

Cell adhesion to collagen presented on beads activates Ca2+ entry and promotes the formation of phagosomes enriched with NMMIIA and gelsolin. The Ca2+-dependent interaction of gelsolin and NMMIIA in turn enables actin remodeling and enhances collagen degradation by phagocytosis.
Abstract
We examine how collagen substrate topography, free intracellular calcium ion concentration ([Ca2+]i, and the association of gelsolin with nonmuscle myosin IIA (NMMIIA) at collagen adhesions are regulated to enable collagen phagocytosis. Fibroblasts plated on planar, collagen-coated substrates show minimal increase of [Ca2+]i, minimal colocalization of gelsolin and NMMIIA in focal adhesions, and minimal intracellular collagen degradation. In fibroblasts plated on collagen-coated latex beads there are large increases of [Ca2+]i, time- and Ca2+-dependent enrichment of NMMIIA and gelsolin at collagen adhesions, and abundant intracellular collagen degradation. NMMIIA knockdown retards gelsolin recruitment to adhesions and blocks collagen phagocytosis. Gelsolin exhibits tight, Ca2+-dependent binding to full-length NMMIIA. Gelsolin domains G4–G6 selectively require Ca2+ to interact with NMMIIA, which is restricted to residues 1339–1899 of NMMIIA. We conclude that cell adhesion to collagen presented on beads activates Ca2+ entry and promotes the formation of phagosomes enriched with NMMIIA and gelsolin. The Ca2+ -dependent interaction of gelsolin and NMMIIA in turn enables actin remodeling and enhances collagen degradation by phagocytosis.
INTRODUCTION
Deregulation of collagen degradation leads to imbalances of matrix homeostasis (Perez-Tamayo, 1978). These imbalances can manifest as destruction of normal matrix structure, tissue overgrowth, or fibrosis in a wide variety of connective tissue lesions that include, respectively, osteoarthritis, gingival hyperplasia, or heart failure. Collagen degradation is mediated by an extracellular matrix metalloproteinase–dependent extracellular pathway and by a poorly defined, intracellular phagocytic pathway that involves fibroblasts (Everts et al., 1996). During phagocytosis, fibroblasts bind collagen via integrins, which is followed by collagen fibril segregation and then partial digestion of the fibrils by extracellular matrix metalloproteinases (Murphy and Nagase, 2008). Short fibrils are internalized and further digested in lysosomal compartments (ten Cate, 1972; Melcher and Chan, 1981; Everts et al., 1996). It is not understood how cells sequester collagen fibrils and focus their proteolytic machinery to enable collagen digestion by phagocytosis. Fibroblasts in vivo exhibit marked alterations of cell shape around collagen fibrils (ten Cate, 1972; Svoboda et al., 1979; Melcher and Chan, 1981) that might be important for enabling collagen digestion, but it is not known how cells recruit cytoskeletal proteins to collagen fibril attachment sites for initiating and enabling phagocytosis.
We considered that the complex topography exhibited by collagen fibrils in vivo might hamper our ability to define the control systems that regulate collagen phagocytosis. Examination of collagen remodeling by phagocytosis has been primarily studied on cells plated on rigid, planar substrates. The flattened appearance of fibroblasts plated on planar surfaces differs substantially from their in vivo appearance, which is a more dendritic and stellate shape than that of cultured cells (Grinnell, 2003). The differences in the shape of fibroblasts cultured on planar surfaces compared with fibroblasts in mammalian connective tissues reflect, in part, the response to substrate stiffness (Yeung et al., 2005) and substrate topography (Curtis and Wilkinson, 1998). Indeed, substrate stiffness is an important determinant of the shape and function of adhesions that form when cells attach to extracellular matrices (Vicente-Manzanares and Horwitz, 2011). In the context of substrate topography, when avidly phagocytic cells are plated on flat surfaces, they spread and engage the surface in an attempt to engulf it or until all of the adhesion receptors are engaged or internalized (Takemura et al., 1986). A state of “frustrated” phagocytosis can also be achieved by incubating cells with polystyrene beads of sufficient size so that the cells are unable to engulf the beads (Cannon and Swanson, 1992). Accordingly, the topographical presentation of collagen to phagocytic cells might affect the nature of adhesion formation and possibly collagen phagocytic processes (Grinnell and Geiger, 1986). We consider here that the topological complexity of collagen fibril presentation to cells might also affect calcium signaling, which is known to be important for phagocytic processes (Stendahl et al., 1994; Zimmerli et al., 1996). In particular, previous data show the existence of stretch-sensitive, cation-permeable channels in fibroblasts from periodontium (Arora et al., 1994; Glogauer et al., 1995). Accordingly, the topology of collagen fibril presentation, which depends on surface characteristics, might affect the opening probability of stretch-activated, calcium-permeable channels that regulate intracellular calcium concentration.
Initial cell attachment to collagen in cultured cells occurs via specialized adhesions and their potential in vivo correlates, fibronexus junctions (Singer et al., 1984). Focal adhesions are paradigmatic examples of the complex molecular architectures that enable adhesion receptors like integrins to connect the extracellular matrix to the actin cytoskeleton (Burridge and Chrzanowska-Wodnicka, 1996). In cultured cells, adhesions form initially near the leading edge of the lamellipodium, a region of the cell in which rapid actin polymerization drives membrane protrusion (Pollard and Borisy, 2003). The earliest adhesions that form after cell contact with matrix ligands are largely immobile (Alexandrova et al., 2008), but these adhesions can mature and enlarge over time as the cells become more adherent to the substrate and more structural and signaling molecules are recruited to the adhesions. In this context, focal adhesions are a heterogeneous group of adhesions with a wide spectrum of shapes and sizes that reflect the temporal continuum of adhesion maturation (Geiger and Yamada, 2011). They are often small, highly circumscribed structures that contain integrins, actin-binding proteins such as talin and vinculin, and signaling molecules such as FAK and p130CAS (Geiger et al., 2009).
The maturation of focal complexes to focal adhesions is a process that involves the interactions of actin filaments with a large number of actin-binding proteins such as α-actinin and nonmuscle myosin IIA (NMMIIA; Choi et al., 2008). Of note, the contractile activity of NMMIIA is dispensable for the adhesion maturation process (Choi et al., 2008) and is also not required for collagen phagocytosis (Arora et al., 2008a). Although most of our understanding of adhesion formation and maturation has been obtained from the study of cells cultured on planar surfaces, cells in vivo adhere to and remodel collagen (Melcher and Chan, 1981) that is organized into complex surfaces in three dimensions. Indeed, adhesions formed in three-dimensional matrices tend to be small and highly dynamic and are NMMIIA dependent (Fraley et al., 2010; Kubow and Horwitz, 2011). It is not understood how NMMIIA regulates adhesion formation to collagen in more topologically complex environments than planar cell culture surfaces.
NMMIIA is an actin-based motor protein that is important for cell migration because of its effect on adhesion, lamellar protrusion, and polarity (Conti and Adelstein, 2008; Vicente-Manzanares et al., 2009). Myosin II molecules are hexamers composed of myosin heavy-chain dimers and two pairs of myosin light chains (Conti and Adelstein, 2008). The heavy chains include the α-helical coiled-coil rod domain, which contributes in part to the assembly of nonmuscle myosin monomers into filaments. The heavy chains also contain phosphorylation sites for several kinases (Dulyaninova et al., 2005). In the context of phagocytosis, myosin-superfamily motor proteins interact with actin filaments to generate contractile forces, which are required for phagosome development (Groves et al., 2008). It is not understood how NMMIIA contributes to the regulation of cell adhesion in collagen phagocytosis.
Efficient collagen phagocytosis requires actin assembly at matrix adhesion sites (Arora et al., 2008a), a process that depends on the function of the actin-severing and -capping protein gelsolin (Arora et al., 2003, 2005). Gelsolin is an actin-binding protein that nucleates filament assembly in a Ca2+-dependent manner (Yin and Stossel, 1979; Yin et al., 1981; Weeds and Maciver, 1993) and plays an important role in FcγR-mediated phagocytosis but not in complement-receptor–mediated phagocytosis (Serrander et al., 2000). Gelsolin associates with NMMIIA at collagen-binding sites; this association is required for actin assembly and adhesion to collagen (Arora et al., 2011), but it is not known whether gelsolin interacts with NMMIIA directly and how this interaction is regulated to affect adhesion to collagen.
In this study we examine the effect of collagen substrate topography on adhesion formation. We define the role of Ca2+ in regulating the interaction of NMMIIA with gelsolin to enable adhesion formation and collagen degradation by phagocytosis. With the use of purified, full-length, and isolated domains of NMMIIA and gelsolin we show that gelsolin interacts with the coiled-coil domain of NMMIIA in a Ca2+-dependent manner. This interaction controls actin filament assembly (Arora et al., 2011) and contributes to adhesion and phagocytosis of collagen.
RESULTS
Gelsolin and NMMIIA localize selectively to collagen-coated beads
We determined whether cell adhesion to collagen was affected by topology. Wild-type fibroblasts plated on collagen-coated planar surfaces exhibited prominent vinculin staining at focal adhesions. Colocalization of gelsolin or NMMIIA with vinculin was minimal, as quantified by the Pearson r (Figure 1A). In contrast, cells plated on collagen-coated beaded surfaces (2 μm diameter) showed marked colocalization of gelsolin or NMMIIA with vinculin at bead adhesion sites (Figure 1B). We estimated the relative abundance of β-actin, gelsolin, and NMMIIA associated with collagen beads by first counting the number of beads associated with cells, preparing collagen bead–associated proteins (Glogauer et al., 1998), and then comparing blot densities of the bead-associated proteins with known amounts of purified gelsolin and NMMIIA that were analyzed on the same gels. These data indicated that on a molar basis, there was approximately twofold more gelsolin than NMMIIA that was associated with the collagen adhesions (Figure 1C; p < 0.05). Gelsolin and vinculin localized to collagen beads in NMMIIA wild-type embryonic stem cells but not in NMMIIA-null embryonic stem cells (Figure 1D), indicating that gelsolin localization to collagen bead adhesions might involve NMMIIA. We immunostained gelsolin-null and wild-type (WT) cells for the α2 integrin, which is an important integrin subunit for collagen binding. These results showed colocalization of α2 integrin with NMMIIA in gelsolin WT cells (but not gelsolin-null cells) at collagen bead–binding sites (Supplemental Figure 1A).
FIGURE 1:

(A) Representative images of gelsolin and NMMIIA show minimal colocalization with vinculin-stained focal adhesions in wild-type cells plated on collagen-coated planar substrates. There was 30 and 40% colocalization, respectively, between gelsolin and vinculin and between NMMIIA and vinculin. (B) Wild-type cells plated on 2-μm collagen-coated beads show colocalization of gelsolin and NMMIIA with vinculin at collagen bead adhesion sites. Insets show targeting of gelsolin and NMMIIA to developing phagosomes. (C) Quantitative immunoblot analysis of collagen bead–associated proteins. Data are mean ± SEM (in picomoles per cell) of β-actin, gelsolin, and NMMIIA. (D) Gelsolin strongly colocalizes with vinculin at collagen bead–binding sites in wild-type embryonic stem cells but not in NMMIIA-null embryonic stem cells.
Interaction of NMMIIA with gelsolin
The finding of colocalization of gelsolin and NMMIIA suggested potential interactions between these proteins. We examined these interactions using purified full-length NMMIIA (Supplemental Figure 1B) and full-length gelsolin. In the preparation of purified NMMIIA, treatment with MgATP dissociated actin contaminants, as shown by immunoblotting of the purified fraction for β-actin (Supplemental Figure 1C). From the purified preparations, negatively stained NMMIIA filaments were visualized by transmission electron microscopy (Supplemental Figure 1D). Pelleting assays showed that full-length NMMIIA associates with full-length gelsolin in calcium buffer but not in ethylene glycol tetraacetic acid (EGTA) buffer (Figure 2A). In Coomassie-stained SDS–polyacrylamide gels of the pelleting assays, high-salt (600 mM KCl) buffer prevented NMMIIA binding to gelsolin that was bound to glutathione S-transferase (GST)–Sepharose beads (Figure 2B). We extended these studies by mapping the gelsolin-binding domain on NMMIIA filaments using GST pull downs. GST-gelsolin domains G1–G3 exhibited Ca2+-independent binding to full-length NMMIIA, whereas gelsolin domains G4–G6 displayed Ca2+-dependent binding to NMMIIA filaments (Figure 2C). Preparations of NMMIIA and gelsolin that were prepared for immuno–electron microscopy showed increased immunogold labeling of gelsolin on NMMIIA filaments compared with preparations with no primary antibody (Supplemental Figure 1E).
FIGURE 2:

(A) Pelleting assays show association of full-length NMMIIA with full-length gelsolin in presence or absence of EGTA. (B) Coomassie-stained SDS-polyacrylamide gel shows that high-salt (600 mM KCl) buffer prevents NMMIIA binding to gelsolin bound to GST-Sepharose beads. (C) Association between full-length NMMIIA and gelsolin domains G1–G3 and G4–G6 in pull-down assays. Pellets were visualized on Coomassie-stained SDS–PAGE gel and show that the interaction between GST-gelsolin domains G4–G6 and full-length NMMIIA requires Ca2+. (D) Quantification of the binding signal of gelsolin to NMMIIA shows that gelsolin (G1–G6) binds to NMMIIA efficiently with Kd = 0.158 ± 0.082 μM. (E) In the presence of EGTA the binding was reduced (Kd = 0.423 ± 0.152 μM). (F, G) The N- and C-terminal halves G1–G3 and G4–G6 showed binding with Kd = 0.220 ± 0.049 and 0.212 ± 0.063 μM, respectively. (H, I) In the presence of EGTA, N- and C-terminal halves G1–G3 and G4–G6 showed reduced binding with Kd = 0.495 ± 0.178 and 1.312 ± 0.256 μM, respectively, suggesting that binding of G4–G6 to NMMIIA requires calcium. (J) Pull-down assays show pellets in Coomassie-stained gels. GST-Sepharose bead–bound gelsolin did not bind to HMM IIA (1–1337) in the presence or absence of EGTA. (K) NMMIIA-His-thioredoxin residues 1339–1891 associated with full-length gelsolin G1-G6–Sepharose beads in absence of EGTA. (L) NMMIIA residues 1899–1960 bound to GST-Sepharose beads did not bind to gelsolin in the presence or absence of EGTA.
We performed more-detailed binding experiments in which we used full-length G1–G6, the N-terminal half of gelsolin (G1–G3), the C-terminal half of gelsolin (G4–G6), and the rod domain of NMMIIA. Binding experiments were performed in the presence or absence of EGTA (1 mM). Quantification of the binding of gelsolin to NMMIIA showed that gelsolin G1–G6 binds tightly to NMMIIA (dissociation constant Kd = 0.158 ± 0.082 μM; Figure 2D). In the presence of EGTA the binding was reduced (Kd = 0.423 ± 0.152 μM; Figure 2E). The N- and C-terminal halves of gelsolin (G1–G3, G4–G6) showed binding with Kd = 0.220 ± 0.049 and 0.212 ± 0.063 μM, respectively (Figure 2, F and G). In the presence of EGTA, the N- and C-terminal halves of gelsolin (G1–G3, G4–G6) showed reduced binding, with Kd = 0.495 ± 0.178 and 1.312 ± 0.256 μM, respectively, suggesting that binding of G4–G6 to NMMIIA requires calcium ions (Figure 2, H and I).
Mapping of binding domains in gelsolin and NMMIIA filaments
With pull-down assays we examined the binding of truncated forms of NMMIIA to full-length gelsolin (G1–G6). In pull-down assays, gelsolin bound to GST-Sepharose beads did not associate with residues 1–1338 of NMMIIA (heavy meromyosin [HMM]) in the presence or absence of EGTA (Figure 2J). GST-Sepharose bead–bound gelsolin cosedimented with NMMIIA–histidine (His)-thioredoxin residues 1339–1891 in the presence of calcium ions (Figure 2K). GST-Sepharose beads bound with the tail region of NMMIIA (residues 1899–1960) and that were incubated with full-length gelsolin did not sediment gelsolin in the presence or absence of EGTA (Figure 2L). Taken together, these data suggest that the rod region enables binding of NMMIIA to gelsolin. In a control experiment purified GST did not cosediment with full-length NMMIIA filaments, indicating specific binding of gelsolin to NMMIIA (Supplemental Figure 1F). Similarly, gelsolin bound to GST-Sepharose did not cosediment with His-thioredoxin (Supplemental Figure 1G).
Effect of NMMIIA on gelsolin-induced actin polymerization
We determined whether the interaction of gelsolin with NMMIIA affects the nucleating, capping, and severing functions of gelsolin (Yin, 1987). To assess the effects of gelsolin on actin polymerization, we used a pyrene-labeled actin assay. The addition of gelsolin to actin monomers sharply increased the rate of pyrene fluorescence, but the addition of gelsolin/NMMIIA complexes at various NMMIIA concentrations to pyrene–actin monomers exerted no effect on the actin nucleation function of gelsolin (Supplemental Figure 2A). NMMIIA alone exerted no effect on actin polymerization (Supplemental Figure 2B).
Effect of NMMIIA on gelsolin's capping activity
We measured the capping activity of gelsolin/NMMIIA complexes by incubating this complex with pyrene actin monomers in polymerization buffer for 18 h. In the presence of gelsolin/NMMIIA complexes, the final pyrene–actin fluorescence was similar for both gelsolin alone and the gelsolin/NMMIIA complexes (Supplemental Figure 2C). The addition of NMMIIA alone had no effect on pyrene–actin fluorescence (Supplemental Figure 2D). These data indicate that when NMMIIA is bound to gelsolin, there is no effect of NMMIIA on the actin-capping activity of gelsolin.
Effect of NMMIIA on gelsolin's severing activity
We investigated the effect of purified gelsolin/NMMIIA complexes on actin filament severing by gelsolin. Full-length NMMIIA and gelsolin were coincubated and were added to pyrene-labeled actin filaments; the rate of fluorescence loss was compared with the addition of gelsolin alone. When gelsolin was added to pyrene-labeled actin filaments, there was an initial loss of fluorescence due to severing of actin filaments by gelsolin. When various concentrations of the gelsolin/NMMIIA complex were added to pyrene-labeled actin filaments, there were small reductions of F-actin severing, which was slightly lower (30% reduction) than the severing generated by gelsolin alone (Figure 3A). Collectively these data indicate that gelsolin/NMMIIA complexes had no effect on the polymerization or capping activity of gelsolin, but that binding of NMMIIA to gelsolin minimally reduced the severing activity of gelsolin. In experiments of similar design, when the rod domain of NMMIIA (residues 1339–1891) was incubated with gelsolin, there was also reduced severing of actin filaments compared with severing by gelsolin alone (Figure 3C). In contrast, when NMMIIA residues 1–1338 (Figure 3B) or NMMIIA residues 1899–1960 (Figure 3D) were incubated with gelsolin, there was no effect on actin severing.
FIGURE 3:

(A) Histogram shows loss of fluorescence of pyrene-labeled actin filaments (0.3 μM) as a result of severing by gelsolin (0.1 μM) in the presence of 1 mM CaCl2. Addition of purified NMMIIA (0.01, 0.02, 0.05, 0.1, 0.2, and 0.4 μM) and gelsolin (0.1 μM) complex show decreased reduction of fluorescence compared with gelsolin alone. (B–D) In experiments of similar design, NMMIIA domains consisting of residues 1–1338, 1339–1891, or 1899–1960 were incubated with gelsolin, and actin severing was measured. (E) Lysates prepared from cells spreading on collagen-coated beads show increase in severing of pyrene-labeled actin filaments compared with cells grown overnight on collagen-coated tissue culture plates. Inset, G-purified gelsolin standard; S, Q, gelsolin expression levels in cell lysates prepared from spreading (S) and quiescent (Q) cells. (F) Total cell lysates from gelsolin-null and wild-type cells were immunoblotted for the indicated proteins. (G) Cells transfected with NMMIIA siRNA show 75% reduction in NMMIIA protein levels compared with cells treated with nontargeted siRNA. (H) Wild-type or gelsolin-null cells transfected with control siRNA or NMMIIA siRNA were maintained in quiescence or were spread on collagen. Cell lysates were analyzed for actin severing by loss of pyrene fluorescence. Wild-type cells transfected with NMMIIA siRNA show enhanced severing of actin filaments in actively spreading cells compared with cells transfected with irrelevant, control siRNA.
Gelsolin's severing activity in cells
Because our in vitro data showed that gelsolin severing may be affected by binding to NMMIIA, we determined whether gelsolin severing in intact cells is also affected by NMMIIA. We performed severing assays on lysates prepared from cells that had been plated for 1 h on planar collagen (actively spreading cells [S]) or after overnight incubation on collagen (quiescent cells [Q]). The severing activity of cells was slightly (30%) lower in cells plated overnight on collagen (quiescent) compared with actively spreading cells plated for 1 h on collagen (p < 0.1; Figure 3E).
We transfected wild-type and gelsolin-null cells with NMMIIA small interfering RNA (siRNA) or a control siRNA and examined whether NMMIIA knockdown affected the severing activity of wild-type and gelsolin-null cells. The protein expression levels of NMMIIA were similar in gelsolin-null and WT cells (Figure 3F). Cells transfected with NMMIIA siRNA showed 75% reduction of NMMIIA expression levels, but gelsolin protein levels in these cells were unchanged (Figure 3G).
In quiescent cells (overnight plating on collagen), the severing activity of the cell lysates was ∼50% lower in the gelsolin-null cells compared with wild-type cells (Figure 3H), indicating that about one-half of the actin-severing activity in these cells is attributable to gelsolin. NMMIIA knockdown did not affect the severing activity in actively spreading gelsolin-null cells compared with gelsolin-null cells transfected with control siRNA (Figure 3H). In gelsolin wild-type cells after NMMIIA knockdown there was a small increase of severing activity (by ∼25%; p < 0.1) compared with control cells (Figure 3H). Therefore in both intact cells and in assays of purified proteins, NMMIIA exerted a small inhibitory effect on gelsolin-mediated actin severing.
Collagen phagocytosis on planar and beaded surfaces
Because gelsolin and NMMIIA did not localize markedly to adhesion complexes on planar surfaces but are known to be important for phagocytosis of collagen-coated beads (Arora et al., 2004, 2008b), we examined the effect of substrate topography on collagen degradation. We analyzed the relative abundance of fluorescein isothiocyanate (FITC)–collagen in four cellular compartments that are associated with collagen degradation in cells plated on planar surfaces or on 2-μm-diameter latex beads (Figure 4A). We first determined the total area and relative abundance of FITC-collagen coated on planar surfaces (47.1 μg/cm2) and on 2-μm beads (49.6 μg/cm2), indicating that the abundance of collagen on these different surfaces was very similar. In cells plated on planar surfaces, ∼10% of the substrate collagen was degraded and released into the medium, ∼35% of collagen was associated with the cell surface, <10% was internalized, and 45% was not degraded (i.e., remained attached to the substrate; Figure 4A). For cells plated on 2-μm collagen-coated beads, 18% of collagen was degraded and released into the medium, 22% was associated with the cell surface, 45% was internalized, and 15% was not degraded (Figure 4A). Given that the percentage of internalized collagen was approximately fourfold higher in cells plated on beaded compared with planar substrates (p < 0.001), substrate topography evidently affects collagen internalization.
FIGURE 4:
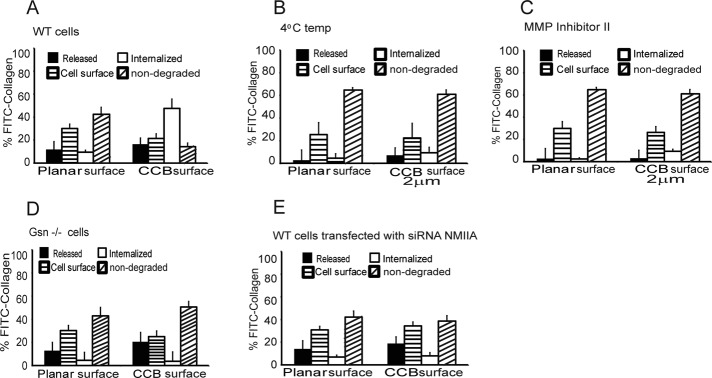
(A) Comparison of collagen degradation on collagen-coated planar surface and collagen-coated 2-μm beads. Histogram shows FITC-collagen associated with discrete cellular compartments. The data are percentage of FITC-collagen released into media, cell-surface bound, internalized, or remaining (nondegraded) adherent to substrate (planar or bead surfaces). (B, C) Collagen internalization was retarded at low temperatures and in the presence of MMP inhibitor. (D, E) Histograms show collagen on planar or irregular surface metabolized by gelsolin-null, WT cells transfected with myosin IIA siRNA.
We performed similar experiments at 4ºC and observed greatly decreased collagen degradation in cells plated on planar or beaded surfaces (Figure 4B). Inhibition of collagenase activity with N-hydroxy-1,3-di-(4-methoxybenzenesulfonyl)-5,5-dimethyl-[1,3]-piperazine-2-carboxamide, an MMP-1 inhibitor (Pikul et al., 1998), reduced collagen internalization to low levels for cells plated on collagen-coated planar or beaded surfaces. In these experiments most of the collagen was either bound to the cell surface or was not degraded (Figure 4C).
As shown earlier, in cells plated on planar collagen substrates, collagen internalization was minimal, and gelsolin and NMMIIA did not colocalize with vinculin-stained adhesions (Figure 1). We determined whether cells plated on larger collagen-coated beads (2, 20, and 45 μm; Supplemental Figure 3A) would exhibit collagen internalization when the beads were sufficiently large that they could not be internalized. We found that cells adhered to all sizes of beads but did not ingest the larger beads (20 and 45 μm), as determined by trypan blue quenching of FITC-collagen on the bead surface (Arora et al., 2008a; unpublished data). Of note, for cells plated on 20- or 45-μm collagen-coated beads, the compartmentalization of collagen degradation was similar to the pattern observed in cells that were plated on planar surfaces; in these preparations there was minimal internalized collagen, as measured with FITC-collagen (<4%; Supplemental Figure 3B).
Because the immunostaining data (Figure 1) suggested that gelsolin and NMMIIA localize to adhesions around collagen beads but not on planar substrates, we studied the role of gelsolin and NMMIIA in collagen remodeling in cells plated on the same planar or beaded collagen substrates. Gelsolin-null cells or wild-type cells or gelsolin wild-type cells transfected with NMMIIA siRNA were plated on FITC-collagen–coated planar surfaces or FITC-collagen–coated beaded (2 μm) surfaces. Compared with wild-type cells, there was >10-fold reduction in collagen internalization for gelsolin-null cells or cells treated with NMMIIA siRNA (Figure 4, D and E), but there was no difference of collagen release into the medium. Further, in gelsolin-null cells or cells with NMMIIA knockdown, there was reduced collagen internalization only in cells plated on beaded substrates and not on planar substrates. These data suggested that gelsolin and NMMIIA are important proteins in mediating collagen internalization on topologically complex surfaces.
Because collagen internalization was inhibited in gelsolin-null cells, we examined whether collagen proteolysis in intracellular compartments was different in gelsolin-null cells compared with gelsolin wild-type cells. Cells were plated on beads coated with exogenous biotinylated collagen for 2 or 8 h and detached with trypsin from the plates, and exogenous collagen that had been internalized was probed in cell lysates with streptavidin peroxidase (Supplemental Figure 3C). These data showed markedly more collagen degradation in cells expressing gelsolin than in gelsolin-null cells. Of note, the vacuolar hydrolases that cleave collagen in the phagocytic pathway produce multiple collagen cleavage fragments of widely varying molecular mass, in contrast to the interstitial collagenases, which typically degrade collagen into one-fourth and three-fourths fragments.
Ca2+ mobilization in response to surface topology
The importance of gelsolin and NMMIIA in collagen internalization and the requirement for increased Ca2+ for gelsolin-NMMIIA interactions motivated us to determine whether substrate topology affects intracellular calcium responses during cell spreading. In cells loaded with Fura-2/AM and plated on collagen-coated beads, there were large increases of free intracellular calcium ion concentration ([Ca2+]i) over time. The increase in [Ca2+]i was fourfold higher (Figure 5A) in cells plated on collagen-coated beads than in cells plated on planar collagen-coated surfaces (Figure 5D). We considered that cells very likely undergo membrane stretch during spreading over collagen-coated beads but less so on planar surfaces. Accordingly, we found that treatment of cells with the stretch-activated channel inhibitor Grammostola spatulata mechanotoxin 4 (GsMTx-4; 10 μM), reduced collagen-induced increases of [Ca2+]i more markedly in cells plated on beaded surfaces than on planar surfaces (Figure 5, B and E). In cells incubated in an EGTA-containing buffer, there was minimal increase of [Ca2+]i in cells plated on collagen beads or collagen-coated planar surfaces (Figure 5, C and F). Similarly, chelation of intracellular Ca2+ with BAPTA/AM reduced Ca2+ transients in these cells (Figure 5, C and F), suggesting that Ca2+ flux through stretch-activated channels might be in part responsible for the Ca2+ transients exhibited by cells spreading on beaded collagen surfaces.
FIGURE 5:

(A, D) Fura-2/AM–loaded cells plated on collagen-coated beads or collagen-coated planar surface show increase in intracellular calcium levels ([Ca2+]i) over time. Images show estimates of intracellular calcium levels in pseudocolor. (B, E) Treatment of cells with the stretch-activated channel (SAC) inhibitor GsMTx-4 (10 μM) reduced [Ca2+]i increases in response to plating on collagen. (C, F) In the presence of EGTA-containing buffer, the increase in [Ca2+]i was minimal in cells plated on collagen beads or collagen-coated planar surface. Chelation of intracellular Ca2+ with BAPTA/AM also reduced Ca2+ transients. (G, H) Lack of effect of GsMTx-4 on store-operated channels. Cells were subjected to store depletion of intracellular calcium stores with low Ca2+ buffer, followed by thapsigargin (1 μM), and treated with either vehicle (G) or GsMTx-4 (10 μM; H).
We determined whether the effect of GsMTx-4 on [Ca2+]i was restricted to entry of Ca2+ through stretch-activated channels, since there is the possibility that GsMTx-4 might inhibit store-operated Ca2+-permeable channels in the plasma membrane (Wang et al., 2001). First we depleted cytoplasmic Ca2+ by incubation of cells in buffer with very low Ca2+, followed by thapsigargin treatment, and then we activated store-operated channels by addition of extracellular Ca2+ in the presence or absence of GsMTx-4. These experiments showed no inhibition of Ca2+ entry (Figure 5, G and H), indicating that GsMTx-4 did not cause global inactivation of plasma membrane Ca2+ channels.
NMMIIA- and calcium-dependent enrichment of adhesion sites with gelsolin
Because immunostaining showed that gelsolin and NMMIIA were localized to collagen-bead adhesion sites (Figure 1), we determined the temporal sequence of the enrichment of these proteins at collagen-bead adhesions. Wild-type fibroblasts were incubated with collagen-coated beads, fixed after discrete time points, and immunostained for gelsolin and NMMIIA. Fluorescence intensity was measured in regions of interest around beads that exhibited staining intensities above background thresholds for gelsolin and NMMIIA; these data were scored as percentage of positive regions of interest (50 cells in each experimental condition). There were 25% NMMIIA positively stained regions of interest around collagen beads within 2 min after bead incubation, which preceded the appearance of positive immunostaining for gelsolin (not seen until 5–15 min). Regions of interest that were stained for gelsolin were always positive for NMMIIA. Thus NMMIIA might function in the recruitment of gelsolin to sites of bead adhesion (Figure 6A): <1% of gelsolin-positive regions of interest did not show corresponding NMMIIA fluorescence.
FIGURE 6:

(A, B) Quantitative analysis of localized fluorescent regions of interest around bead attachment sites. The data show the percentage of positively stained regions of interest around bound collagen beads for cells immunostained with gelsolin and NMMIIA. Data were collected over time in the presence or absence of EGTA. (C) Pretreatment of cells with GsMTx-4 (10 μM) blocked colocalization of NMMIIA and gelsolin at bead sites. (D) Loss of fluorescence of pyrene-labeled actin filaments (0.3 μM) as a result of severing by wild-type gelsolin (0.1 μM) or the severing mutant (Gsn 100/210). (E) Pelleting assays show association of full-length NMMIIA with gelsolin severing mutant (Gsn 100/210) in the presence or absence of EGTA in a Coomassie-stained, SDS-polyacrylamide gel. (F) Gelsolin null cells transfected with gelsolin cDNA and severing mutant showing localization of gelsolin and NMMIIA to collagen beads. (G, H) Histogram showing requirement of severing activity of gelsolin for collagen internalization during compartmentalization of FITC-collagen coated on beads. Severing mutant (Gsn 100/210) shows retarded collagen internalization. (I) Stretch-activated channel inhibitor (GsMTx-4) prevents internalization and collagen degradation.
The percentage of positively stained regions of interest for NMMIIA and gelsolin increased over time after plating, and this process was strongly dependent on the presence of extracellular Ca2+, since chelation of extracellular Ca2+ (1 mM EGTA-containing buffer) reduced gelsolin recruitment (Figure 6B). Compared with gelsolin, the percentage of positively stained regions for NMMIIA was much less affected by the chelation of Ca2+. Of note, pretreatment of cells with GsMTx-4 (10 μM) blocked colocalization of NMMIIA and gelsolin at bead sites (Figure 6C), indicating that Ca2+ transients are likely important for association of NMMIIA and gelsolin at collagen adhesion sites. We also examined the potential functional significance of NMMIIA activity by treating cells with blebbistatin (50 μM) but found that targeting of NMMIIA and gelsolin to collagen-coated beads was not affected by blebbistatin (Supplemental Figure 3D).
Role of gelsolin and NMMIIA in collagen degradation
Because NMMIIA had no effect on gelsolin polymerization and capping functions, we examined whether the severing activity of gelsolin was important for collagen degradation mediated by the intracellular pathway. We used a gelsolin severing mutant (Gsn 100/210; Arora et al., 2005) that exhibits marked reduction of actin severing compared with wild-type gelsolin, as measured by fluorescence loss of pyrene-labeled actin filaments (Figure 6D). In vitro pelleting assays showed association of full-length NMMIIA with the gelsolin severing mutant (Gsn 100/210) in calcium-containing buffer (Figure 6E). Further, in gelsolin-null cells transfected with the severing mutant, although there were reduced numbers of bound collagen beads, gelsolin immunostaining colocalized with NMMIIA at collagen bead–binding sites (Figure 6F).
We examined the collagen degradation patterns in cells plated on planar or beaded substrates in gelsolin-null cells that were transfected with a wild-type gelsolin expression construct or in gelsolin-null cells transfected with the gelsolin severing mutant (Gsn 100/210). In gelsolin-null cells transfected with cDNA for wild-type gelsolin there was a >10-fold increase in internalized collagen in cells plated on beaded collagen (but not planar substrates) compared with gelsolin-null cells (Figure 4D). When gelsolin-null cells were transfected with the gelsolin severing mutant construct (Arora et al., 2005), collagen internalization and the amount of degraded collagen were similar to that of gelsolin-null cells (Figure 6, G and H), and there was no difference in the pattern of collagen degradation in cells plated on beaded or planar substrates. Taken together, these data indicate that gelsolin's severing activity is important for collagen phagocytosis, but this process operates only when collagen is presented on a beaded surface.
As described here, we found that in cells binding to collagen beads, there was a marked increase of [Ca2+]i that was inhibited by treatment with the stretch-activated cation channel inhibitor GsMTx-4 (Figure 5). Because Ca2+ was evidently crucial for binding of NMMIIA to gelsolin (Figure 2) and is known to be important for gelsolin's severing activity (Yin et al., 1981), we treated cells plated on collagen with GsMTx-4 or vehicle and examined collagen internalization. There was ninefold-reduced collagen internalization in cells treated with GsMTx-4 compared with vehicle controls (p < 0.001; Figure 6I).
DISCUSSION
The potential interactions involving gelsolin and NMMIIA that may regulate early steps of collagen phagocytosis are not defined. Because the flattened appearance of cultured fibroblasts plated on planar surfaces differs from their in vivo appearance (Grinnell, 2003), which tends to be of a more complex dendritic shape, we examined whether collagen substrate topography affects the recruitment of gelsolin and NMMIIA to the adhesions, their interactions, and the nature of collagen degradation. We found that gelsolin and NMMIIA are enriched in cell adhesions that mediate collagen phagocytosis but only when cells are plated on a beaded and not on planar collagen substrates. Further, NMMIIA binds gelsolin at collagen adhesion sites; this Ca2+-dependent interaction enables actin remodeling at adhesion sites (Arora et al., 2011), which is required for collagen phagocytosis. One of the regulatory systems that enable phagocytosis involves activation of stretch-sensitive, Ca2+-permeable channels when cells attach and stretch over small, collagen-coated beads. The entry of Ca2+ through these channels may be stimulated by the extensive shape changes of fibroblasts that are manifest when engage collagen fibrils in vivo (ten Cate, 1972; Svoboda et al., 1979; Melcher and Chan, 1981). Of note, humans and animals that are treated with drugs that inhibit Ca2+ entry exhibit inhibition of the collagen phagocytosis pathway and fibrosis (McCulloch and Knowles, 1993; McCulloch, 2004).
NMMIIA and gelsolin at adhesion sites
We found that NMMIIA and gelsolin do not colocalize with vinculin-stained focal adhesions in cells spread on planar surfaces but are recruited to adhesions forming on beaded substrates, which may more closely mimic in vivo conditions (Melcher and Chan, 1981). NMMIIA is recruited to nascent adhesions in the lamella in migrating cells (Burnette et al., 2011), whereas NMMIIB associates with actin filament bundles in stable focal adhesions (Vicente-Manzanares et al., 2007). Of note, myosin VI plays specific roles in different intracellular functions such as endocytosis and cell migration (Buss et al., 2002), and myosin X associates with actin filaments at the distal tips of filopodia involved in rapid back and forth oscillations during retrograde flow of bundled actin (Nagy et al., 2008). It is conceivable that NMMIIA may play specific roles in adhesion to and phagocytosis of collagen in fibroblasts.
Cell-spreading processes on flat surfaces are fundamentally similar to the behavior of cells plated on very large fibronectin-coated beads (Grinnell and Geiger, 1986). We observed that collagen degradation was affected by the nature of the adhesions (planar or irregular surfaces), which also determined the recruitment of adhesion-regulating proteins that are required for remodeling by phagocytosis (Everts et al., 1996). The gene product of the Drosophila lethal giant larvae regulates NMMIIA cellular distribution and focal adhesion morphology to optimize cell migration (Dahan et al., 2011), which suggests that NMMIIA might play a fundamental role in determining adhesion function in different cells and cellular processes.
We found that collagen binding increased [Ca2+]i, which might reflect activation of stretch-activated calcium channels. Regulation of cell movement is also mediated by stretch-activated calcium channels (Lee et al., 1999). Our studies of purified proteins showed that gelsolin and NMMIIA interactions are Ca2+ dependent. Ca2+ also enhances the actin-severing function of gelsolin (Kiselar et al., 2003), which in turn is important for initiating actin remodeling at the cytoplasmic side of collagen-binding sites (Arora et al., 2011). Therefore the collagen bead–evoked Ca2+ that occur early on in the binding process might play a fundamental role in enabling subsequent recruitment of critical actin-modifying proteins to the nascent phagosome.
NMMIIA–gelsolin interactions
We identified residues 1339–1891 of the NMMIIA heavy chain as a gelsolin-interacting domain, a region that is also important for NMMIIA filament assembly (Dulyaninova et al., 2005). Examination of gelsolin and NMMIIA interacting domains indicated that gelsolin domains 4 and 6 interact with NMMIIA in a Ca2+-dependent manner. Gelsolin adopts an activated state in the presence of Ca2+ in which three masked actin-binding sites are exposed; these include actin monomer–binding sites on domains G1 and G4, and a filament side–binding site on G2 (Way et al., 1989; Pope et al., 1991). However, crystallization studies of the C-terminal half of gelsolin in the presence of Ca2+ (but in the absence of actin) demonstrated that actin is not a prerequisite for enabling the development of an activated gelsolin conformation (Narayan et al., 2003). Our data show that, unlike the N-terminal half of gelsolin (domains G1–G3), the C-terminal half of gelsolin (G4–G6) does not interact with NMMIIA in the presence of EGTA. It is conceivable that the interaction between NMMIIA and gelsolin domains 4 and 6 requires Ca2+ to enable conformational changes that are consistent with the interaction of the two molecules.
Effect of NMMIIA on gelsolin function
Actin capping, nucleation, and severing are important functions of gelsolin required for regulation of adhesion maturation and collagen matrix remodeling. We considered that the interaction of NMMIIA with gelsolin might affect gelsolin function. Our in vitro data indicated that when NMMIIA was bound to gelsolin, there was no effect on gelsolin-mediated capping or nucleation function and only a small inhibition of gelsolin's severing activity. It is conceivable that a small inhibition of the severing function of gelsolin by binding to NMMIIA might be necessary initially to prevent actin depolymerization in the locale of adhesions. Another, more likely possibility is that since NMMIIA sequesters gelsolin to collagen bead–binding sites, small reductions of gelsolin's severing activity are more than made up for by the abundance of this abundant actin-remodeling protein at the nascent phagosome, where it enables actin polymerization and capping (Arora et al., 2011). Indeed, our experiments on intact cells show that, whereas knockdown of NMMIIA inhibits total cellular actin-severing activity, this same type of knockdown markedly inhibits collagen internalization.
In summary, cell adhesion to topologically complex surfaces such as collagen beads activates Ca2+ entry through stretch-activated channels and promotes the formation of phagosomes enriched with NMMIIA and gelsolin. The Ca2+-dependent interactions of gelsolin with NMMIIA enable actin remodeling at collagen bead adhesion sites, which enhances collagen degradation by the phagocytic pathway (Figure 7).
FIGURE 7:

Schematic model of calcium regulation of gelsolin interaction with NMMIIA. Cell adhesion to topologically complex collagen substrate induces [Ca2+]i flux, which promotes interaction of NMMIIA rods with gelsolin domains G1–G3 and G4–G6. This interaction enhances actin remodeling by gelsolin at collagen adhesion sites to enable collagen phagocytosis. Low [Ca2+]i inhibits binding of NMMIIA with gelsolin domains G4–G6, which results in reduced actin remodeling and inhibition of collagen phagocytosis.
MATERIALS AND METHODS
Reagents
Latex beads (2, 20, and 45 μm in diameter) were purchased from Polysciences (Warrington, PA). Purified, pepsin-digested, bovine type I collagen was purchased from Advanced BioMatrix (San Diego, CA). Antibodies to β-actin (clone AC-15), myosin light chain, glyceraldehyde-3-phosphate dehydrogenase, FITC-conjugated goat anti-mouse antibody, and tetra-methyl rhodamine isothiocyanate–phalloidin were obtained from Sigma-Aldrich (Oakville, Canada). Clostridial collagenase was from Sigma-Aldrich. MMP inhibitor II (N-hydroxy-1,3-di-(4-methoxybenzenesulfonyl)-5,5-dimethyl-[1,3]-piperazine-2-carboxamide) and blebbistatin were obtained from Calbiochem (San Diego, CA). FITC-labeled, bovine type I collagen (5 mg/ml) was purchased from AnaSpec (Fremont, CA). Pyrene-labeled actin was from Cytoskeleton (Denver, CO). Fura-2/AM was from Invitrogen (Mississauga, Canada). FFP-18 was from Teflabs (Austin, TX). GsMTx-4, an inhibitor of stretch channels, was from Alomone (Jerusalem, Israel).
Cells
Fibroblasts were derived from gelsolin wild-type and null mice as described (Arora et al., 2004). NMMIIA wild-type embryonic stem cells and NMMIIA-null embryonic stem cells were obtained from R. Adelstein (National Institutes of Health, Bethesda, MD) and cultured as described (Arora et al., 2008a).
Collagen bead binding and cell plating
Unlabeled collagen-coated latex beads (2 μm diameter) were applied to microbiological (i.e., non–tissue culture) dishes, dried down, and attached as described (Arora et al., 2003), followed by washing with phosphate-buffered saline (PBS). The number of beads plated per dish was adjusted to produce final bead:cell ratios specific for each experiment. Cells were counted electronically, and the cell concentration was adjusted before plating cells on dishes containing collagen-coated beads. The plates were maintained at room temperature for 20 min to allow the cells to settle and subsequently washed with fresh medium at 37°C. Detached cells were removed by repeated washes. In experiments that evaluated sites of collagen degradation (see later description), the amount of collagen bound to beads or to plates was determined using FITC-collagen, fluorimetry, and estimates of the surface areas of either the planar substrates or the beaded substrates that were used in specific experiments.
Collagen degradation
We determined collagen degradation in cells plated on collagen substrates with different topologies. For planar substrates, 48-well cell culture plates were coated with FITC-collagen (30 min). Plates were allowed to dry after removing excess, unbound collagen solution; nonspecific combining sites were blocked with 1% bovine serum albumin (BSA) solution. For beaded substrates, latex beads (2, 20, and 45 μm diameter) were coated with FITC-collagen, plated in the wells, allowed to settle for 2 h, and dried after aspiration of excess fluid, and nonspecific combining sites were blocked with BSA. We measured the amounts of FITC-collagen attached to the respective substrates by treatment with bacterial collagenase, followed by photon counting of the released FITC-collagen. The amounts of collagen on the various substrates were adjusted so that each surface exhibited equivalent collagen loading for equivalent surface areas.
Cells (5 × 104) were plated in each well and incubated for 1 h. After cell incubations, the medium in each well contained released (i.e., extracellularly degraded) collagen that was collected for analysis. After collection of the medium, PBS (200 μl × 3) was used to wash each well, and washes were collected for further analysis. These washes contained FITC-collagen that was loosely associated with the cell surface and was removed by washing. Trypsin (1%; 200 μl) was used to detach cells from planar or beaded substrates, and the FITC-collagen in these washes was added to the previously described cell surface–associated collagen. Of note, trypsinization did not remove beads from the bottom of the wells, as assessed by microscopy of the trypsin fraction. The detached cells in the trypsin fraction were centrifuged (5 min at 14,000 × g) and then lysed in Triton-X (1%) buffer; this fraction contained FITC-collagen that had been internalized by cells. Finally, bacterial collagenase (1%; 200 μl) was added to solubilize the remaining collagen attached to the bottom of the wells or to the bead surfaces (nondegraded collagen fraction). All fractions were measured separately in a spectrofluorimeter (absorbance/emission = 492/520 nm; PTI, London, Canada). For each of the different types of substrates, FITC-collagen photon counts were determined on a percentage basis for each of the released, cell-surface, internalized, and nondegraded fractions. In some experiments, a collagenase inhibitor (N-hydroxy-1,3-di-(4-methoxybenzenesulfonyl)-5,5-dimethyl-[1,3]-piperazine-2-carboxamide) was used at 1 μM. This inhibitor potently blocks MMP-1, with IC50 = 24 nM (Pikul et al., 1998).
Gelsolin and nonmuscle myosin constructs
Human full-length gelsolin cDNA (Kwiatkowski et al., 1986) was provided by H. L. Yin (University of Texas, Dallas, TX). A primer (GenBank number X04412.1) pair (forward, 5′-GCGCAAGCTTGTGGTGGAACACCCCGAGTTCCTCAAGGCA-3′; reverse, 5′-GCGCGGATCCCTAAGACCAGTAATCATCATCCCAGCCAAG-3′) was designed to amplify full-length gelsolin (G1–G6, 2.151 kb, encoding gelsolin residues 26–742). A primer pair (forward, 5′-GCGCAAGCTTGTGGTGGAACACCCCGAGTTCCTCAAGGCA-3′; reverse, 5′-GCGCGGATCCCTAGTCCCGCCAGTTCTTGAAGAACTGCTT-3′) was used to amplify the N-terminal three domains (G1–G3, 1.038 kb, encoding gelsolin residues 26–371; Burtnick et al., 2004) and a primer pair (forward, 5′-GCGCAAGCTTGACGATGGCACAGGCCAGAAACAGATCTGG-3′; reverse, 5′-GCGCGGATCCCTAAGACCAGTAATCATCATCCCAGCCAAG-3′) was used to amplify the C-terminal three domains (G4–G6, 0.987 kb, encoding gelsolin residues 414–742). The resulting PCR products were ligated into the HindIII/BamHI sites (underlined sequences in the primers) of N3×FLAG-pCMV5c (constructed by Miriam Barrios-Rodiles, Samuel Lunenfeld Research Institute, Toronto, Canada) and sequenced (ACGT Corp., Toronto, Canada).
Expression constructs
To prepare the GST-tagged terminal gelsolin half (GST-G1-3) construct, a primer pair (forward, 5′-GCGCGAATTCAAGTGGTGGAACACCCCGAGTTCCTCAAGG-3′; reverse, 5′-GCGCCTCGAGACTAGTCCCGCCAGTTCTTGAAGAACTGCT-3′) was designed to amplify the N-terminal three domains (G1–G3, 1.038 kb, encoding gelsolin residues 26–371) of human gelsolin cDNA, flanked by EcoRI and XhoI restriction sites (underlined). The resulting PCR product was ligated into the corresponding sites of pGEX-4T-2 (Amersham, Oakville, Canada) and transformed into DH5α-competent Escherichia coli cells (Invitrogen, Burlington, Canada). The construct was sequenced (ACGT Corp.) and transformed into BL21(DE3)-competent E. coli cells for GST-tagged protein expression and purification.
The GST-gelsolin C-terminal half (G4–G6) construct was provided by H. L Yin. Briefly, the gene fragment coding for residues 414–746 of gelsolin (domains G4–G6) was ligated into a modified PGEX-6P-1 plasmid (Amersham) using the PCR (Narayan et al., 2003). Domains G4–G6 were expressed in E. coli XL-1 Blue cells grown in Luria broth (LB) containing 100 μg/ml ampicillin and induced with 0.5 mM isopropyl-β-d-thiogalactoside (IPTG)for 3 h at 30ºC.
Purified HMM of NMMIIA (1–1339) was provided by J. Sellers (National Institutes of Health, Bethesda, MD). GST-tagged 1899–1960 NMMIIA was expressed and purified as described previously (Li et al., 2003).
To prepare the myosin construct consisting of amino acids 1339–1891, a primer pair (forward, 5′-GCGCGAATTCAATTCCTTCCGGGAGCAGCT-3′; reverse, 5′-GCGCGCGGCCGCACTAGGCGTTGGCCCGCTGG-3′) was designed according to GenBank number P35579.4 to generate EcoRI and NotI restriction sites (underlined) that flanked human NMMIIA (amino acids 1339–1891). We used pET-28a (+)-hNMMIIA 1339-end from one of us (A.B.) as the template. The resulting PCR product was ligated into pET-32a (+) (Novagen, Gibbstown, NJ; provided by Y. Yao and M. F. Manolson, University of Toronto, Toronto, Canada) digested by EcoRI and NotI. The sequence of the insert was confirmed by sequencing (ACGT). This construct was transformed into BL21 (DE3) cells for protein expression and purification. His-thioredoxin expressed by the original vector in BL21 (DE3) was used as the control. Nickel–nitriloacetic acid superflow columns (Qiagen, Valencia, CA) and nondenaturing conditions were used for protein purification.
GST-tagged proteins expressed in bacterial expression systems were isolated and purified as described previously (Puius et al., 2000; Arora et al., 2005). Briefly, for production of gelsolin proteins, BL21(DE3) cells were transformed with wild-type constructs. Luria broth (250 ml) containing ampicillin (100 μg/ml) was inoculated overnight at 37ºC, followed by induction with IPTG (1 mM) for 4 h. Equivalent expression levels of the mutants was examined by immunoblotting. Proteins isolated from inclusion bodies were dialyzed overnight and loaded onto glutathione–Sepharose 4B columns (Pharmacia, Sigma-Aldrich, Oakville, ON, Canada). The fusion proteins were cleaved on the column with thrombin (20 U in 2 ml of PBS) after overnight incubation. The eluate containing gelsolin and thrombin was separated by Centricon 50 filters (Amicon, Millipore, Billerica, MA). Protein yields were determined by optical density and protein analysis.
Purification of full-length nonmuscle myosin
Full-length NMMIIA was isolated from J744 macrophages as described (Trotter and Adelstein, 1979). Briefly, cells (107) were washed, collected by low-speed centrifugation, and resuspended in ice-cold extraction buffer (340 mM sucrose, 15 mM Tris-HCl, pH 7.5, 1 mM EDTA, 10 mM dithiothreitol [DTT], and 10 mM Na4P2O7 plus protease inhibitors) equivalent to two times the weight of the pellet. The cell suspension was stirred for 45 min in the cold, and cells were broken in a Dounce homogenizer. The volume of the cell suspension was brought to three times the weight of the pellet. The homogenate was centrifuged at 40,000 × g for 40 min at 4ºC. The cell extract was equilibrated to 10 mM in MgATP and then brought to 35% saturation by slow addition of saturated ammonium sulfate containing 10 mM EDTA, pH 8.2. The precipitate was collected by centrifugation at 15,000 × g for 30 min at 4ºC; the supernatant was brought to 55% saturation in ammonium sulfate. The precipitated proteins (35–55%) were collected by centrifugation and dissolved in 10 ml of high–ionic strength buffer (0.6 M KCl, 15 mM Tris HCl, pH 7.5, 1 mM EDTA, 5 mM DTT). The solution was centrifuged, and the clarified solution was dialyzed against low–ionic strength buffer (60 mM KCl, 15 mM Tris HCl, pH 7.5, 1 mM EDTA, 5 mM DTT). The precipitated protein was collected by centrifugation and dissolved in 1–2 ml of high–ionic strength buffer. This crude fraction, designated as actomyosin, was made in 10 mM MgATP to dissociate actin from myosin and clarified by centrifugation at 100,000 × g for 45 min. The clear solution was dialyzed to remove traces of MgATP. To obtain a well-distributed suspension of myosin filaments, we reduced the salt concentration during the final dilution to 150 mM or by dialysis into 150 mM KCl, 0.1 mM EGTA, 2 mM MgCl2, and 10 mM 3-(N-morpholino)propanesulfonic acid, pH 7.0.
In vitro binding assays
Protein concentrations were determined by running standards on SDS polyacrylamide gels or by the bicinchoninic acid protein determination method. To assess binding of NMMIIA, we dialyzed rod filaments (residues 1338–1960) to GST–gelsolin full-length (G1–G6), GST–gelsolin N-terminal half (G1–G3), or GST–gelsolin C-terminal half (G4–G6) proteins against assembly buffer (20 mM Tris, pH 7.5, 20 mM NaCl, 2 mM MgCl2, 1 mM DTT) overnight before binding assays. Various concentrations of purified GST-gelsolin proteins (0.25–6 μM) were incubated with assembled NMMIIA rods (1 μM) at 23ºC for 45 min in the reaction buffer containing 20 mM Tris-HCl, pH 7.5, 20 mM NaCl, 2 mM MgCl2, 0.3 mM CaCl2, and 1 mM DTT. Samples were centrifuged for 15 min at 80,000 rpm. Supernatant and pellet samples were separated by SDS–PAGE and stained with Coomassie blue. Similar experiments were done with GST-gelsolin proteins bound to glutathione–Sepharose in the presence of Ca2+ (0.3 mM) or in the presence of EGTA (2 mM).
Actin-severing, -nucleation, and -capping assays
Lyophilized rabbit skeletal muscle actin or human platelet nonmuscle actin (both from Cytoskeleton) were resuspended in G-buffer containing actin monomers (0.3 μM; 30% pyrenyl-G-actin) and was polymerized in polymerization buffer for 2 h at room temperature. The decrease in fluorescence due to actin severing by cell proteins was measured in a fluorimeter (PTI; excitation, 365 nm; emission, 386 nm). For assessment of actin polymerization, lyophilized actin (Cytoskeleton) was resuspended and dialyzed in G-buffer (2 mM Tris, pH 8.0, 0.1 mM CaCl2, 0. 2 mM ATP, 0.5 mM β-mercaptoethanol) overnight. G-actin (2 μM; 15% pyrenyl–G-actin) was treated with polymerization buffer (25 mM Tris, pH 7.0, 50 mM KCl, 2 mM MgCl2, 0.1 mM ATP). The increase in fluorescence was measured in a PTI fluorimeter (excitation, 365 nm; emission, 386 nm).
We measured the severing activity of lysates prepared from gelsolin wild-type and null cells that were previously transfected with NMMIIA siRNA or a control siRNA. Cell lysates were collected with detergent plus protease inhibitors in buffer containing 50 mM KCl, 2 mM MgCl2, 0.5 mM ATP, 2 mM Tris, pH 8.0, 1 mM EGTA, and 1% Triton X-100. Lysates were dialysed with several changes of buffer containing 2 mM MgCl2, 50 mM KCl, 2 mM Tris-HCl, and 1 mM EGTA and 0.5 mM β-mercaptoethanol. Purified gelsolin and gelsolin incubated with various concentrations of NMMIIA protein were incubated with pyrene-labeled actin filaments in F-buffer (4 mM Tris, pH 8.0, 50 mM KCl, 2 mM MgCl2, and 1 mM CaCl2), and the rate of fluorescence loss was compared with that for gelsolin.
Immunofluorescence and confocal and total internal reflection fluorescence microscopy
We determined the spatial distribution of endogenous gelsolin and NMMIIA with respect to bound collagen beads. Cells were incubated with collagen-coated beads, fixed with 3% formaldehyde in PBS, permeabilized with 0.2% Triton X-100, and immunostained. The spatial distribution of staining around beads was determined by confocal microscopy (Leica, Heidelberg, Germany; 40× oil immersion lens). Transverse optical sections were obtained at 0.5-μm nominal thickness using Leica software and analyzed with Photoshop (Adobe, San Jose, CA). Colocalization was analyzed three to six fields per cell depending, on the number of bound beads by using the ImageJ plug-in (National Institutes of Health, Bethesda, MD; Bolte and Cordelieres, 2006). At least 20 cells were examined for each experimental condition. Pearson's r of colocalizing proteins was expressed as the mean ± SD. In some experiments we used total internal reflection fluorescence (Leica) microscopy to selectively illuminate fluorophores within ∼100 nm of the interface of collagen bead attachments to the cell surface.
Intracellular Ca2+ measurements
Cells were loaded with Fura-2/AM (3 μM) or with FFP-18 (5 μM; Teflabs, Austin, TX) and plated on collagen-coated beads or collagen-coated planar substrates. Intracellular Ca2+ concentration ([Ca2+]i) in single, attached cells was estimated with a microscope-based, ratio fluorimeter (Arora et al., 1994). In some experiments that involved Ca2+ chelation, cells were treated with BAPTA/AM (2 μM) for 30 min before experiments. In experiments to examine the influence of stretch-sensitive channels on collagen-induced increases of [Ca2+]i, GsMTx-4 (10 μM; Suchyna et al., 2000) was used.
Statistical analysis
For all continuous-variable data, means and SDs were computed. When appropriate, comparisons between two samples were made by Student's t test with statistical significance set at p < 0.05. For multiple comparisons, analysis of variance was used. All experiments were performed at least three times in triplicate.
Supplementary Material
Acknowledgments
Research was supported by a Canadian Institutes of Health Research operating grant to C.A.M. (MOP-36332), whose salary is provided by a Canada Research Chair (Tier 1).
Abbreviations used:
- [Ca2+]i
free intracellular calcium ion concentration
- EGTA
ethylene glycol tetraacetic acid
- GsMTx-4
Grammostola spatulata mechanotoxin 4
- GST
glutathione S-transferase
- NMMIIA
nonmuscle myosin IIA
- Q
quiescent cells
- S
spreading cells
- WT
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-10-0754) on January 16, 2013.
REFERENCES
- Alexandrova AY, Arnold K, Schaub S, Vasiliev JM, Meister JJ, Bershadsky AD, Verkhovsky AB. Comparative dynamics of retrograde actin flow and focal adhesions: formation of nascent adhesions triggers transition from fast to slow flow. PLoS One. 2008;3:e3234. doi: 10.1371/journal.pone.0003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Bibby KJ, McCulloch CA. Slow oscillations of free intracellular calcium ion concentration in human fibroblasts responding to mechanical stretch. J Cell Physiol. 1994;161:187–200. doi: 10.1002/jcp.1041610202. [DOI] [PubMed] [Google Scholar]
- Arora PD, Chan MW, Anderson RA, Janmey PA, McCulloch CA. Separate functions of gelsolin mediate sequential steps of collagen phagocytosis. Mol Biol Cell. 2005;16:5175–5190. doi: 10.1091/mbc.E05-07-0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Conti MA, Ravid S, Sacks DB, Kapus A, Adelstein RS, Bresnick AR, McCulloch CA. Rap1 activation in collagen phagocytosis is dependent on nonmuscle myosin II-A. Mol Biol Cell. 2008a;19:5032–5046. doi: 10.1091/mbc.E08-04-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Fan L, Sodek J, Kapus A, McCulloch CA. Differential binding to dorsal and ventral cell surfaces of fibroblasts: effect on collagen phagocytosis. Exp Cell Res. 2003;286:366–380. doi: 10.1016/s0014-4827(03)00096-x. [DOI] [PubMed] [Google Scholar]
- Arora PD, Glogauer M, Kapus A, Kwiatkowski DJ, McCulloch CA. Gelsolin mediates collagen phagocytosis through a rac-dependent step. Mol Biol Cell. 2004;15:588–599. doi: 10.1091/mbc.E03-07-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Marignani PA, McCulloch CA. Collagen phagocytosis is regulated by the guanine nucleotide exchange factor Vav2. Am J Physiol Cell Physiol. 2008b;295:C130–C137. doi: 10.1152/ajpcell.00168.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora PD, Wang Y, Janmey PA, Bresnick A, Yin HL, McCulloch CA. Gelsolin and non-muscle myosin IIA interact to mediate calcium-regulated collagen phagocytosis. J Biol Chem. 2011;286:34184–34198. doi: 10.1074/jbc.M111.247783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microscopy. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, Lippincott-Schwartz J. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 2011;13:371–381. doi: 10.1038/ncb2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Burtnick LD, Urosev D, Irobi E, Narayan K, Robinson RC. Structure of the N-terminal half of gelsolin bound to actin: roles in severing, apoptosis and FAF. EMBO J. 2004;23:2713–2722. doi: 10.1038/sj.emboj.7600280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss F, Luzio JP, Kendrick-Jones J. Myosin VI, an actin motor for membrane traffic and cell migration. Traffic. 2002;3:851–858. doi: 10.1034/j.1600-0854.2002.31201.x. [DOI] [PubMed] [Google Scholar]
- Cannon GJ, Swanson JA. The macrophage capacity for phagocytosis. J Cell Sci. 1992;101:907–913. doi: 10.1242/jcs.101.4.907. [DOI] [PubMed] [Google Scholar]
- Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti MA, Adelstein RS. Nonmuscle myosin II moves in new directions. J Cell Sci. 2008;121:11–18. doi: 10.1242/jcs.007112. [DOI] [PubMed] [Google Scholar]
- Curtis AS, Wilkinson CD. Reactions of cells to topography. J Biomater Sci Polym Ed. 1998;9:1313–1329. doi: 10.1163/156856298x00415. [DOI] [PubMed] [Google Scholar]
- Dahan I, Yearim A, Touboul Y, Ravid S. The tumor suppressor Lgl1 regulates NMII-A cellular distribution and focal adhesion morphology to optimize cell migration. Mol Biol Cell. 2011;23:591–601. doi: 10.1091/mbc.E11-01-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulyaninova NG, Malashkevich VN, Almo SC, Bresnick AR. Regulation of myosin-IIA assembly and Mts1 binding by heavy chain phosphorylation. Biochemistry. 2005;44:6867–6876. doi: 10.1021/bi0500776. [DOI] [PubMed] [Google Scholar]
- Everts V, van der Zee E, Creemers L, Beertsen W. Phagocytosis and intracellular digestion of collagen, its role in turnover and remodelling. Histochem J. 1996;28:229–245. doi: 10.1007/BF02409011. [DOI] [PubMed] [Google Scholar]
- Fraley SI, Feng Y, Krishnamurthy R, Kim DH, Celedon A, Longmore GD, Wirtz D. A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat Cell Biol. 2010;12:598–604. doi: 10.1038/ncb2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- Geiger B, Yamada KM. Molecular architecture and function of matrix adhesions. Cold Spring Harb Perspect Biol. 2011;3:005033. doi: 10.1101/cshperspect.a005033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glogauer M, Arora P, Chou D, Janmey PA, Downey GP, McCulloch CA. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. J Biol Chem. 1998;273:1689–1698. doi: 10.1074/jbc.273.3.1689. [DOI] [PubMed] [Google Scholar]
- Glogauer M, Ferrier J, McCulloch CA. Magnetic fields applied to collagen-coated ferric oxide beads induce stretch-activated Ca2+ flux in fibroblasts. Am J Physiol. 1995;269:C1093–C1104. doi: 10.1152/ajpcell.1995.269.5.C1093. [DOI] [PubMed] [Google Scholar]
- Grinnell F. Fibroblast biology in three-dimensional collagen matrices. Trends Cell Biol. 2003;13:264–269. doi: 10.1016/s0962-8924(03)00057-6. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Geiger B. Interaction of fibronectin-coated beads with attached and spread fibroblasts. Binding, phagocytosis, and cytoskeletal reorganization. Exp Cell Res. 1986;162:449–461. doi: 10.1016/0014-4827(86)90349-6. [DOI] [PubMed] [Google Scholar]
- Groves E, Dart AE, Covarelli V, Caron E. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell Mol Life Sci. 2008;65:1957–1976. doi: 10.1007/s00018-008-7578-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar JG, Janmey PA, Almo SC, Chance MR. Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc Natl Acad Sci USA. 2003;100:3942–3947. doi: 10.1073/pnas.0736004100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubow KE, Horwitz AR. Reducing background fluorescence reveals adhesions in 3D matrices. Nat Cell Biol. 2011;13:3–5. doi: 10.1038/ncb0111-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DJ, Stossel TP, Orkin SH, Mole JE, Colten HR, Yin HL. Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature. 1986;323:455–458. doi: 10.1038/323455a0. [DOI] [PubMed] [Google Scholar]
- Lee J, Ishihara A, Oxford G, Johnson B, Jacobson K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature. 1999;400:382–386. doi: 10.1038/22578. [DOI] [PubMed] [Google Scholar]
- Li ZH, Spektor A, Varlamova O, Bresnick AR. Mts1 regulates the assembly of nonmuscle myosin-IIA. Biochemistry. 2003;42:14258–14266. doi: 10.1021/bi0354379. [DOI] [PubMed] [Google Scholar]
- McCulloch CA. Drug-induced fibrosis: interference with the intracellular collagen degradation pathway. Curr Opin Drug Discov Devel. 2004;7:720–724. [PubMed] [Google Scholar]
- McCulloch CA, Knowles GC. Deficiencies in collagen phagocytosis by human fibroblasts in vitro: a mechanism for fibrosis. J Cell Physiol. 1993;155:461–471. doi: 10.1002/jcp.1041550305. [DOI] [PubMed] [Google Scholar]
- Melcher AH, Chan J. Phagocytosis and digestion of collagen by gingival fibroblasts in vivo: a study of serial sections. J Ultrastruct Res. 1981;77:1–36. doi: 10.1016/s0022-5320(81)80064-0. [DOI] [PubMed] [Google Scholar]
- Murphy G, Nagase H. Progress in matrix metalloproteinase research. Mol Aspects Med. 2008;29:290–308. doi: 10.1016/j.mam.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy S, Ricca BL, Norstrom MF, Courson DS, Brawley CM, Smithback PA, Rock RS. A myosin motor that selects bundled actin for motility. Proc Natl Acad Sci USA. 2008;105:9616–9620. doi: 10.1073/pnas.0802592105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan K, Chumnarnsilpa S, Choe H, Irobi E, Urosev D, Lindberg U, Schutt CE, Burtnick LD, Robinson RC. Activation in isolation: exposure of the actin-binding site in the C-terminal half of gelsolin does not require actin. FEBS Lett. 2003;552:82–85. doi: 10.1016/s0014-5793(03)00933-5. [DOI] [PubMed] [Google Scholar]
- Perez-Tamayo R. Pathology of collagen degradation. A review. Am J Pathol. 1978;92:508–566. [PMC free article] [PubMed] [Google Scholar]
- Pikul S, et al. Discovery of potent, achiral matrix metalloproteinase inhibitors. J Med Chem. 1998;41:3568–3571. doi: 10.1021/jm980253r. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Borisy GG. Cellular motility driven by assembly and disassembly of actin filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- Pope B, Way M, Weeds AG. Two of the three actin-binding domains of gelsolin bind to the same subdomain of actin. Implications of capping and severing mechanisms. FEBS Lett. 1991;280:70–74. doi: 10.1016/0014-5793(91)80206-i. [DOI] [PubMed] [Google Scholar]
- Puius YA, Fedorov EV, Eichinger L, Schleicher M, Almo SC. Mapping the functional surface of domain 2 in the gelsolin superfamily. Biochemistry. 2000;39:5322–5331. doi: 10.1021/bi992364d. [DOI] [PubMed] [Google Scholar]
- Serrander L, Skarman P, Rasmussen B, Witke W, Lew DP, Krause KH, Stendahl O, Nusse O. Selective inhibition of IgG-mediated phagocytosis in gelsolin-deficient murine neutrophils. J Immunol. 2000;165:2451–2457. doi: 10.4049/jimmunol.165.5.2451. [DOI] [PubMed] [Google Scholar]
- Singer II, Kawka DW, Kazazis DM, Clark RA. In vivo co-distribution of fibronectin and actin fibers in granulation tissue: immunofluorescence and electron microscope studies of the fibronexus at the myofibroblast surface. J Cell Biol. 1984;98:2091–2106. doi: 10.1083/jcb.98.6.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stendahl O, Krause KH, Krischer J, Jerstrom P, Theler JM, Clark RA, Carpentier JL, Lew DP. Redistribution of intracellular Ca2+ stores during phagocytosis in human neutrophils. Science. 1994;265:1439–1441. doi: 10.1126/science.8073285. [DOI] [PubMed] [Google Scholar]
- Suchyna TM, Johnson JH, Hamer K, Leykam JF, Gage DA, Clemo HF, Baumgarten CM, Sachs F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J Gen Physiol. 2000;115:583–598. doi: 10.1085/jgp.115.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda EL, Melcher AH, Brunette DM. Stereological study of collagen phagocytosis by cultured periodontal ligament fibroblasts: time course and effect of deficient culture medium. J Ultrastruct Res. 1979;68:195–208. doi: 10.1016/s0022-5320(79)90154-0. [DOI] [PubMed] [Google Scholar]
- Takemura R, Stenberg PE, Bainton DF, Werb Z. Rapid redistribution of clathrin onto macrophage plasma membranes in response to Fc receptor-ligand interaction during frustrated phagocytosis. J Cell Biol. 1986;102:55–69. doi: 10.1083/jcb.102.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tenCate AR. Morphological studies of fibrocytes in connective tissue undergoing rapid remodelling. J Anat. 1972;112:401–414. [PMC free article] [PubMed] [Google Scholar]
- Trotter JA, Adelstein RS. Macrophage myosin. Regulation of actin-activated ATPase, activity by phosphorylation of the 20,000-dalton light chain. J Biol Chem. 1979;254:8781–8785. [PubMed] [Google Scholar]
- Vicente-Manzanares M, Horwitz AR. Adhesion dynamics at a glance. J Cell Sci. 2011;124:3923–3927. doi: 10.1242/jcs.095653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Ko KS, Kapus A, McCulloch CA, Ellen RP. A spirochete surface protein uncouples store-operated calcium channels in fibroblasts: a novel cytotoxic mechanism. J Biol Chem. 2001;276:23056–23064. doi: 10.1074/jbc.M011735200. [DOI] [PubMed] [Google Scholar]
- Way M, Gooch J, Pope B, Weeds AG. Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J Cell Biol. 1989;109:593–605. doi: 10.1083/jcb.109.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeds A, Maciver S. F-actin capping proteins. Curr Opin Cell Biol. 1993;5:63–69. doi: 10.1016/s0955-0674(05)80009-2. [DOI] [PubMed] [Google Scholar]
- Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- Yin HL. Gelsolin: calcium- and polyphosphoinositide-regulated actin-modulating protein. Bioessays. 1987;7:176–179. doi: 10.1002/bies.950070409. [DOI] [PubMed] [Google Scholar]
- Yin HL, Albrecht JH, Fattoum A. Identification of gelsolin, a Ca2+-dependent regulatory protein of actin gel-sol transformation, and its intracellular distribution in a variety of cells and tissues. J Cell Biol. 1981;91:901–906. doi: 10.1083/jcb.91.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HL, Stossel TP. Control of cytoplasmic actin gel-sol transformation by gelsolin, a calcium-dependent regulatory protein. Nature. 1979;281:583–586. doi: 10.1038/281583a0. [DOI] [PubMed] [Google Scholar]
- Zimmerli S, Majeed M, Gustavsson M, Stendahl O, Sanan DA, Ernst JD. Phagosome-lysosome fusion is a calcium-independent event in macrophages. J Cell Biol. 1996;132:49–61. doi: 10.1083/jcb.132.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.