

Tbc1d1 is involved in AICAR-dependent GLUT4 liberation. Tbc1d1 acquires temporal insulin responsiveness with AICAR pretreatment. This shift in regulatory mode requires Ser- 237 phosphorylation and the PTB1 domain. PTB1 mutants exhibit no shift in regulatory mode and thus no insulin responsiveness.
Abstract
Tbc1d1 is key to skeletal muscle GLUT4 regulation. By using GLUT4 nanometry combined with a cell-based reconstitution model, we uncover a shift in the regulatory mode of Tbc1d1 by showing that Tbc1d1 temporally acquires insulin responsiveness, which triggers GLUT4 trafficking only after an exercise-mimetic stimulus such as aminoimidazole carboxamide ribonucleotide (AICAR) pretreatment. The functional acquisition of insulin responsiveness requires Ser-237 phosphorylation and an intact phosphotyrosine-binding (PTB) 1 domain. Mutations in PTB1, including R125W (a natural mutant), thus result in complete loss of insulin-responsiveness acquisition, whereas AICAR-responsive GLUT4-liberation activity remains intact. Thus our data provide novel insights into temporal acquisition/memorization of Tbc1d1 insulin responsiveness, relying on the PTB1 domain, possibly a key factor in the beneficial effects of exercise on muscle insulin potency.
INTRODUCTION
Insulin and exercise are two important physiological stimuli of GLUT4 translocation from its intracellular storage compartment(s) to the cell surface (Goodyear and Kahn, 1998). The upstream signaling pathways of these two stimuli differ from each other: insulin's actions are mediated by phosphatidylinositol 3-kinase/Akt, whereas exercise might involve AMP-activated protein kinase (AMPK). Therefore these two biochemical signaling pathways should converge somewhere in order to be deciphered into the physical process of GLUT4 translocation. Recent studies suggest that two tre-2/USP6, BUB2, cdc16 domain (Tbc1d)–family Rab GTPase–activating proteins (RabGAPs)—the Akt substrate of 160 kDa (AS160)/Tbc1d4 (hereafter AS160) and Tbc1d1—act as convergence sites for insulin- and exercise-responsive GLUT4 translocation. These proteins share ∼50% identical amino acid sequences with similar domain structures, such as two phosphotyrosine-binding (PTB) domains, a putative calmodulin-binding domain (CBD), a RabGAP domain (Roach et al., 2007; Park et al., 2011), and multiple Akt-phosphorylation sites (Sano et al., 2003; Peck et al., 2009). The substrate specificities of these RabGAPs in vitro are also similar. One notable difference between these two proteins is tissue distribution: AS160 is expressed in multiple tissues at similar levels, whereas Tbc1d1 is highly expressed in skeletal muscle as compared with other tissues (Taylor et al., 2008; Castorena et al., 2011). Another key difference is the presence of an AMPK-phosphorylation site (Ser-237) in Tbc1d1 (Nedachi et al., 2008; Taylor et al., 2008; Vichaiwong et al., 2010), which apparently contributes to the unique regulatory aspects of Tbc1d1, such as contraction-induced glucose uptake. Studies such as these have provided a detailed understanding of the biochemical properties of AS160 and Tbc1d1, but the functional aspects of these proteins remain ambiguous due to the technical limitations of traditional biochemical assays for analyzing GLUT4 trafficking, which can assess only cell surface amounts of GLUT4 and cannot directly describe intracellular GLUT4 behavior.
To overcome the methodological limitations, we recently developed a novel nanometrological method for quantifying intracellular GLUT4 behavior based on single-molecule imaging with quantum dot (QD) fluorescent nanocrystals, which can dissect the intracellular GLUT4-trafficking process into discrete, experimentally traceable steps (Fujita et al., 2010; Hatakeyama and Kanzaki, 2011). With this approach, we revealed the “static retention” property governed by sortilin to be the major sequestration mechanism for GLUT4 (Hatakeyama and Kanzaki, 2011). Moreover, we clearly identified insulin-responsive “liberation” of static GLUT4 as a major functional aspect of insulin action and also demonstrated that AS160 governed this process in an Akt-mediated phosphorylation-dependent manner (Fujita et al., 2010). Of importance, by virtue of our GLUT4 nanometry, we successfully established an “experimental reconstitution model” of mature insulin-responsive GLUT4 trafficking systems in 3T3L1 fibroblasts by exogenously expressing only two proteins—sortilin and AS160. This cell-based reconstitution model has contributed to analyzing regulatory mechanisms of intracellular GLUT4 behavior in further detail by focusing on specific trafficking processes, which cannot be done with traditional biochemical assays.
Unlike AS160, the functional roles of its closest relative, Tbc1d1, remain uncertain. A major obstacle in Tbc1d1 functional research is its predominant expression in skeletal muscles, making it difficult to investigate GLUT4 regulation. Revealing Tbc1d1 function would presumably be important for understanding both physiological and pathophysiological glucose homeostasis, since the R125W mutation of Tbc1d1 can cause severe obesity (Stone et al., 2006; Meyre et al., 2008), and this mutation reportedly decreases insulin-dependent glucose transport (An et al., 2010). Therefore, in the present study, by using GLUT4 nanometry combined with a cell-based experimental reconstitution model, we examine the functional roles of Tbc1d1 in great detail, focusing especially on GLUT4-liberation processes. Our findings reveal a unique memorization ability for insulin-responsive activity, relying on the PTB1 domain of Tbc1d1. This might explain the defect caused by natural mutations such as R125W.
RESULTS
Tbc1d1 serves as a key regulator of aminoimidazole carboxamide ribonucleotide–induced GLUT4 liberation
With QD-based GLUT4 nanometry, we can track the movements of individual intracellular GLUT4 molecules with high precision (Figure 1, A and B). Using this approach, we describe intracellular GLUT4 behavior based on two parameters—velocities and representative diffusion coefficient maps (Figure 1, C–E). The calculations of these values were performed as previously described (Fujita et al., 2010; Hatakeyama and Kanzaki, 2011; see Materials and Methods). Note that rightward shift of the velocity distribution or increases in spots having hotter colors in representative diffusion coefficient maps indicate increases in fast-moving molecules. With this approach, we reconfirmed that the static GLUT4 storage compartment was generated by exogenous sortilin expression and found that the resulting stationary GLUT4 behavior was not influenced by additional coexpression of Tbc1d1, whereas expression of Tbc1d1 alone generated no static GLUT4 storage compartment (Figure 1F and Supplemental Figure S1A). Because the calculated velocities are “apparent” values representing a combination of true movement and inevitable instrumental noise, instead of raw data we use the “corrected” velocities, obtained by subtracting apparent velocities in fixed cells in the figures, to represent mean velocities (see Materials and Methods).
FIGURE 1:
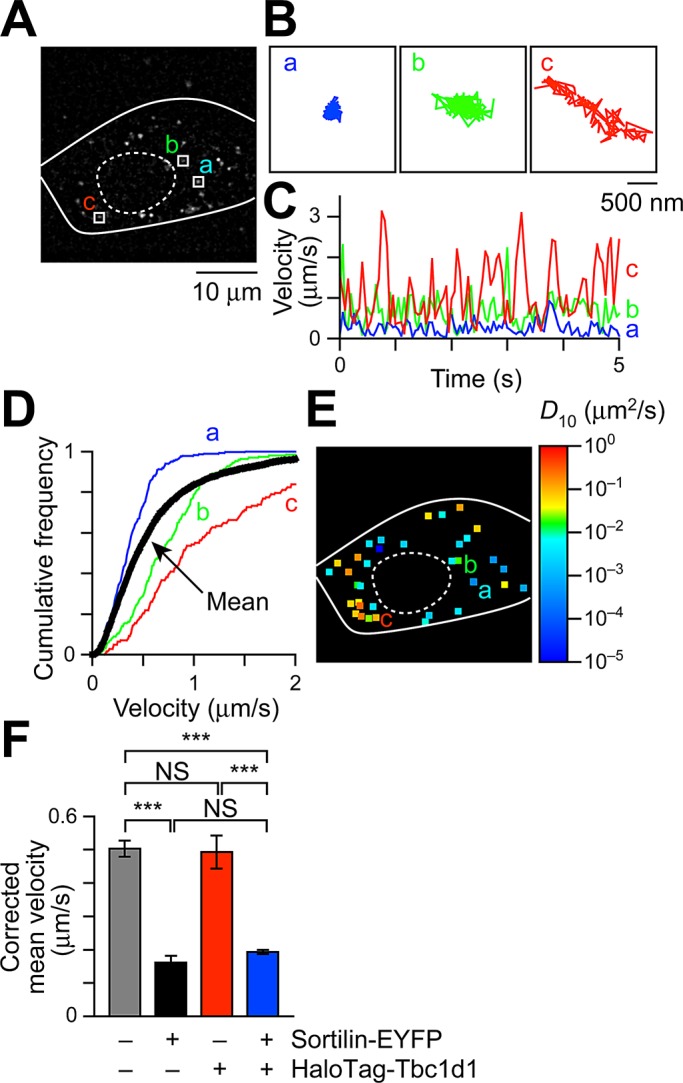
Intracellular GLUT4 nanometry based on single-molecule imaging. (A) Snapshot of QD fluorescence in 3T3L1 fibroblasts. Trajectories (B), velocity vs. time plots (C), and cumulative velocity distributions (D) of three particles shown in A. In D, the mean velocity distribution calculated from all particles in the cell is also shown (black), with error bars omitted for clarity. Mean velocities (in μm/s) of the three particles shown in A are 0.38 (a), 0.50 (b), and 1.13 (c), and the mean velocity of all particles within the cell is 0.62 μm/s. (E) Diffusion coefficient (D10) map in a cell shown in A. The three particles shown in A are also indicated. Right, pseudocolor coding used to represent the diffusion coefficients of the molecules estimated by linear fitting of MSD values (see Materials and Methods). (F) Mean velocities in cells expressing the indicating proteins. The values were obtained from 6–84 cells. ***p < 0.001 by Tukey–Kramer multiple comparison.
Using fibroblasts exogenously expressing both sortilin and Tbc1d1 (Supplemental Figure S1E), we first analyzed insulin effects and found that insulin failed to liberate GLUT4 from its static state (Figure 2, A and D), which differed from the response of fibroblasts expressing sortilin and AS160 (Hatakeyama and Kanzaki, 2011). Instead, AMPK activators, either aminoimidazole carboxamide ribonucleotide (AICAR) or A-769642, induced GLUT4 liberation, displaying rightward shift of the velocity distribution and increases in spots with hotter colors in diffusion coefficient maps (Figure 2, B and D, and Supplemental Figure S1B; see also Figure 3H), along with increased phosphorylation of Ser-237 (AMPK site), but not with that of Thr-596 (Akt site; Figure 2E). In contrast, insulin increased Thr-596, but not Ser-237, phosphorylation (Figure 2E). We also found that a forced increase of intracellular Ca2+ concentration ([Ca2+]i) by ionomycin induced no significant GLUT4 liberation (Figure 2, C and D), despite this treatment augmenting phosphorylation of Ser-237 but not Thr-596 (Figure 2E) as in AICAR-treated cells. AICAR-induced GLUT4 liberation was dependent on exogenously expressed Tbc1d1, as there was no liberation in fibroblasts expressing sortilin alone (Supplemental Figure S1C). Consistent with our previous observations (Hatakeyama and Kanzaki, 2011), insulin also failed to liberate static GLUT4 in these fibroblasts (Supplemental Figure S1D). These results indicate that fibroblasts have no functionally significant expression of either AS160 or Tbc1d1, at least in terms of GLUT4-liberation activity.
FIGURE 2:
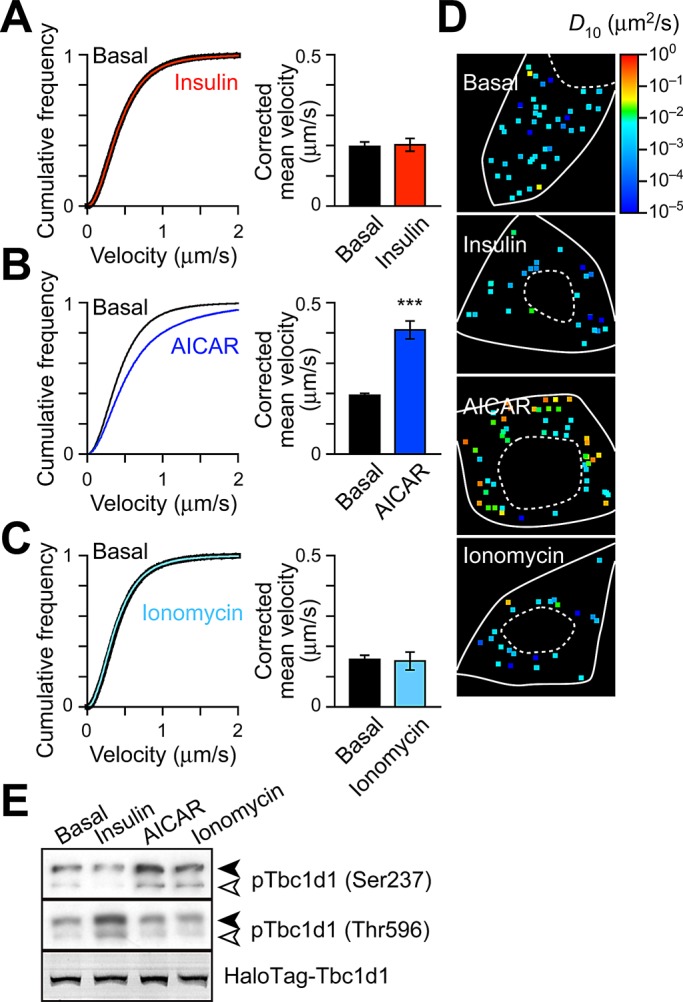
Tbc1d1 mediates AICAR-dependent GLUT4 liberation. Velocity distributions (left) and corrected mean velocities (right) of GLUT4 movement (A) before (black) and after (red) insulin stimulation (100 nM, 30 min, n = 12), (B) without (black, n = 67) or with (blue, n = 39) AICAR (1 mM, 30 min), and (C) before (black) and after (cyan) ionomycin treatment (0.1 μg/ml, 10 min, n = 4). ***p< 0.001. (D) Representative diffusion coefficient maps of GLUT4 movement in cells under the indicated conditions. (E) Tbc1d1 phosphorylation induced by insulin (100 nM, 5 min), AICAR (1 mM, 30 min), or ionomycin (0.1 μg/ml, 5 min). Solid and open arrowheads represent exogenously expressed HaloTag-Tbc1d1 and endogenous Tbc1d1, respectively. Exogenously expressed HaloTag-Tbc1d1 was also detected by labeling with fluorescent HaloTag ligand.
FIGURE 3:
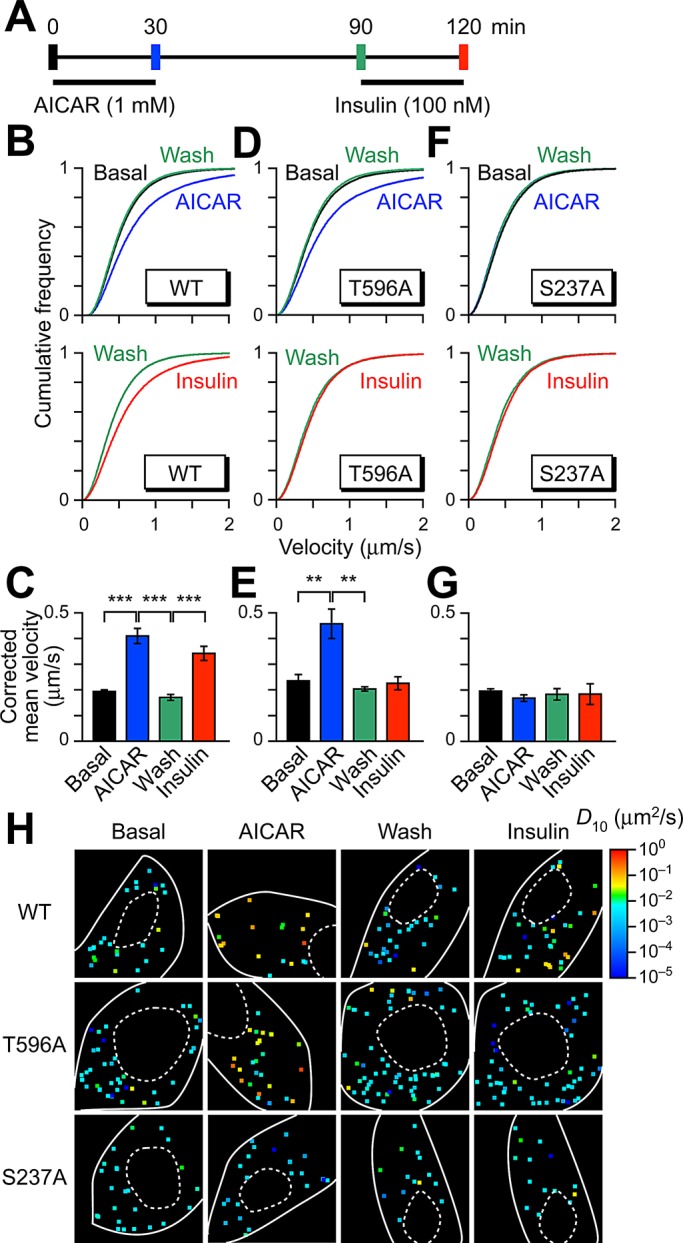
Acquisition of insulin responsiveness of Tbc1d1 by sequential treatment with AICAR and insulin. (A) Treatment protocol. Imaging was performed at the times indicated by vertical bars. Mean velocity distributions (B, D, and F) and corrected mean velocities (C, E, and G) of GLUT4 movement in cells expressing wild-type (B, C), T596A (D, E), and S237A (F, G) Tbc1d1. In B and C, the data in basal and AICAR-treated cells are the same as in Figure 2B. Data were obtained from at least three cells. **p < 0.01, ***p < 0.001. Statistical analyses were performed with Tukey–Kramer multiple comparison. (H) Representative diffusion coefficient maps of GLUT4 movement in cells under the indicated conditions.
Tbc1d1 acquires its insulin-responsive GLUT4-liberation ability after AICAR pretreatment
We next examined whether sequential multiple stimuli liberated GLUT4 in a Tbc1d1-dependent manner (Figure 3A). Intriguingly, we found that, although insulin alone failed to release static GLUT4 as noted earlier (Figure 2A), insulin did trigger GLUT4 liberation when Tbc1d1-expressing cells had been treated previously with AICAR (Figure 3, B, C, and H). The static GLUT4 behavior was restored at 1 h after AICAR removal (Figure 3, B, C, and H), presumably due to sortilin-mediated retrograde trafficking of GLUT4 (Hatakeyama and Kanzaki, 2011). Although the static characteristics of their GLUT4-inducing behaviors were indistinguishable, insulin displayed a potent ability to liberate GLUT4 only in the AICAR-exposed requiescent cells (Figure 3, B, C, and H; cf. Figure 2A), and this insulin action was still observed even 3 h after AICAR withdrawal (unpublished data). Acquired insulin responsiveness was abolished by wortmannin, a phosphatidylinositol 3-kinase inhibitor (Supplemental Figure S2), and a Tbc1d1/T596A mutant displayed no insulin-responsive GLUT4 liberation (Figure 3, D, E, and H). However, this mutant was fully functional in terms of introductory AICAR-induced GLUT4 liberation (Figure 3, D, E, and H).
On the other hand, a Tbc1d1/S237A mutant completely lacked GLUT4-liberation activity regardless of the stimuli used, including combinatorial (Figure 3, F–H), indicating phosphorylation of Ser-237 to be crucial not only for AICAR-dependent GLUT4 liberation, but also for AICAR-induced temporal acquisition of insulin-responsive activity. We confirmed that AICAR-induced Ser-237 phosphorylation was restored to basal levels 1 h after AICAR removal and that insulin-induced Thr-596 phosphorylation was not significantly influenced by AICAR treatment (Figure 4). In addition, mutational analysis of other potential phosphorylation sites around the crucial Thr-596, including Ser-565 and Ser-566 (Peck et al., 2009), demonstrated that these serine residues are irrelevant to GLUT4-liberation activity (unpublished data), although the possibility that other phosphorylation sites are involved cannot be ruled out. It should be noted that the acquisition of insulin responsiveness was observed in the Tbc1d1-expressing fibroblasts treated with AS160 siRNA (Supplemental Figure S3).
FIGURE 4:
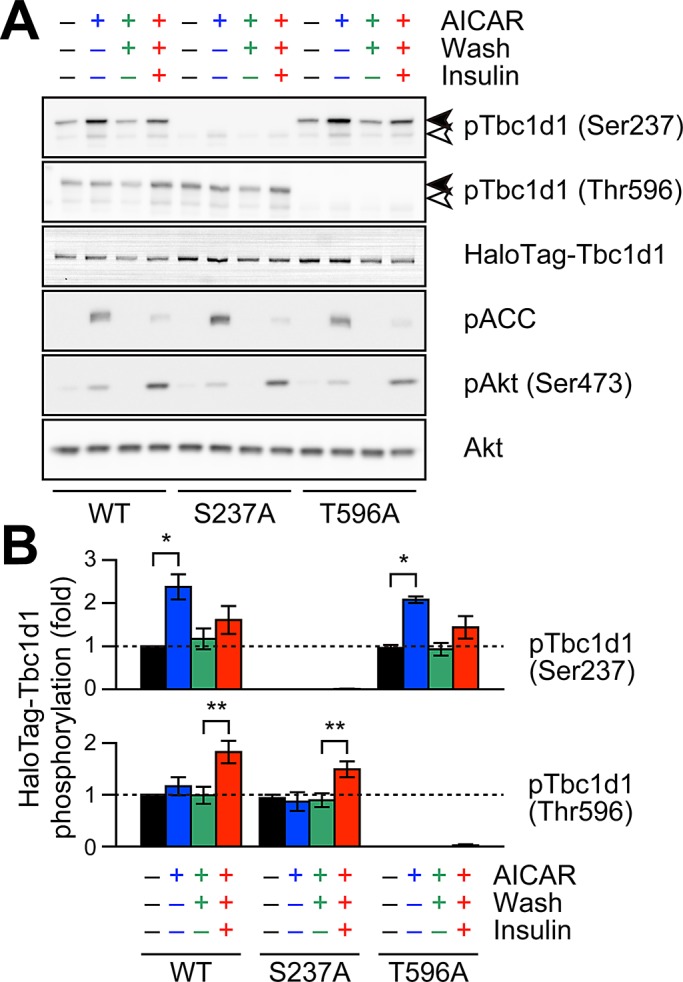
Effects of Tbc1d1 mutants on AICAR- or insulin-induced phosphorylation. Representative Western blotting (A) and quantification (B) of HaloTag-Tbc1d1 phosphorylation. In A, pACC and pAkt are also shown in order to confirm the activations of AMPK and insulin signaling pathways. In B, the values were calculated from three independent assays. The data are shown as ratios (phosphorylation/total) and as fold increases compared with untreated wild type. *p < 0.05, **p < 0.01.
Taken together, the results indicate that phosphorylation processes of both Ser-237 and Thr-596, even if sequential rather than simultaneous, appeared to be crucial for acquiring insulin-responsive activity. More specifically, phosphorylation of Ser-237 was essential for endowing Tbc1d1 with insulin responsiveness, whereas subsequent insulin-induced GLUT4 liberation required phosphorylation of Thr-596, but not Ser-237, under these specific conditions.
Repackaging into a distinct GLUT4-storage compartment is not necessary for insulin responsiveness
Tbc1d1 appears to become insulin responsive in certain situations. However, given that the initial AICAR treatment liberates GLUT4, resulting in its drastic redistribution, it is possible that after AICAR withdrawal the released GLUT4 was restored to a distinct compartment, one with the insulin-responsive property, different from that one in which GLUT4 was originally localized. Alternatively, it is also possible that Tbc1d1 somehow acquired insulin responsiveness after AICAR treatment. To address this important question, we showed that insulin was also capable of triggering Tbc1d1-dependent GLUT4 liberation when [Ca2+]i was temporally or simultaneously increased. For rapid and transient increases in [Ca2+]i, we applied photolysis of caged Ca2+ compound o-nitrophenyl ethylene glycol tetraacetic acid (NPE)–loaded cells (Figure 5A). The increases in [Ca2+]i alone failed to liberate GLUT4 at least for 10 min (Figure 5B), whereas with insulin, acute increases in [Ca2+]i released GLUT4 just 30 s after photolysis (Figure 5C, p < 0.001 by Kolmogorov–Smirnov test). Similarly, immediately after increases in [Ca2+]i, insulin stimulation liberated GLUT4 in wild type–expressing cells (Figure 5D) but not in T596A or S237A mutant cells (Supplemental Figure S4, A and B). The effect of [Ca2+]i in causing Tbc1d1 insulin responsiveness lasted for 5 min, and the insulin gradually lost this ability (Supplemental Figure S4, C and D). Ionomycin showed no significant changes in insulin-induced phosphorylation of Tbc1d1 (Supplemental Figure S4E).
FIGURE 5:
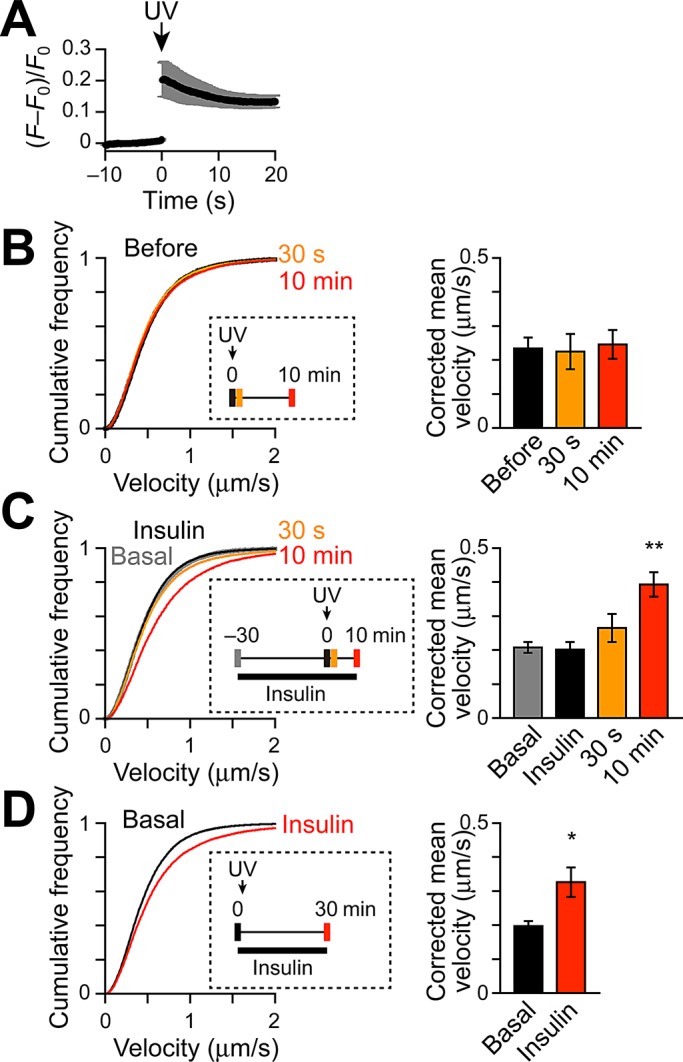
Acquisition of insulin responsiveness of Tbc1d1 by combined stimulation with Ca2+ and insulin. (A) Changes in [Ca2+]i induced by NPE photolysis in cells monitored by Ca2+ indicator fluo-4 fluorescence (F). Photolysis was induced at time 0 (UV, arrow). F0 represents mean fluorescence intensity before photolysis. Velocity distributions (left) and corrected mean velocities (right) of GLUT4 movement (B) before (black) and after (orange: 30 s; red: 10 min) NPE photolysis (n = 6), (C) basal (gray), insulin-stimulated (black), and after (orange: 30 s; red: 10 min) photolysis (n = 7), and (D) before (black) and after (red) insulin stimulation (100 nM, n = 6). Insets show detailed treatment and acquisition protocols. *p < 0.05 and ** p < 0.01 by Dunnett's multiple comparison vs. basal.
These observations show that drastic GLUT4 repackaging is not always necessary to endow insulin-responsive Tbc1d1 activity, although AICAR pretreatment endowed Tbc1d1 with much longer potency than did [Ca2+]i transients. Similar to AICAR-exposed cells, phosphorylation sites of both Ser-237 (AMPK site) and Thr-596 (Akt site) were crucial for generating the temporal insulin-responsive Tbc1d1 activity. It is intriguing, however, that Tbc1d1/W725G, a mutant version of the putative CBD in which AS160 possesses a similar CBD (Kane and Lienhard, 2005), exhibited no obvious defect (unpublished data), suggesting that the CBD was minimally involved, at least under these experimental conditions.
Temporal endowment of insulin-responsive potency requires intact PTB domain
Our GLUT4 nanometry monitoring of intracellular behaviors of single GLUT4 molecules provided compelling evidence that Tbc1d1 does indeed function as a key nexus deciphering biochemical phosphorylation events of Ser-237 and Thr-596, with multiple, even sequential, stimuli being involved in the physical process of GLUT4 liberation. However, our finding that their phosphorylation did not always need to be simultaneous to exert full liberation activity was rather surprising since phosphorylated Thr-596 alone, unlike phosphorylated Ser-237, was reportedly insufficient for 14-3-3 binding (Chen et al., 2008; Pehmoller et al., 2009), and previous studies on AS160 showed the importance of 14-3-3 association in GLUT4 regulation (Ramm et al., 2006; Chen et al., 2011; Koumanov et al., 2011).
To further explore molecular mechanisms underlying temporal endowment of insulin responsiveness, we attempted to identify responsible Tbc1d1 domain(s), other than these key phosphorylation sites, that might be involved in functional acquisition/memorization enabling Tbc1d1 insulin responsiveness. Mutational analysis of several major domains in Tbc1d1 revealed that the functional PTB domain is essential, since a point mutation in the putative PTB sequence in Tbc1d1 (Tbc1d1/F169V), believed to interact with NPxY asparagine (Zhou et al., 1995), showed no acquired ability to liberate GLUT4 in response to sequential insulin stimulation, despite introductory AICAR-dependent GLUT4 liberation being accomplished (Figure 6, A, B, and E). Insulin-responsive GLUT4 liberation with a combined [Ca2+]i transient was also impaired in the Tbc1d1/F169V mutant (Supplemental Figure S5, A and B). We also found that a natural mutation (R125W) in the same PTB domain of Tbc1d1 (Stone et al., 2006; Meyre et al., 2008) exhibits essentially the same defects in acquisition/memorization of insulin-responsive ability after AICAR-pretreatment as with the combinatorial increase of [Ca2+]i (Figure 6, C–E, and Supplemental Figure S5). Tbc1d1 phosphorylation profiles at Ser-237 and Thr-596 did not differ significantly among these mutants and wild type (Figure 7).
FIGURE 6:
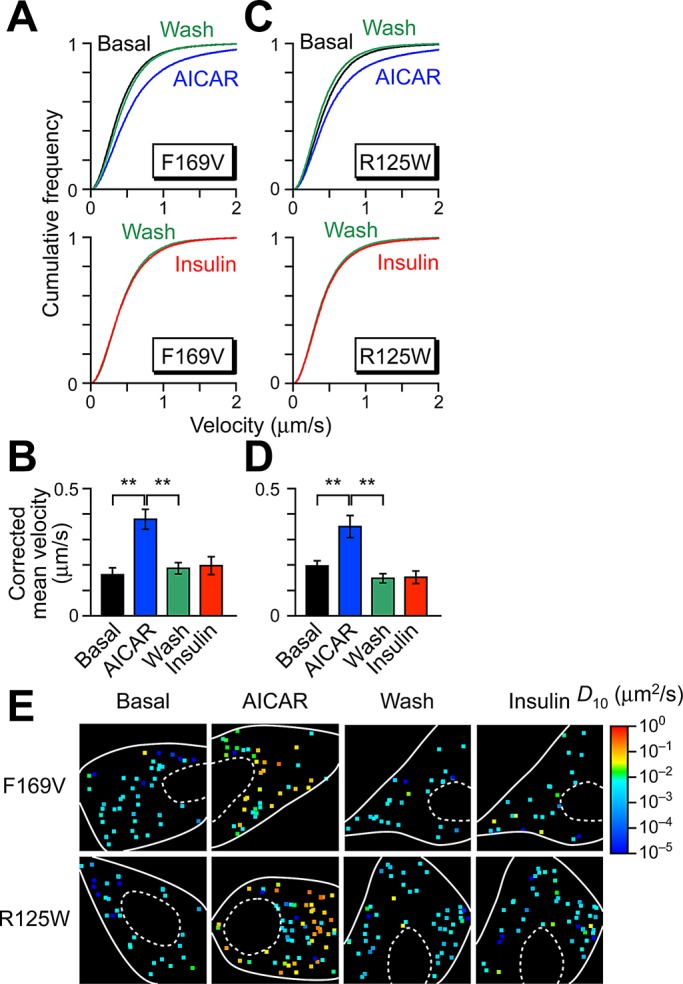
Intact PTB1 domain is necessary for temporal acquisition of insulin responsiveness. Sequential stimulation with AICAR and insulin in cells expressing F169V (A, B) and R125W (C, D). Stimulation protocols and colors are the same as in Figure 3. Data were obtained from at least four cells. **p < 0.01. Statistical analyses were performed with Tukey–Kramer multiple comparison. (E) Representative diffusion coefficient maps of GLUT4 movement in cells under indicated conditions.
FIGURE 7:
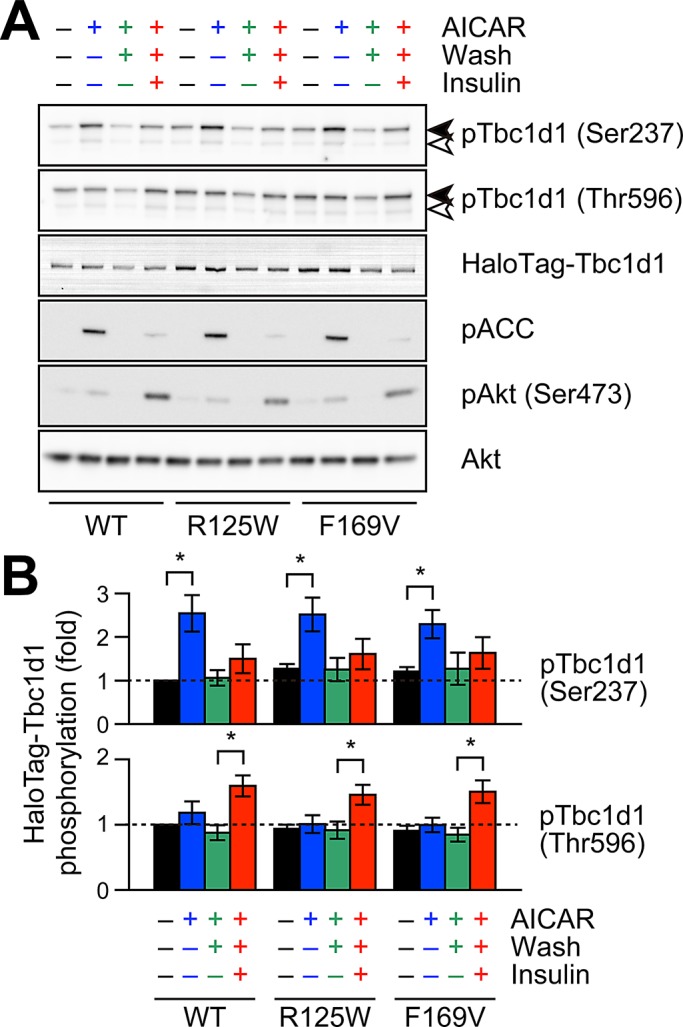
Effects of Tbc1d1 PTB1-domain mutants on AICAR- and insulin-induced phosphorylation. Representative Western blotting (A) and quantification (B) of HaloTag-Tbc1d1 phosphorylation. Data were obtained and represented as in Figure 4. *p < 0.05.
Taken together, these data provide important insights into a unique regulatory mechanism of Tbc1d1 that relies on an intact PTB1 domain, which is essential for Ser-237 phosphorylation–dependent acquisition of insulin responsiveness with AICAR pretreatment.
DISCUSSION
Our GLUT4 nanometry, combined with a cell-based experimental reconstitution model, allowed the clear demonstration of Tbc1d1 function and its unique regulatory mechanisms in GLUT4 liberation (Figure 8). Our findings provide novel mechanistic insights into the regulatory mode shift of Tbc1d1 and conceptual frameworks for understanding insulin-responsive GLUT4 translocation involving Tbc1d1, as in skeletal muscle, where Tbc1d1 is abundantly expressed (Castorena et al., 2011). It is noteworthy that mutations in the PTB1 domain, including R125W, result in complete loss of the acquisition of insulin responsiveness, which may explain the defect identified in human patients with this natural mutation (Stone et al., 2006; Meyre et al., 2008) and also suggests that this regulatory mode shift resulting in the acquisition of insulin responsiveness after exercise-mimetic stimuli is related to the beneficial effects of exercise on muscle insulin potency, particularly augmented GLUT4 translocation.
FIGURE 8:
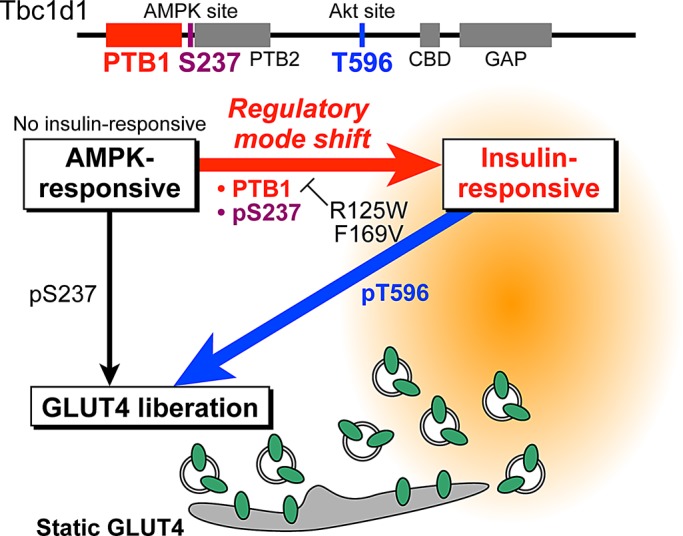
Schematic depiction of the shift in regulatory mode of Tbc1d1. Tbc1d1 is required for AICAR-dependent GLUT4 liberation from the static GLUT4 storage compartment. Whereas naive Tbc1d1 has no ability to respond to the initial insulin stimulation, Tbc1d1 acquires insulin responsiveness only after exposure to exercise-mimetic stimuli. This shift in regulatory mode of Tbc1d1 for GLUT4 liberation requires Ser-237 phosphorylation and the PTB1 domain. PTB1-domain mutants including R125W and F169V exhibit no regulatory mode shift and thus no insulin responsiveness.
Superiority of GLUT4 nanometry for analyzing Tbc1d1 functions
Two significant advantages of our experimental approach contribute to such clear determinations of functional roles of Tbc1d1. First, our experimental reconstitution model can definitively describe the actions of specific regulatory factors that are inherently up-regulated only upon cellular differentiation, that is, sortilin and Tbc1d-family RabGAPs, for GLUT4 trafficking by manipulating ectopic expression levels or combinations of these key factors in undifferentiated fibroblastic cells (Hatakeyama and Kanzaki, 2011). On the contrary, due to various technical limitations, previous studies attempting to understand functional aspects of Tbc1d1 in GLUT4 translocation, which used a cell-surface GLUT4 exposure assay, were performed in mature 3T3L1 adipocytes overexpressing exogenous Tbc1d1 (Roach et al., 2007; Chavez et al., 2008), making it difficult to exclusively examine the functional roles of Tbc1d1 versus those of AS160 despite detailed knowledge of their biochemical properties. Second, our single-molecule imaging system can dissect complex GLUT4 trafficking itineraries in each experimentally traceable step, including liberation, transport, and tethering/fusion with the plasma membrane (Fujita et al., 2010).
We focused on the stimulus-responsive liberation of GLUT4 from the static compartment because of the similar functional context of Tbc1d1 with AS160, which is required for insulin-responsive liberation. Although the possibility of Tbc1d1 being involved in other GLUT4-trafficking processes cannot be ruled out, the liberation process must be critical for the entire GLUT4 itinerary since liberation from the static compartment is indispensable for GLUT4 to reach the plasma membrane.
One striking observation in the present study is that Tbc1d1 itself, unaccompanied by AS160, can function similarly to AS160, facilitating GLUT4 liberation, although their proximal regulatory signals differ. Tbc1d1 can facilitate GLUT4 liberation in response to AICAR treatment. As expected, AICAR-induced Ser-237 phosphorylation was found to be important, whereas insulin-induced Thr-596 phosphorylation was insufficient for GLUT4 liberation, possibly due to failure of 14-3-3 binding to solely phosphorylated Thr-596 (Chen et al., 2008). However, a key unexpected observation was failure of GLUT4 liberation by ionomycin despite obvious phosphorylation of Tbc1d1 at Ser-237, implying a further layer of complexity in Tbc1d1 regulation, apparently involving mechanisms unrelated or parallel to Ser-237 phosphorylation (as discussed in detail later).
Regulatory mode shift of Tbc1d1
To our surprise, Tbc1d1 acquires temporal insulin-responsive ability for GLUT4 liberation only after exercise-mimetic stimuli such as AICAR pretreatment. This observation strongly suggested that Tbc1d1 molecules have at least two distinct regulatory modes (AMPK responsive and insulin responsive) and that Tbc1d1 undergoes the regulatory mode shift in response to a Ser-237 phosphorylation process, even though this phosphorylation is not necessarily persistent (Figure 8). Of greater importance, we found the necessity of the PTB1 domain of Tbc1d1 for its shift of regulatory mode by taking advantage of Tbc1d1 being phosphorylated by two distinct inputs governed by different protein kinases (AMPK and Akt).
Both AS160 and Tbc1d1 have two PTB domains, which are classified as disabled like (Uhlik et al., 2005). A truncation mutant in AS160 (R363X) has been identified as being involved in postprandial hyperinsulinemia (Dash et al., 2009), and a recent report demonstrated overexpression of the PTB-domain fragments of AS160 to affect 14-3-3 binding and in vitro fusion activities of GLUT4 vesicles (Koumanov et al., 2011), highlighting the importance of PTB domains. Thus AS160 might also have a similar regulatory mechanism involving PTB domains, although both crucial phosphorylation sites of AS160 are governed by Akt and thus have yet to be discovered. In any case, the regulatory mode shift of Tbc1d1 was found to be dependent on the PTB1 domain, and malfunction in this regulatory mode shift might be rather widely involved in human pathophysiological states such as insulin resistance and type 2 diabetes, as exemplified by the aforementioned genetic disorders.
In general, PTB domains are adaptors or scaffolds for proteins to recruit signaling complexes by binding with NPxY motifs in both phosphotyrosine-dependent and -independent manners (Uhlik et al., 2005). Low-density-lipoprotein receptor–related protein 1 (LRP1), a recently identified component of GLUT4-containing vesicles (Jedrychowski et al., 2010), has two NPxY motifs in its C-terminal cytoplasmic domain and reportedly binds with PTB domain–containing proteins such as ShcA, Fe65, and disabled-like ones (Stolt and Bock, 2006). In 3T3L1 adipocytes, knockdown of LRP1 resulted in reduced GLUT4 expression and decreased insulin-stimulated glucose uptake, suggesting the importance of LRP1 in appropriate GLUT4 behavior. In addition, the cytoplasmic region of LRP1 has the ability to bind with AS160 by glutathione S-transferase pull down (Jedrychowski et al., 2010). Therefore we analyzed the possibility of the LRP1 cytoplasmic region being a binding partner of Tbc1d1 PTB domains. However, our preliminary results suggest that the cytoplasmic region of LRP1 may not be a major regulator for Tbc1d1 actions since the cytoplasmic region of LRP1 shows neither detectable binding with Tbc1d1 nor augmented tyrosine phosphorylation in response to various stimuli, including AICAR and insulin. Furthermore, overexpression of the cytoplasmic region of LRP1 lacking a luminal domain failed to modify any GLUT4 behaviors under basal or various stimulated conditions (unpublished observations). Insulin-regulated aminopeptidase (IRAP) is another major constituent of GLUT4-containing vesicles and is reported to be a binding partner of AS160 PTB domain–containing regions (Larance et al., 2005; Peck et al., 2006; Park et al., 2012), although IRAP has no canonical NPxY motifs. In 3T3L1 adipocytes, microinjection of the cytoplasmic fraction of IRAP resulted in GLUT4 translocation (Waters et al., 1997), suggesting that IRAP modulates GLUT4 behavior.
In the regulatory mode shift of Tbc1d1, since only temporal Ser-237 phosphorylation with AICAR pretreatment was sufficient for subsequent insulin-induced GLUT4 liberation along with Thr-596 (but not Ser-237) phosphorylation, it is tempting to speculate that the PTB1 domain is responsible for the functionally crucial status of Tbc1d1 without relying on the phosphorylation status of Ser-237 upon executing the regulatory mode shift of Tbc1d1 that might be achieved by associating with unspecified PTB-binding partner(s). Future studies are warranted to determine such interacting molecule(s) for understanding GLUT4 behavior and glucose metabolism. It is also worth noting that the Tbc1d1 subcellular localization detected by conventional immunofluorescence analysis was not apparently changed during exposure to various stimuli (unpublished observations), suggesting further complexity in Tbc1d1 regulation. It is also possible that other phosphorylation sites of Tbc1d1 undergo more prolonged or slow-onset phosphorylation in response to AICAR treatment and are involved in the regulatory mode shift of Tbc1d1 in conjunction with the PTB1 domain. Future detailed investigations of the molecular mechanisms underlying the actions of Tbc1d1 are anticipated to unveil these regulatory complexities.
GLUT4 behavior in cells coexpressing AS160 and Tbc1d1
Unlike adipocytes predominantly expressing AS160, skeletal muscles express both AS160 and Tbc1d1 (Taylor et al., 2008; Castorena et al., 2011). Therefore it is conceivable that GLUT4 behavior in skeletal muscle cells is more complex than that in adipocytes. Previous observations suggested that actions of Tbc1d1 dominate over those of AS160 since ectopic expression of Tbc1d1 strongly inhibited insulin-responsive GLUT4 translocation in 3T3L1 adipocytes (Roach et al., 2007; Chavez et al., 2008). This inhibitory action partially diminished in the presence of AICAR, indicating that Tbc1d1 and AS160 have distinct modes of action in regulating GLUT4 translocation, which is consistent with the present observations. As mentioned before, AS160 also has PTB domains that may modulate in vivo glucose metabolism. Therefore it is possible that AS160 PTB domains also have similar regulatory mechanisms and that the distinct binding properties of 14-3-3 with AS160 and Tbc1d1, in concert with the regulatory mode shift of Tbc1d1, which appear to be dominant over AS160 actions, are key factors determining GLUT4 behavior after various stimuli in skeletal muscle.
Both AS160 and Tbc1d1 have RabGAP activities with nearly similar specificities in vitro (Miinea et al., 2005; Roach et al., 2007). Rab GTPases are major regulators of vesicle transport and have been implicated in multiple steps of vesicle trafficking, including vesicle budding, movement, and tethering/docking and fusion with the plasma membrane (Stenmark, 2009). The human genome encodes >60 members of the Rab family, which are localized to distinct intracellular compartments, and many Rab proteins have been implicated in GLUT4 trafficking (Stockli et al., 2011). Although it is difficult to pinpoint the functions of individual Rab proteins in GLUT4 trafficking, a recent live-cell total internal reflection fluorescence (TIRF) imaging study obtained direct evidence suggesting that different Rab proteins regulate distinct GLUT4-trafficking steps (Chen et al., 2012). Therefore it is possible that different Rab proteins are involved in distinct regulatory modes of Tbc1d1. Although TIRF microscopy is a superior method for analyzing cellular events beneath the plasma membrane, this approach cannot examine the events occurring deeper within the cells. Because our GLUT4 nanometry can easily detect intracellular GLUT4 behavior with high precision, our approach has the potential to be a powerful tool for unveiling the roles of specific Rab proteins in intracellular trafficking.
In summary, on the basis of our previous and present results, we have clearly demonstrated the similarities and differences between functional roles of two key RabGAPs, AS160 and Tbc1d1, in static GLUT4 liberation, using GLUT4 nanometry and a cell-based reconstitution model, respectively. Based on these lines of evidence, further analysis in cells coexpressing both RabGAPs will provide knowledge promoting better understanding of GLUT4 behavior in skeletal muscle and therefore exercise physiology.
MATERIALS AND METHODS
Plasmids and antibodies
The HaloTag-Tbc1d1 expression vector was from Promega (Madison, WI). The sequence was based on human Tbc1d1. All mutations were generated by PCR-based site-directed mutagenesis and confirmed by sequencing (PRISM 3130; Applied Biosystems, Foster City, CA). Antibodies against phospho-Tbc1d1/S237 and phospho-Tbc1d1/T596 of Tbc1d1 were generated by immunizing rabbits with KLH-conjugated peptides (H-CRPMRKSFpSQPGLRS-OH for phospho-Tbc1d1/S237 or H-CRRANpTLSHFP-OH for phospho-Tbc1d1/T596), followed by immunoaffinity purification using SulfoLink Coupling Gel (Pierce, Rockford, IL). Anti-myc monoclonal antibody (9E10) was purified from hybridoma culture supernatants. Antibodies against pACC, pAkt (Ser473), and Akt were purchased from Cell Signaling (Beverly, MA).
Cell culture
All experiments were performed in 3T3L1 fibroblasts exogenously expressing myc-GLUT4-ECFP, sortilin–enhanced yellow fluorescent protein (EYFP), and/or HaloTag-Tbc1d1. Cells were plated onto glass-bottom dishes (thickness, 0.17 mm; Matsunami Glass, Osaka, Japan) or six-well culture plates and then transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) with plasmid DNA (each 1–2 μg) according to the manufacturer's instructions. For imaging experiments, the cells were immersed in a solution containing 150 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid–NaOH (pH 7.4), and 5.5 mM d-glucose.
Protein detections
Before electrophoresis, cell lysates were incubated with HaloTag TMR Ligand (Bio-Rad, Hercules, CA) for 1 h to fluorescently label HaloTag-fused proteins. Fluorescence of HaloTag TMR Ligand and EYFP were detected with PharosFX Molecular Imager (Bio-Rad) equipped with 488- and 532-nm lasers. Western blotting analyses were performed following standard procedures, and chemiluminescence was detected with ImageQuant LAS4000 mini (GE Healthcare Life Sciences, Piscataway, NJ). Quantification was performed with ImageJ (National Institutes of Health, Bethesda, MD) or ImageQuant TL (GE Healthcare Life Sciences). We calculated the ratio of intensities between phosphorylated and total HaloTag-Tbc1d1, and mean data obtained from three independent assays are shown.
QD labeling of myc-GLUT4-ECFP
QD-conjugated antibody was prepared as follows. First, the Fab fragment of anti-myc antibody was prepared with a Fab Preparation Kit (Pierce). Then the Fab fragment of anti-myc antibody was conjugated with QD655 using a QD antibody conjugation kit (Invitrogen). The final concentration of the QD655-conjugated antibody was determined by optical density at 632 nm according to the manufacturer's instructions. For labeling, the cells were serum starved and incubated in the presence of 1.5–5 nM QD655-conjugated antibodies for 1 h. The cells were then extensively washed to remove unbound QD-labeled antibodies, followed by an additional incubation for at least 3 h.
Single-particle tracking and movement analysis
Imaging experiments were performed with a homemade microscope consisting of an inverted microscope (IX71, Olympus), an electron-multiplying charge-coupled device camera (iXon 887; Andor Technology, Belfast, United Kingdom), a Nipkow disk confocal unit (CSU10; Yokogawa, Tokyo, Japan), and an oil-immersion objective lens (UPLSAPO100xO, numerical aperture 1.4; Olympus, Tokyo, Japan) at ∼30°C. QD655 fluorescence was excited at 532 nm with a solid-state laser (Spectra-Physics, Santa Clara, CA) and detected through a 655/12 bandpass filter (Semrock, Rochester, NY). In these experiments, expressions of sortilin-EYFP and HaloTag-Tbc1d1 were confirmed by EYFP fluorescence and by staining of cells with HaloTag Ligand TMR, respectively. Single-particle tracking was performed with G-Count (G-Angstrom, Sendai, Japan) with a two-dimensional Gaussian fitting mode. We tracked each particle successfully fitted within an 8 × 8 pixel region of interest for at least 30 frames. When the signal in a frame was lost because of blinking, no fitting was performed until reappearance of the bright spot. When a bright spot did not reappear within 10 frames, tracking was aborted. We typically tracked 50–150 particles per cell and obtained the images in at least three independent experiments. We evaluated movements with velocity distributions, mean velocities, and diffusion coefficient maps. The velocities for individual particle movements were calculated by linear fit of the displacement during four frames. Mean velocities and frequency distributions were first calculated in a cell and then averaged among cells under the same treatment conditions. We found that QDs in fixed cells showed nonnegligible velocities (∼0.29 μm/s), believed to be derived from inevitable instrumental noise (Fujita et al., 2010). Thus, in figures representing mean velocities, instead of raw data we used “corrected” values obtained by subtracting the noise. Diffusion coefficients of individual molecules were estimated as follows. First, mean-square displacement (MSD) values of individual particles were calculated with
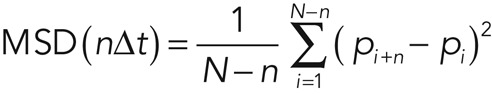 (1) (1)
|
where N, n, Δt, and p are the total number of positions measured, the measurement index (going from 1 to N − 1), the time interval between two consecutive image sequences, and the positions of the molecule, respectively. The diffusion coefficient of the molecule was calculated by fitting the first 10 time points of the MSD values with
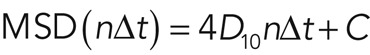 (2) (2)
|
where D10 and C are the diffusion coefficient of the first 10 time points and instrumental noise, respectively. The MSD and D10 calculations and D10 map constructions were performed with a custom-written program based on LabVIEW and Vision (National Instruments, Austin, TX).
Photolysis of caged compounds and monitoring of [Ca2+]i
The acetoxymethyl (AM) ester forms of Ca2+ indicator fluo-4 (Invitrogen) and caged Ca2+ compound NPE (Invitrogen) were first dissolved in dimethyl sulfoxide at 10 mM and then diluted in serum-free DMEM. Cells were incubated for 30 min at 37°C in serum-free DMEM containing 10 μM NPE-AM and 0.03% Cremophor EL (Sigma-Aldrich, St. Louis, MO) in the presence or absence of 10 μM fluo-4-AM and then washed with imaging buffer. Photolysis of NPE was induced with a mercury lamp (U-ULS100HG, Olympus) through a 360-nm bandpass filter. The mercury lamp radiation was gated with an electric shutter (SSH-R; Sigma Koki, Tokyo, Japan) with an opening duration of 0.1 s. Uncaging experiments were performed under yellow light illumination to prevent unintended photolysis of NPE. Fluo-4 fluorescence was excited at 488 nm with a solid-state laser (Spectra-Physics) and detected through a 530/40 bandpass filter (Olympus). Fluorescence intensities were obtained by ImageJ.
Statistical analyses
Data are represented as mean ± SEM. The Kolmogorov–Smirnov test was used to compare velocity distributions, the Mann–Whitney U test or Wilcoxon signed-rank test was used for mean velocities unless otherwise indicated, and p < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank Fumie Wagatsuma and Natsumi Emoto for their technical assistance. This study was supported by grants from the Japan Society for the Promotion of Science (22590969 and 20001007). This study was also partially supported by the Banyu Life Science Foundation International and the Takeda Science Foundation. H.H. is a Research Fellow of the Japan Society for the Promotion of Science.
Abbreviations used:
- AM
acetoxymethyl
- AMPK
AMP-activated protein kinase
- AS160
Akt substrate of 160 kDa
- [Ca2+]i
intracellular Ca2+ concentration
- CBD
calmodulin-binding domain
- IRAP
insulin-responsive aminopeptidase
- LRP1
low-density-lipoprotein receptor–related protein 1
- MSD
mean-square displacement
- NPE
o-nitrophenyl ethylene glycol tetraacetic acid
- PTB
phosphotyrosine binding
- QD
quantum dot
- RabGAP
Rab GTPase-activating protein
- Tbc1d
tre-2/UPS6, BUB2, cdc16 domain
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-10-0725) on January 16, 2013.
REFERENCES
- An D, Toyoda T, Taylor EB, Yu H, Fujii N, Hirshman MF, Goodyear LJ. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes. 2010;59:1358–1365. doi: 10.2337/db09-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castorena CM, Mackrell JG, Bogan JS, Kanzaki M, Cartee GD. Clustering of GLUT4, TUG and RUVBL2 protein levels correlate with myosin heavy chain isoform pattern in skeletal muscles, but AS160 and TBC1D1 levels do not. J Appl Physiol. 2011;111:1106–1117. doi: 10.1152/japplphysiol.00631.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JA, Roach WG, Keller SR, Lane WS, Lienhard GE. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J Biol Chem. 2008;283:9187–9195. doi: 10.1074/jbc.M708934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, Mackintosh C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J. 2008;409:449–459. doi: 10.1042/BJ20071114. [DOI] [PubMed] [Google Scholar]
- Chen S, Wasserman DH, MacKintosh C, Sakamoto K. Mice with AS160/TBC1D4-Thr649Ala knockin mutation are glucose intolerant with reduced insulin sensitivity and altered GLUT4 trafficking. Cell Metab. 2011;13:68–79. doi: 10.1016/j.cmet.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. Rab10 and myosin-Va mediate insulin-stimulated GLUT4 storage vesicle translocation in adipocytes. J Cell Biol. 2012;198:545–560. doi: 10.1083/jcb.201111091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash S, et al. A truncation mutation in TBC1D4 in a family with acanthosis nigricans and postprandial hyperinsulinemia. Proc Natl Acad Sci USA. 2009;106:9350–9355. doi: 10.1073/pnas.0900909106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Hatakeyama H, Watanabe TM, Sato M, Higuchi H, Kanzaki M. Identification of three distinct functional sites of insulin-mediated GLUT4 trafficking in adipocytes using quantitative single molecule imaging. Mol Biol Cell. 2010;21:2721–2731. doi: 10.1091/mbc.E10-01-0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyear LJ, Kahn BB. Exercise, glucose transport, and insulin sensitivity. Annu Rev Med. 1998;49:235–261. doi: 10.1146/annurev.med.49.1.235. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Kanzaki M. Molecular basis of insulin-responsive GLUT4 trafficking systems revealed by single molecule imaging. Traffic. 2011;12:1805–1820. doi: 10.1111/j.1600-0854.2011.01279.x. [DOI] [PubMed] [Google Scholar]
- Jedrychowski MP, Gartner CA, Gygi SP, Zhou L, Herz J, Kandror KV, Pilch PF. Proteomic analysis of GLUT4 storage vesicles reveals LRP1 to be an important vesicle component and target of insulin signaling. J Biol Chem. 2010;285:104–114. doi: 10.1074/jbc.M109.040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane S, Lienhard GE. Calmodulin binds to the Rab GTPase activating protein required for insulin-stimulated GLUT4 translocation. Biochem Biophys Res Commun. 2005;335:175–180. doi: 10.1016/j.bbrc.2005.07.056. [DOI] [PubMed] [Google Scholar]
- Koumanov F, Richardson JD, Murrow BA, Holman GD. AS160 phosphotyrosine-binding domain constructs inhibit insulin-stimulated GLUT4 vesicle fusion with the plasma membrane. J Biol Chem. 2011;286:16574–16582. doi: 10.1074/jbc.M111.226092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larance M, et al. Characterization of the role of the Rab GTPase-activating protein AS160 in insulin-regulated GLUT4 trafficking. J Biol Chem. 2005;280:37803–37813. doi: 10.1074/jbc.M503897200. [DOI] [PubMed] [Google Scholar]
- Meyre D, et al. R125W coding variant in TBC1D1 confers risk for familial obesity and contributes to linkage on chromosome 4p14 in the French population. Hum Mol Genet. 2008;17:1798–1802. doi: 10.1093/hmg/ddn070. [DOI] [PubMed] [Google Scholar]
- Miinea CP, Sano H, Kane S, Sano E, Fukuda M, Peranen J, Lane WS, Lienhard GE. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J. 2005;391:87–93. doi: 10.1042/BJ20050887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedachi T, Fujita H, Kanzaki M. Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am J Physiol Endocrinol Metab. 2008;295:E1191–E1204. doi: 10.1152/ajpendo.90280.2008. [DOI] [PubMed] [Google Scholar]
- Park S, Kim KY, Kim S, Yu YS. Affinity between TBC1D4 (AS160) phosphotyrosine-binding domain and insulin-regulated aminopeptidase cytoplasmic domain measured by isothermal titration calorimetry. BMB Rep. 2012;45:360–364. doi: 10.5483/bmbrep.2012.45.6.030. [DOI] [PubMed] [Google Scholar]
- Park SY, Jin W, Woo JR, Shoelson SE. Crystal structures of human TBC1D1 and TBC1D4 (AS160) RabGTPase-activating protein (RabGAP) domains reveal critical elements for GLUT4 translocation. J Biol Chem. 2011;286:18130–18138. doi: 10.1074/jbc.M110.217323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck GR, Chavez JA, Roach WG, Budnik BA, Lane WS, Karlsson HK, Zierath JR, Lienhard GE. Insulin-stimulated phosphorylation of the Rab GTPase-activating protein TBC1D1 regulates GLUT4 translocation. J Biol Chem. 2009;284:30016–30023. doi: 10.1074/jbc.M109.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck GR, Ye S, Pham V, Fernando RN, Macaulay SL, Chai SY, Albiston AL. Interaction of the Akt substrate, AS160, with the glucose transporter 4 vesicle marker protein, insulin-regulated aminopeptidase. Mol Endocrinol. 2006;20:2576–2583. doi: 10.1210/me.2005-0476. [DOI] [PubMed] [Google Scholar]
- Pehmoller C, Treebak JT, Birk JB, Chen S, Mackintosh C, Hardie DG, Richter EA, Wojtaszewski JF. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab. 2009;297:E665–E675. doi: 10.1152/ajpendo.00115.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem. 2006;281:29174–29180. doi: 10.1074/jbc.M603274200. [DOI] [PubMed] [Google Scholar]
- Roach WG, Chavez JA, Miinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J. 2007;403:353–358. doi: 10.1042/BJ20061798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Kane S, Sano E, Miinea CP, Asara JM, Lane WS, Garner CW, Lienhard GE. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278:14599–14602. doi: 10.1074/jbc.C300063200. [DOI] [PubMed] [Google Scholar]
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- Stockli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci. 2011;124:4147–4159. doi: 10.1242/jcs.097063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt PC, Bock HH. Modulation of lipoprotein receptor functions by intracellular adaptor proteins. Cell Signal. 2006;18:1560–1571. doi: 10.1016/j.cellsig.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Stone S, et al. TBC1D1 is a candidate for a severe obesity gene and evidence for a gene/gene interaction in obesity predisposition. Hum Mol Genet. 2006;15:2709–2720. doi: 10.1093/hmg/ddl204. [DOI] [PubMed] [Google Scholar]
- Taylor EB, et al. Discovery of TBC1D1 as an insulin-, AICAR-, and contraction-stimulated signaling nexus in mouse skeletal muscle. J Biol Chem. 2008;283:9787–9796. doi: 10.1074/jbc.M708839200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlik MT, Temple B, Bencharit S, Kimple AJ, Siderovski DP, Johnson GL. Structural and evolutionary division of phosphotyrosine binding (PTB) domains. J Mol Biol. 2005;345:1–20. doi: 10.1016/j.jmb.2004.10.038. [DOI] [PubMed] [Google Scholar]
- Vichaiwong K, Purohit S, An D, Toyoda T, Jessen N, Hirshman MF, Goodyear LJ. Contraction regulates site-specific phosphorylation of TBC1D1 in skeletal muscle. Biochem J. 2010;431:311–320. doi: 10.1042/BJ20101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters SB, D'Auria M, Martin SS, Nguyen C, Kozma LM, Luskey KL. The amino terminus of insulin-responsive aminopeptidase causes Glut4 translocation in 3T3-L1 adipocytes. J Biol Chem. 1997;272:23323–23327. doi: 10.1074/jbc.272.37.23323. [DOI] [PubMed] [Google Scholar]
- Zhou MM, Ravichandran KS, Olejniczak EF, Petros AM, Meadows RP, Sattler M, Harlan JE, Wade WS, Burakoff SJ, Fesik SW. Structure and ligand recognition of the phosphotyrosine binding domain of Shc. Nature. 1995;378:584–592. doi: 10.1038/378584a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.