Western Blotting Detection Reagents (RPN2106; GE Healthcare, Piscataway, NJ) or Amersham ECL Plus (RPN2132; GE Healthcare).
Supplementary Material
Acknowledgments
We thank Christopher Caughman, Sean Coyer, David Dumbauld, and Krystalyn Hudson for their technical assistance and advice. This work was supported by funding from the National Institutes of Health through Grants T32EY007092 (R.O.), F30HL110447 (B.A.N.), R01HL77870 (P.A.V.), R01GM065918 (A.J.G.), and R01AR050501 (A.P.K.), the National Science Foundation through Grant BES0827719 (A.J.G.), and a National Science Foundation Graduate Research Fellowship Award (C.E.).
Abbreviations used:
- IL-2R
interleukin-2 receptor
- MEC
microvascular endothelial cell
- VE
vascular endothelial
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0471) on January 16, 2013.
REFERENCES
- Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J Invest Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- Anastasiadis PZ. p120-ctn: a nexus for contextual signaling via Rho GTPases. Biochim Biophys Acta. 2007;1773:34–46. doi: 10.1016/j.bbamcr.2006.08.040. [DOI] [PubMed] [Google Scholar]
- Anastasiadis PZ, Moon SY, Thoreson MA, Mariner DJ, Crawford HC, Zheng Y, Reynolds AB. Inhibition of RhoA by p120 catenin. Nat Cell Biol. 2000;2:637–644. doi: 10.1038/35023588. [DOI] [PubMed] [Google Scholar]
- Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- Bodeau AL, Berrier AL, Mastrangelo AM, Martinez R, LaFlamme SE. A functional comparison of mutations in integrin beta cytoplasmic domains: effects on the regulation of tyrosine phosphorylation, cell spreading, cell attachment and beta1 integrin conformation. J Cell Sci. 2001;114:2795–2807. doi: 10.1242/jcs.114.15.2795. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–157. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Cattelino A, et al. The conditional inactivation of the {beta}-catenin gene in endothelial cells causes a defective vascular pattern and increased vascular fragility. J Cell Biol. 2003;162:1111–1122. doi: 10.1083/jcb.200212157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung LW, Leung PC, Wong AS. Cadherin switching and activation of p120 catenin signaling are mediators of gonadotropin-releasing hormone to promote tumor cell migration and invasion in ovarian cancer. Oncogene. 2010;29:2427–2440. doi: 10.1038/onc.2009.523. [DOI] [PubMed] [Google Scholar]
- Chiasson CM, Wittich KB, Vincent PA, Faundez V, Kowalczyk AP. p120-catenin inhibits VE-cadherin internalization through a Rho-independent mechanism. Mol Biol Cell. 2009;20:1970–1980. doi: 10.1091/mbc.E08-07-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163:525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin–beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld DW, Shin H, Gallant ND, Michael KE, Radhakrishna H, García AJ. Contractility modulates cell adhesion strengthening through focal adhesion kinase and assembly of vinculin-containing focal adhesions. J Cell Physiol. 2010;223:746–756. doi: 10.1002/jcp.22084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich JS, Hansen MD, Nelson WJ. Spatio-temporal regulation of Rac1 localization and lamellipodia dynamics during epithelial cell-cell adhesion. Dev Cell. 2002;3:259–270. doi: 10.1016/s1534-5807(02)00216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant ND, Garcia AJ. Model of integrin-mediated cell adhesion strengthening. J Biomech. 2007;40:1301–1309. doi: 10.1016/j.jbiomech.2006.05.018. [DOI] [PubMed] [Google Scholar]
- Gallant ND, Michael KE, García AJ. Cell adhesion strengthening: contributions of adhesive area, integrin binding, and focal adhesion assembly. Mol Biol Cell. 2005;16:4329–4340. doi: 10.1091/mbc.E05-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García A, Gallant N. Stick and grip. Cell Biochem Biophys. 2003;39:61–73. doi: 10.1385/CBB:39:1:61. [DOI] [PubMed] [Google Scholar]
- García AJ, Huber F, Boettiger D. Force required to break alpha5beta1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state. J Biol Chem. 1998a;273:10988–10993. doi: 10.1074/jbc.273.18.10988. [DOI] [PubMed] [Google Scholar]
- García AJ, Takagi J, Boettiger D. Two-stage activation for alpha5beta1 integrin binding to surface-adsorbed fibronectin. J Biol Chem. 1998b;273:34710–34715. doi: 10.1074/jbc.273.52.34710. [DOI] [PubMed] [Google Scholar]
- Goodwin M, Kovacs EM, Thoreson MA, Reynolds AB, Yap AS. Minimal mutation of the cytoplasmic tail inhibits the ability of E-cadherin to activate Rac but not phosphatidylinositol 3-kinase: direct evidence of a role for cadherin-activated Rac signaling in adhesion and contact formation. J Biol Chem. 2003;278:20533–20539. doi: 10.1074/jbc.M213171200. [DOI] [PubMed] [Google Scholar]
- Gory-Faure S, Prandini MH, Pointu H, Roullot V, Pignot-Paintrand I, Vernet M, Huber P. Role of vascular endothelial-cadherin in vascular morphogenesis. Development. 1999;126:2093–2102. doi: 10.1242/dev.126.10.2093. [DOI] [PubMed] [Google Scholar]
- Herron CR, Lowery AM, Hollister PR, Reynolds AB, Vincent PA. p120 regulates endothelial permeability independently of its NH2 terminus and Rho binding. Am J Physiol Heart Circ Physiol. 2011;300:H36–H48. doi: 10.1152/ajpheart.00812.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S, Ferreri DM, DeCocco NC, Minnear FL, Vincent PA. VE-cadherin-p120 interaction is required for maintenance of endothelial barrier function. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1143–L1153. doi: 10.1152/ajplung.00305.2003. [DOI] [PubMed] [Google Scholar]
- Kalman D, Gomperts SN, Hardy S, Kitamura M, Bishop JM. Ras family GTPases control growth of astrocyte processes. Mol Biol Cell. 1999;10:1665–1683. doi: 10.1091/mbc.10.5.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs EM, Ali RG, McCormack AJ, Yap AS. E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J Biol Chem. 2002;277:6708–6718. doi: 10.1074/jbc.M109640200. [DOI] [PubMed] [Google Scholar]
- Martinez-Rico C, Pincet F, Thiery JP, Dufour S. Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J Cell Sci. 2010;123:712–722. doi: 10.1242/jcs.047878. [DOI] [PubMed] [Google Scholar]
- Nanes BA, Chiasson-Mackenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199:365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanes BA, Kowalczyk AP. Adherens junction turnover: regulating adhesion through cadherin endocytosis, degradation, and recycling. In: Harris T., editor. Adherens Junctions: From Molecular Mechanisms to Tissue Development and Disease. Dordrecht, Netherlands: Springer; 2012. pp. 197–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ. Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans. 2008;36:149–155. doi: 10.1042/BST0360149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91:691–731. doi: 10.1152/physrev.00004.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren NK, Liu BP, Burridge K, Kreft B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J Cell Biol. 2000;150:567–580. doi: 10.1083/jcb.150.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren NK, Niessen CM, Gumbiner BM, Burridge K. Cadherin engagement regulates Rho family GTPases. J Biol Chem. 2001;276:33305–33308. doi: 10.1074/jbc.C100306200. [DOI] [PubMed] [Google Scholar]
- O'Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oas RG, Xiao K, Summers S, Wittich KB, Chiasson CM, Martin WD, Grossniklaus HE, Vincent PA, Reynolds AB, Kowalczyk AP. p120-catenin is required for mouse vascular development. Circ Res. 2010;106:941–951. doi: 10.1161/CIRCRESAHA.109.207753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu Rev Cell Dev Biol. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Tan W, Palmby TR, Gavard J, Amornphimoltham P, Zheng Y, Gutkind JS. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–1838. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- Xiao K, Allison DF, Buckley KM, Kottke MD, Vincent PA, Faundez V, Kowalczyk AP. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163:535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Garner J, Buckley KM, Vincent PA, Chiasson CM, Dejana E, Faundez V, Kowalczyk AP. p120-catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol Biol Cell. 2005;16:5141–5151. doi: 10.1091/mbc.E05-05-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Pruschy M, Gumbiner BM. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol. 1997;7:308–315. doi: 10.1016/s0960-9822(06)00154-0. [DOI] [PubMed] [Google Scholar]
- Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol. 1998;141:779–789. doi: 10.1083/jcb.141.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.