

DIAPH1, the RhoA effector protein, forms a complex in adrenocortical cells and is phosphorylated by a cAMP/PKA-dependent pathway. Phosphorylation differentially modulates protein–protein interactions, regulates the stability of the protein, and facilitates sumoylation.
Abstract
Diaphanous homologue 1 (DIAPH1) is a Rho effector protein that coordinates cellular dynamics by regulating microfilament and microtubule function. We previously showed that DIAPH1 plays an integral role in regulating the production of cortisol by controlling the rate of mitochondrial movement, by which activation of the adrenocorticotropin (ACTH)/cAMP signaling pathway stimulates mitochondrial trafficking and promotes the interaction between RhoA and DIAPH1. In the present study we use mass spectrometry to identify DIAPH1 binding partners and find that DIAPH1 interacts with several proteins, including RhoA, dynamin-1, kinesin, β-tubulin, β-actin, oxysterol-binding protein (OSBP)–related protein 2 (ORP2), and ORP10. Moreover, DIAPH1 is phosphorylated in response to dibutyryl cAMP (Bt2cAMP) at Thr-759 via a pathway that requires extracellular signal-related kinase (ERK). Alanine substitution of Thr-759 renders DIAPH1 more stable and attenuates the interaction between DIAPH1 and kinesin, ORP2, and actin but has no effect on the ability of the protein to interact with RhoA or β-tubulin. Finally, overexpression of a DIAPH1 T759A mutant significantly decreases the rate of Bt2cAMP-stimulated mitochondrial movement. Taken together, our findings establish a key role for phosphorylation in regulating the stability and function of DIAPH1.
INTRODUCTION
Cortisol biosynthesis occurs in two cellular organelles, where both the first and final reactions occur in mitochondria and the intermediary enzymatic steps take place in the endoplasmic reticulum (ER). We previously reported that adrenocorticotropin (ACTH) regulates cortisol production by stimulating an increase in the rate of mitochondrial trafficking (Li and Sewer, 2010). Inhibition of microtubule polymerization attenuated ACTH/cAMP-stimulated mitochondrial movement and attenuated the secretion of cortisol, whereas agents that perturb the polymerization of actin increased the rate of mitochondrial movement and cortisol secretion both basally and in response to ACTH/cAMP. We also identified key roles for RhoA and the RhoA effector, diaphanous homologue 1 (DIAPH1), in regulating ACTH/cAMP-stimulated mitochondrial movement and cortisol production. Overexpression of a constitutively active DIAPH1 prevented mitochondrial movement and cortisol secretion, suggesting a role for DIAPH1 in coordinating the trafficking of mitochondria in response to ACTH stimulation.
DIAPH1 is a member of the formin family of effector proteins that regulate cytoskeletal dynamics by interacting with actin, microtubules, and other cytoskeletal-associated regulatory proteins (Watanabe et al., 1997; Ishizaki et al., 2001; Palazzo et al., 2001a; Bartolini and Gundersen, 2010; DeWard et al., 2010; Schonichen and Geyer, 2010; Young and Copeland, 2010). The ability of DIAPH1 to promote changes in cell shape and mobility is regulated by signal transduction cascades that are mediated by the small Rho GTPases Rho, Rac, and Cdc42. For example, lysophosphatidic acid (LPA)–stimulated activation of Rho promotes the activation of DIAPH1 and subsequent formation of actin filaments (Watanabe et al., 1997; Palazzo et al., 2001b; Copeland and Treisman, 2002; Wen et al., 2004). DIAPH1 binds directly to actin via its formin homology (FH)-2, thus facilitating actin nucleation (Shimada et al., 2004; Schonichen and Geyer, 2010). The ability of DIAPH1 to interact with actin (and other binding partners) is controlled by a diaphanous autoregulatory domain (DAD) in the carboxy terminus of the protein. In the absence of activated signaling, the DAD binds to the amino terminus–localized Rho-binding domain, thereby rendering the FH1 and FH2 domains inaccessible for proteins such as actin, tubulin, and SH3 domain–containing proteins (e.g., Src kinase) to bind to DIAPH1 (Watanabe et al., 1999; Alberts, 2001). On GTP-bound Rho binding, the autoinhibitory conformation is relieved and DIAPH1 is able to bind to downstream effector proteins. Thus truncation mutants that lack the DAD are constitutively active variants that are capable of nucleating actin in the absence of GTPase activation (Alberts et al., 1998; Watanabe et al., 1999).
In addition to regulating the actin polymerization, DIAPH1 also controls microtubule dynamics via binding to the FH2 domain (Ishizaki et al., 2001; Palazzo et al., 2001a; Bartolini et al., 2008). DIAPH1 facilitates the stabilization of microtubules in response to LPA (Nagasaki and Gundersen, 1996; Cook et al., 1998) and is required for generating stable microtubules in multiple cell types, including fibroblasts (Eng et al., 2006; Bartolini and Gundersen, 2010), glioma cells (Yamana et al., 2006), and platelets (Higashi et al., 2008). DIAPH1 has also been found to be required for broad lamellipodia formation in response to growth factor stimulation (Zaoui et al., 2008) and for LPA-stimulated phosphorylation of glycogen synthase kinase 3β in migrating fibroblasts (Eng et al., 2006). Finally, roles for DIAPH1 in regulating serum response factor target gene transcription (Copeland and Treisman, 2002; Geneste et al., 2002; Gopinath et al., 2007) and the PP2C phosphatase family member POPX2 (Xie et al., 2008) have been described. On the basis of our previous studies demonstrating a role for RhoA and DIAPH1 in controlling mitochondrial movement and cortisol production in human adrenocortical cells (Li and Sewer, 2010) and the ability of DIAPH1 to interact with multiple cellular proteins (Bartolini and Gundersen, 2010; Schonichen and Geyer, 2010; Young and Copeland, 2010), we used mass spectrometry to identify proteins that interact with DIAPH1 in H295R adrenocortical cells. In addition to identifying several DIAPH1 binding partners, we show that DIAPH1 is a phosphoprotein and that phosphorylation regulates the stability of the protein, its ability to interact with several binding partners, and the rate of mitochondrial movement.
RESULTS
Identification of DIAPH1 binding partners
We previously identified a role for DIAPH1 in regulating glucocorticoid biosynthesis in human adrenocortical cells by controlling the rate of mitochondrial movement (Li and Sewer, 2010). These studies revealed that ACTH activated a signal transduction cascade that led to the acute activation of the small GTPase RhoA and an increased interaction between RhoA and its effector DIAPH1. On the basis of these data, we hypothesized that DIAPH1 coordinates ACTH-stimulated mitochondrial trafficking and cortisol production by forming a larger protein complex that facilitates the exchange of substrate between the ER and mitochondria. To test this hypothesis, we immunoprecipitated DIAPH1 from H295R adrenocortical cells and subjected the immunoprecipitated proteins to SDS–PAGE and mass spectrometry. As shown in Figure 1A, DIAPH1 copurifies with multiple proteins. Tandem mass spectrometric analysis of the tryptic peptide digests identified several putative DIAPH1 binding partners (Table 1). Next we sought to define the role of cAMP signaling in conferring the interaction of DIAPH1 with binding proteins. DIAPH1 was immunoprecipitated from either untreated H295R cells or cells that were treated for 1 h with dibutyryl cAMP (Bt2cAMP). Unexpectedly, when comparing the spectral counts of proteins isolated from control or Bt2cAMP-treated cells, we found that activation of the cAMP signaling pathway decreased the ability of DIAPH1 to copurify with most proteins (Table 2). For example, there was less tubulin, myosin, and vimentin that copurified with DIAPH1 in response to cAMP. In contrast, there was an increase in the interaction between DIAPH1 and actin and the guanine nucleotide exchange factor p523 HERC1 (Table 2). To further examine the ability of proteins that were identified by mass spectrometry to interact with DIAPH1, we carried our coimmunoprecipitation assays. We first focused on proteins known to play a role in regulating cytoskeletal dynamics. As shown in Figure 1B, DIAPH1 interacts with kinesin heavy chain, dynamin-1, β-tubulin, RhoA, and vimentin.
FIGURE 1:

(A) Representative gel of H295R cell lysates that were immunoprecipitated using an anti-DIAPH1 antibody and subjected to SDS–PAGE and Coomassie staining. Immunoprecipitation and mass spectrometric analysis was performed on three separate occasions. Asterisks denote bands that were excised for mass spectrometry. (B) GFP-tagged or endogenous DIAPH1 was immunoprecipitated from control or Bt2cAMP-treated (0.4 mM, 30 min) samples and the immobilized proteins were washed, separated by SDS–PAGE, and analyzed by Western blotting using antibodies against kinesin (heavy chain), dynamin-1, β-tubulin, RhoA, or vimentin. Five percent of inputs were subjected to SDS–PAGE and Western blotting using an anti-GFP antibody. Blots shown are representative, and each immunoprecipitation was performed on at least three separate occasions, each time in duplicate. (C) Lysates from control or Bt2cAMP-treated H295R cells were immunoprecipitated using an anti-OPR2 antibody (input; second from top), and protein A/G agarose or H295R cells were transfected with pEGFP-DIAPH1 and control or Bt2cAMP-stimulated lysates immunoprecipitated using an anti-GFP antibody and protein A/G agarose (input; bottom). Immobilized proteins were washed, separated by SDS–PAGE, and analyzed by Western blotting using antibodies against DIAPH1 (output; top) or ORP10 (output, second from bottom). Immunoprecipitations were carried out three times, each time in duplicate. (D) H295R cells were plated onto 100-mm dishes and the lysates immunoprecipitated with 5 μg of rabbit IgG, anti-ORP2, or ORP4 and the purified proteins separated by SDS–PAGE. Western blotting was performed on 5% of the inputs for ORP2 (middle) or ORP4 (bottom) and the outputs for DIAPH1 (top). Shown are representative blots for studies that were carried out on three separate occasions, each time in duplicate.
TABLE 1:
Mass spectrometric identification of DIAPH1 binding partners.
| Gene | Description | Mass (kDa) |
|---|---|---|
| DIAPH1 | Diaphanous protein homologue 1 | 140 |
| VIM | Vimentin | 54 |
| TUBA1C | Tubulin, α6 | 48 |
| ACTG1 | Actin, γ1 | 42 |
| DNAH7 | Dynein, heavy chain | 461 |
| ACTB | Actin, β | 42 |
| MYH9 | Myosin, nonmuscle | 226 |
| KIF1B | Kinesin family member 1B | 398 |
| TUBB | Tubulin, β | 50 |
| ORP2 | Oxysterol-binding protein–related protein 2 | 55 |
| TLN2 | Talin 2 | 271 |
| MAP4 | Microtubule-associated protein 4 | 121 |
| DYN1 | Dynamin-1 | 97 |
| SOS1 | Son of sevenless protein homologue 1 | 152 |
| PACSIN3 | Protein kinase C and casein kinase substrate | 48 |
| FGD6 | FYVE, rhoGEF, and PH domain–containing protein 6 | 161 |
| TUBA3D | Tubulin α3d | 48 |
| HSP9A | Heat shock protein 70 kDa | 74 |
| AKAP13 | A-kinase anchor protein 13 isoform 1 | 307 |
| ORP10 | Oxysterol-binding protein–related protein 10 | 49 |
| HERC1 | Guanine nucleotide exchange factor p532 | 532 |
| VCP | Valosin-containing protein | 89 |
| HSP90B1 | Heat shock protein 90 kDa β, member 1 | 83 |
TABLE 2:
Effect of Bt2cAMP on the interaction of DIAPH1 with binding partners.
| Control | Bt2cAMP | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Total peptides | SC1 | SC2 | Total | SC1 | SC2 | Total | Description |
| TLN2 | 6 | 2 | 0 | 2 | 1 | 3 | 4 | Talin 2 |
| HERC1 | 10 | 5 | 3 | 8 | 3 | 11 | 14 | Guanine nucleotide exchange factor p532 |
| ACTG1 | 17 | 27 | 11 | 38 | 24 | 22 | 46 | Actin, γ1 propeptide |
| ACTB | 17 | 27 | 11 | 38 | 24 | 22 | 46 | Actin, β |
| HSP90B1 | 6 | 2 | 1 | 3 | 2 | 1 | 3 | Heat shock protein 90 kDa β, member 1 |
| DIAPH1 | 78 | 1158 | 294 | 1452 | 517 | 627 | 1144 | Diaphanous 1 isoform 1 |
| PACSIN3 | 15 | 64 | 25 | 89 | 24 | 40 | 64 | Protein kinase C and casein kinase substrate in neurons 3 |
| DNAH7 | 8 | 4 | 1 | 5 | 1 | 2 | 3 | Dynein, axonemal, heavy chain 7 |
| KIF1B | 4 | 3 | 2 | 5 | 1 | 1 | 2 | Kinesin family member 1B isoform b |
| TUBB | 9 | 19 | 5 | 24 | 1 | 3 | 4 | Tubulin, β |
| TUBA1C | 9 | 22 | 29 | 51 | 5 | 11 | 16 | Tubulin, α6 |
| VIM | 12 | 17 | 12 | 29 | 2 | 6 | 8 | Vimentin |
| MYH9 | 82 | 155 | 7 | 162 | 6 | 11 | 17 | Myosin, heavy polypeptide 9, nonmuscle |
| VCP | 12 | 15 | 1 | 16 | 1 | 0 | 1 | Valosin-containing protein |
Proteins are sorted based on ratio of Bt2cAMP/control spectral counts (from high to low).
Because we postulated that ACTH-dependent mitochondrial movement regulates the exchange of substrate between the ER and mitochondria, we were intrigued by the mass spectrometric data demonstrating that DIAPH1 copurifies with members of the oxysterol related–binding protein (ORP) family. There are 11 members of the ORP family. Although the functional role of most family members is unknown, these proteins are predicted to bind various classes of lipids (Olkkonen and Levine, 2004; Fairn and McMaster, 2008; Ridgway, 2010). For example, ORP2 binds phospholipids and sterols (Xu et al., 2001; Ridgway, 2010). As shown in Figure 1C, DIAPH1 coimmunoprecipitates with both ORP2 and ORP10. In contrast, DIAPH1 was unable to interact with ORP4, another member of the ORP family that is expressed in the H295R cell line, but was not identified by mass spectrometry (Figure 1D).
DIAPH1 is a phosphoprotein
In addition to identifying DIAPH1 binding partners, tandem mass spectrometric analysis also provided site-specific evidence that DIAPH1 is posttranslationally modified. We next determined which amino acid(s) were phosphorylated in response to cAMP signaling by mutating the sites identified by mass spectrometry. H295R cells were transfected with wild-type or phosphomutant DIAPH1 and the levels of phosphorylation assessed by probing Western blots with an antibody that recognizes proteins that are phosphorylated at either Ser or Thr. These studies revealed that mutation of S145 partially decreased the amount of phospho-Ser/Thr DIAPH1, whereas alanine substitution at T1229, T1082, and T759 substantially reduced the amount of phospho-Ser/Thr DIAPH1 detected (Figure 2A). As shown in Figure 2B, DIAPH1 contains a Rho-binding domain in the N-terminus and two formin homology domains toward the C-terminus of the protein. Several researchers used truncation mutants of DIAPH1 to gain information about the functional properties of the domains of the protein (Watanabe et al., 1997; Narumiya et al., 2009; Bartolini and Gundersen, 2010). These studies revealed that the FH2 domain is required for interaction with microtubules and actin nucleation (Watanabe et al., 1999; Palazzo et al., 2001a). One of the phosphorylation sites that we identified, Thr-759, lies in the FH2 domain and is conserved across species (Figure 2C). Of importance, proteomic studies also identified this amino acid residue as being targeted by cyclin-dependent kinase 1 activity (Blethrow et al., 2008).
FIGURE 2:
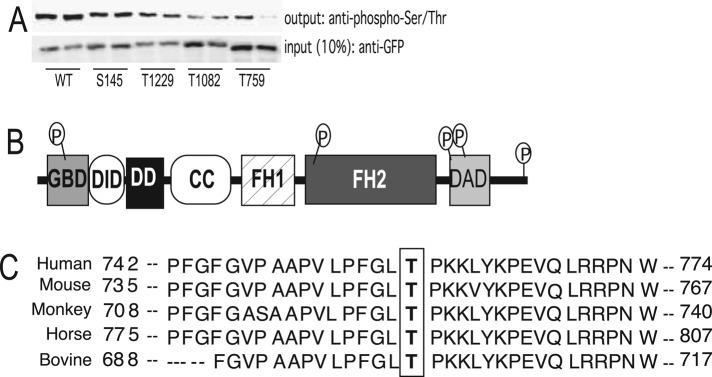
(A) H295R cells were transfected with WT or phosphomutant pEGFP-DIAPH1 and lysates immunoprecipitated using an anti-GFP antibody and protein A/G agarose. Immobilized proteins were subjected to SDS–PAGE, followed by Western blotting using an anti–phospho-Ser/Thr antibody. Ten percent of inputs were subjected to SDS–PAGE and Western blotting using an anti-GFP antibody. Densitometric analysis of phosphor-Ser/Thr and GFP expression is expressed as fold change compared with wild type, and data represent mean ± SD of four experiments, each performed in duplicate. (B) Location of the putative phosphorylation sites on the primary structure of DIAPH1. (C) T-Coffee sequence alignment of DIAPH1, highlighting the position of a conserved threonine residue (T759 in humans) across different species.
cAMP stimulates DIAPH1 phosphorylation at T759
To define the functional significance of phosphorylation of DIAPH1 in controlling complex assembly and steroid hormone biosynthesis, we generated a phosphospecific antibody to Thr-759. To test the specificity of the antibody, we transfected cells with green fluorescent protein (GFP)–tagged wild-type or T759A mutant DIAPH1, stimulated them with Bt2cAMP, and subjected them to immunoprecipitation and Western blotting. As shown in the representative Western blots and in the graph of the densitometric analysis (Figure 3A), Bt2cAMP increased the phosphorylation of wild-type DIAPH1 by 3.8-fold at this site, whereas cells expressing the T759A mutant produced a greatly diminished phosphospecific signal and did not exhibit an increase in phosphorylation in response to Bt2cAMP treatment. Bt2cAMP-stimulated phosphorylation of the endogenous protein was inhibited when the cells were pretreated with H89 or U0126, suggesting that both protein kinase A (PKA) and mitogen-activated protein kinase (MAPK) signaling are required for increased DIAPH1 phosphorylation at this site (Figure 3B). The inhibitory effect of U0126 on cAMP-mediated DIAPH1 phosphorylation prompted us to determine the effect of PKA and MAPK inhibition on extracellular signal-regulated kinase (ERK) activity. Consistent with the work of other laboratories that demonstrated a role for ERK in cAMP signaling in adrenocortical cells (Lotfi et al., 2000; Le and Schimmer, 2001; Janes et al., 2008), there was a 4.5-fold increase in the cellular content of phospho-ERK in response to Bt2cAMP (Figure 3C). Moreover, the Bt2cAMP-stimulated increase in ERK activity was decreased by 70% in cells exposed to H89 and completely inhibited by the MAPK inhibitor. Further, incubating lysates isolated from control of Bt2cAMP-treated H295R cells with λ-phosphatase also diminished detection of Thr-759 phosphorylated DIAPH (Figure 3D).
FIGURE 3:

(A) H295R cells were transfected with WT or T759A-mutant pEGFP-DIAPH1 and treated for 30 min with 0.4 mM Bt2cAMP, and lysates were immunoprecipitated using an anti-GFP antibody and protein A/G agarose. Immobilized proteins were washed, separated by SDS–PAGE, and analyzed by Western blotting using an anti–phospho(T759)-DIAPH1 antibody. Five percent of inputs were subjected to SDS–PAGE and Western blotting using an anti-GFP antibody. Left, representative blots; right, densitometric analysis. Graph data represent mean ± SD of three experiments, each in duplicate. (B) H295R cells were pretreated with 10 μM H89 or 10 μM U0126 and then treated with 0.4 mM Bt2cAMP for 30 min. Cell lysates were harvested and separated by SDS–PAGE, followed by Western blotting using anti–phospho(T769)-DIAPH1or anti-DIAPH1 antibodies. Left, representative blots; right, densitometric analysis. Graph data represent the mean ± SD of five experiments, each in duplicate. (C) H295R cells were pretreated with H89 or U0126 and then treated with Bt2cAMP. Cell lysates were harvested and separated by SDS–PAGE, followed by Western blotting using anti–phospho-ERK1/2 or anti-ERK2 antibodies. Left, representative blots; right, densitometric analysis. Graph data represent mean ± SD of five experiments, each in duplicate. (D) Lysates isolated from control or Bt2cAMP-treated cells were incubated with λ-phosphatase and then subjected to SDS–PAGE and Western blotting for pDIAPH1 or DIAPH1. Left, representative blots; right, densitometric analysis. Graph data represent the mean ± SD of two experiments, each in duplicate. In all graphs, asterisks denote statistical significance (p < 0.05) compared with untreated control.
Phosphorylation of DIAPH1 regulates protein stability
We next determined the effect of phosphorylation at T759 on the stability of DIAPH1 by treating cells transfected with wild-type or T759A mutant DIAPH1 with cycloheximide (CHX). As shown in Figure 4A, CHX reduced the expression of wild-type (WT), GFP-tagged DIAPH1, whereas the T759A mutant was resistant to the effect of the translation inhibitor. Six hours of incubation with CHX decreased the expression of the WT protein by 80%, whereas the expression of the T759A mutant was reduced by only ∼35% (Figure 4B). Proteasome inhibition for 6 h increased WT DIAPH1 to levels similar to that observed for T759A (Figure 4C), consistent with a possible resistance of T759A to proteasomal targeting. Of interest, a recent global proteomics study discovered that DIAPH1 is ubiquitinated at five sites (Kim et al., 2001), which prompted us to speculate that phosphorylation at T759 might alter (in this case, increase) the propensity for ubiquitin attachment to the former site. Of note, ubiquitination regulates the degradation of DIAPH2 at the end of mitosis (DeWard and Alberts, 2009), supporting a role for this posttranslational modification in controlling the stability of the DIAPH family of proteins. To determine whether DIAPH1 is ubiquitinated, H295R cells were treated with Bt2cAMP and DIAPH1 immunoprecipitated for analysis by SDS–PAGE and Western blotting. Although we were unable to detect an increase in the ubiquitination of the endogenous protein in response to Bt2cAMP (unpublished data), cAMP stimulation did increase the cellular amount of sumoylated DIAPH1 (Figure 4E). Mutation of T759 significantly reduces the ability of SUMO to be conjugated to the protein (Figure 4F), suggesting a role for cAMP-stimulated phosphorylation in triggering sumoylation.
FIGURE 4:

(A) H295R cells were transfected with WT or T759A-mutant pEGFP-DIAPH1 and treated with 50 μg/ml CHX for 3 or 6 h. Cell lysates were harvested and separated by SDS–PAGE, followed by Western blotting using anti-GFP and anti-GAPDH antibodies. (B) Graphical analysis of data obtained from Western blotting studies of WT and phospho(T579) DIAPH1 protein expression in cells treated with 50 μg/ml CHX for 0–6 h. Data represent mean ± SEM of four separate experiments, each carried out in duplicate. Asterisks and carats denote statistically different (p < 0.05) from WT 0-h and T759A 0-h groups, respectively. (C) H295R cells were transfected with WT or T759A-mutant pEGFP-DIAPH1 and treated with 20 μM MG132 for 6 h. Cell lysates were harvested and separated by SDS–PAGE, followed by Western blotting using anti-GFP and anti-GAPDH antibodies. Left, representative blots; right, densitometric analysis. Graph data represent mean ± SD of three experiments, each in duplicate. (D) H295R cells were treated for 30 min with 0.4 nM Bt2cAMP in the presence or absence of 20 μM MG132. Cell lysates were harvested and separated by SDS–PAGE, followed by Western blotting using anti–phospho(T759)-DIAPH1, anti-DIAPH1, and anti-GAPDH antibodies. Left, representative blots; right, densitometric analysis. Data represent the mean ± SD of four experiments, each in duplicate. Asterisks denote statistical significance (p < 0.05) compared with untreated control. (E) Lysates from control or Bt2cAMP-treated cells transfected with GFP-tagged DIAPH1 were incubated with anti-GFP antibody and protein A/G agarose and the immunoprecipitated protein analyzed by SDS–PAGE and Western blotting for SUMO-1. Representative blots of study performed on three separate occasions, each time in duplicate. (F) Lysates from cells that were transfected with WT or T759A GFP-tagged DIAPH1 were immunoprecipitated with anti-GFP and the immobilized proteins assessed by SDS–PAGE and Western blotting for SUMO-1. Representative blots of study performed on two separate occasions, each time in duplicate.
Phosphorylation at T759 regulates complex formation
We next sought to define the role of phosphorylation in regulating the ability of DIAPH1 to interact with members of the complex that were identified by mass spectrometry (Figure 1). Although there was no difference between the interaction of wild type or T759A mutant with RhoA (Figure 5A), alanine substitution at T759 decreased the interaction of kinesin with DIAPH1 by 65% (Figure 5B). Given that the FH2 domain of DIAPH1 is required for its interaction of DIAPH1 with actin and microtubules (Eng et al., 2006; Bartolini and Gundersen, 2010) and T759 lies in the FH2 domain (Figure 2C), we also determined the effect of phosphorylation on the interaction of DIAPH1 with actin and tubulin. As shown in Figure 5C, there was a 86% decrease in the interaction of the T759A DIAPH1 mutant with β-actin but no significant decrease in the interaction with β-tubulin (Figure 5D). Of interest, whereas mutation of T759 led to a 78% decrease in the interaction between DIAPH1 and ORP-2 (Figure 5E), the interaction between the T759A DIAPH1 mutant and ORP-10 was reduced by only 33% (Figure 5F).
FIGURE 5:

(A–F) H295R cells were transfected with WT or T759A-mutant pEGFP-DIAPH1, and lysates were immunoprecipitated using an anti-GFP antibody and protein A/G agarose. Immobilized proteins were washed, separated by SDS–PAGE, and analyzed by Western blotting using anti-RhoA (A), anti-kinesin (B), anti–β-actin (C), β-tubulin (D), anti-ORP2 (E), or anti-ORP10 (F) antibodies. Five percent of inputs were subjected to SDS–PAGE and Western blotting using an anti-GFP antibody. All coimmunoprecipitations were performed on at least three separate occasions, in at least duplicate. Data above each blot represent mean ± SD, and asterisks denote significantly different from WT (p < 0.05).
Phosphorylation at T759 modulates mitochondrial movement
We previously showed that cells expressing a constitutively active DIAPH1 mutant exhibit an elongated cell shape and impaired mitochondrial trafficking (Li and Sewer, 2010). However, we also found that silencing DIAPH1 renders the cells unable to secrete cortisol. Collectively these data indicate that the actin-nucleating and/or microtubule-stabilizing activity of the protein must be precisely controlled to ensure optimal cortisol output. To define the functional significance of phosphorylation of the protein in modulating mitochondrial movement, we transfected H295R cells with GFP-tagged wild-type or alanine-substituted mutant DIAPH1 and quantified the rate of mitochondrial trafficking by real-time video imaging, as previously described (Li and Sewer, 2010). As shown in Figure 6, the rate of mitochondrial movement in cells expressing the wild-type protein was increased 1.8-fold by Bt2cAMP. In contrast, the rate of mitochondrial movement in unstimulated cells expressing the DIAPH1 T759A mutant was reduced by 70%. Of interest, although the rate of mitochondrial trafficking was significantly decreased in control cells expressing mutant DIAPH1, Bt2cAMP stimulation increased the rate of mitochondrial movement by 1.9-fold (Figure 6), suggesting that cAMP signaling might modulate DIAPH1 function via mechanisms that are independent of phosphorylation at T759.
FIGURE 6:
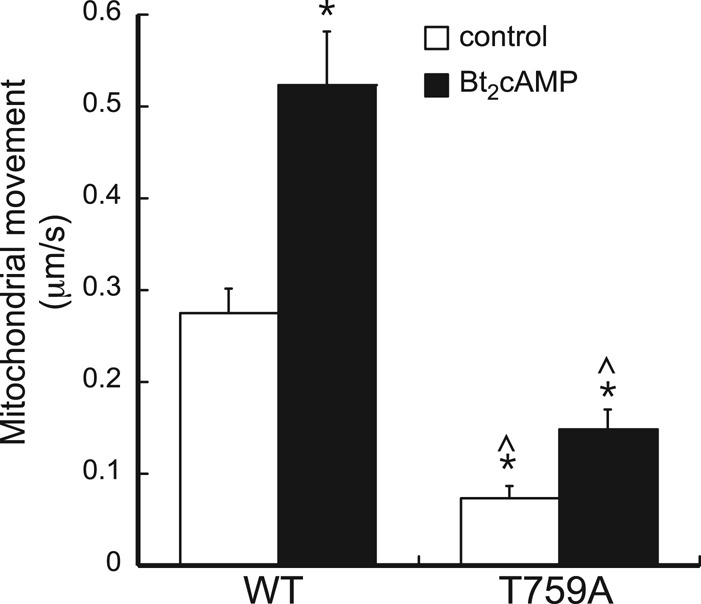
(A) Representative confocal microscopy image of H295R cells that were plated onto cover slips and transfected with WT or T759A-mutant pEGFP-DIAPH1 and Mito Tracker Red. (B) Graphical analysis of mitochondrial movement in H295R cells transfected with WT or T759A-mutant pEGFP-DIAPH1 and treated with 0.4 mM Bt2cAMP. The Zeiss Imaging Physiology platform was used to determine the velocity of movement of individual mitochondrion from video elapsed recordings. Time of exposure of each frame was 1 s, with a 3-s interval between frames. Mitochondrion movement (μm/s) was calculated by subtracting the change in position after each frame interval. Data graphed represents mean ± SEM of three separate experiments. At least 20 mitochondria were analyzed in each experimental replicate. Asterisks and carats denote statistically different (p < 0.05) from WT control and Bt2cAMP-treated groups, respectively.
DISCUSSION
In the human adrenal cortex ACTH coordinates multiple regulatory mechanisms to tightly control the production of cortisol. We previously identified a novel role for ACTH signaling in regulating the rate of mitochondrial movement in H295R human adrenocortical cells (Li and Sewer, 2010). ACTH-stimulated activation of the cAMP/PKA signaling pathway rapidly activates RhoA, promotes the interaction of the GTPase with DIAPH1, and stimulates the subsequent phosphorylation of RhoA. Overexpression of either a dominant-negative RhoA or a constitutively active DIAPH1 mutant attenuated the ability of ACTH or Bt2cAMP to increase the rate of mitochondrial movement and decreased the secretion of cortisol, indicating key roles for these proteins in glucocorticoid production. However, silencing DIAPH1 also reduced cortisol biosynthesis, suggesting that the expression and activity of the protein needs to be precisely fine tuned to achieve optimal steroid hormone capacity. These findings also led us to hypothesize that DIAPH1 may act as a transducer in ACTH/cAMP-stimulated cortisol production in the adrenal cortex and that interaction of the protein with other downstream effectors in adrenocortical cells is required for cortisol biosynthesis. We show here that DIAPH1 copurifies with multiple proteins (Table 1) involved in regulating cytoskeletal dynamics, cell signaling, and lipid homeostasis.
Our present finding that DIAPH1 interacts with RhoA is consistent with both our previous studies (Li and Sewer, 2010) and findings of other laboratories (Watanabe et al., 1997, 1999; Ishizaki et al., 2001; Lammers et al., 2005; Otomo et al., 2005; Yamana et al., 2006; Higashi et al., 2008; Zaoui et al., 2008; Narumiya et al., 2009; Tanizaki et al., 2010). Moreover, the identification of tubulin and actin as DIAPH1 binding partners is consistent with a large body of literature (Palazzo et al., 2001a; Wen et al., 2004; Eng et al., 2006; Bartolini et al., 2008). However, in addition to these proteins, we now show that DIAPH1 copurifies with several other proteins, including vimentin, microtubule-associated protein 4 (MAP4), son of sevenless (SOS), kinesin, dynein, and A-kinase anchor protein 13 (also known as AKAP-Lbc). Coimmunoprecipitation assays confirmed interactions with kinesin, vimentin, and dynamin-1 (Figure 1B). Roles for kinesin and dynamin-1 in regulating organelle positioning and cytoskeletal dynamics are well established. Given that DIAPH1 interacts with microtubules and kinesin regulates the transport of cargo and organelles along microtubules (Allan and Schroer, 1999; Gross, 2004; Frederick and Shaw, 2007; Verhey and Hammond, 2009), the identification of kinesin as a DIAPH1 binding partner is consistent with the role of this motor protein in cellular transport. Of note, targeted disruption of kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria (Tanaka et al., 1998), a phenotype that we observed in cells overexpressing constitutively active DIAPH1 (Li and Sewer, 2010). Our finding that DIAPH1 copurifies with other proteins involved in vesicle and organelle transport, including dynein and dynamin-1 (Table 1), suggests that these proteins act to coordinate communication between mitochondria and the ER for the transport of 11-deoxycortisol during cortisol biosynthesis. Of note, vimentin was recently found to play a pivotal role in ovarian and adrenocortical steroidogenesis, where targeted disruption of the gene resulted in reduced corticosterone production in both male and female mice and impaired progesterone secretion in female mice (Shen et al., 2012). Studies using vimentin-null cells revealed defective movement of cholesterol from lipid droplets to mitochondria (Shen et al., 2012), and the interaction of vimentin with hormone-sensitive lipase regulates lipolysis and lipid droplet homeostasis (Shen et al., 2010). Our findings suggest that in addition to coordinating cholesterol movement, vimentin might also regulate the movement of substrates between the ER and mitochondria, and add to a growing list of roles for vimentin in coordinating communication between vesicles, organelles, and motor proteins (Hall, 1997; Schweitzer and Evans, 1998; Ivaska et al., 2007).
The identification of AKAP13 as a DIAPH1 binding partner (Table 1) also supports a role for ACTH-stimulated assembly of a complex of proteins that facilitate glucocorticoid production. AKAP13 is a member of the A-kinase anchoring protein family that has been shown to bridge PKA and RhoA signaling by virtue of its innate guanine exchange factor activity (Diviani et al., 2001, 2004; Klussmann et al., 2001; Tan et al., 2002; Kino et al., 2006). Because ACTH/cAMP activates both PKA and RhoA (Li and Sewer, 2010) and DIAPH1 coordinates AKAP13 and Rho signaling (Kamasani et al., 2007), we postulate that AKAP13 plays a central role in spatially integrating RhoA and PKA activities to the mitochondria–ER interface.
We were particularly intrigued by the identification of members of the ORP family of lipid-binding proteins (Table 1). ORP2 and ORP10 are members of the oxysterol-related lipid-binding protein family. Members of this protein family are emerging as regulators of lipid metabolism by serving as transporters between organelles, signaling mediators, and sensors of cellular lipid concentrations (Lehto et al., 2001; Olkkonen and Levine, 2004; Fairn and McMaster, 2008; Ridgway, 2010). By virtue of the ability of several members of this protein family to bind to cholesterol and oxysterols, ORPs are implicated in cholesterol metabolism (Wang et al., 2002; Du et al., 2011). However, ORPs have also been shown to bind to phospholipids, notably phosphatidic acid and phosphotidylinositides (Xu et al., 2001; Stefan et al., 2011).
Our finding that DIAPH1 is a phosphoprotein is consistent with published proteomic studies using a Cdk1–cyclin B substrate trap, which demonstrated that DIAPH1 is phosphorylated at Thr-759 in HeLa cells (Blethrow et al., 2008). In H295R cells, Bt2cAMP-stimulated phosphorylation of this site is dependent on ERK kinase activity (Figure 3B). Thr-759 lies in a putative MAP kinase phosphorylation motif, which suggests that other members of this family may target the protein in response to specific cellular cues. Our findings that the T759A mutant is more stable than the wild-type protein and that the protein is sumoylated (Figure 4) support a role for cAMP signaling in activating the phosphorylation and subsequent sumoylation of the protein, potentially phosphorylation triggering the activation of a degradative clock. The ubiquitin-mediated degradation of mDia2 (DIAPH2) is required for completion of mitosis in HeLa and HEK293T cells (DeWard and Alberts, 2009). Moreover, we have preliminary evidence to suggest that silencing DIAPH1 leads to aberrant cortisol secretion (Li and Sewer, 2010), supporting our previous finding that overexpression of a constitutively active DIAPH1 impairs cortisol secretion (Li and Sewer, 2010) and suggesting that tight control of DIAPH1 expression is required for coordinating optimal steroid hormone production in response to ACTH.
In summary, we show that DIAPH1 interacts with multiple proteins in H295R human adrenocortical cells and that cAMP signaling modulates the interaction of a subset of proteins with DIAPH1. We also show that Bt2cAMP promotes the PKA- and ERK-dependent phosphorylation of DIAPH1 at T759 and that mutation of this site alters the stability of the protein and the rate of cAMP-stimulated mitochondrial movement. These findings support a model in which DIAPH1 cAMP signaling triggers posttranslational modification of the protein, thus allowing for tight control of both the expression and activity of the protein, and ultimately cortisol biosynthesis.
MATERIALS AND METHODS
Chemicals
Bt2cAMP was obtained from Sigma-Aldrich (St. Louis, MO). H89, CHX, MG132, and U0126 were purchased from EMD Biosciences (La Jolla, CA).
Cell culture and transient transfection
H295R adrenocortical cells were donated by William E. Rainey (Health Sciences University of Georgia, Augusta, GA) and cultured in Dulbecco's modified Eagle's/F12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% Nu-Serum I (BD Biosciences, Palo Alto, CA), 1% ITS Plus (BD Biosciences, San Diego, CA), and antibiotics.
Mass spectrometry
Five hundred milligrams of total cellular protein isolated from H295R cells was precleared with immunoglobulin G (IgG) and incubated with anti-DIAPH1 (Millipore, Billerica, MA) and protein A/G (Santa Cruz Biotechnology, Santa Cruz, CA) overnight on a tube rotator at 4ºC. Immunoprecipitants were washed twice with RIPA buffer (50 mM Tris-Cl, pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 0.4 mM EDTA, 150 nM aprotinin, 1 mM leupeptin, 1 mM E-64, 500 mM 4-(2-aminoethyl)benzenesulfonylfluoride) and three times with phosphate-buffered saline (PBS) and then subjected to SDS–PAGE and Coomassie staining. Bands were excised, destained, reduced, alkylated, and trypsinized using an IGD Kit (Sigma-Aldrich). Peptides were concentrated using ZipTips (C18; Millipore), mixed with matrix, and spotted onto a matrix-assisted laser desorption ionization plate for tandem time-of-flight mass spectrometric analysis (4700 Proteomics Analyzer; Applied Biosystems, Foster City, CA).
Coimmunoprecipitation
H295R cells were plated onto 100-mm dishes and transfected with 5 μg of murine pEGFP-mDia (generously donated by Shuh Narumiya, Kyoto University Faculty of Medicine, Kyoto, Japan) using GeneJuice (3 μl/μg DNA; EMD Biosciences). At 72 h after transfection, cells were treated for 30 min with 0.4 mM Bt2cAMP and harvested into 100 μl of RIPA buffer. In some experiments, cells were transfected with GFP-tagged wild-type or T751A DIAPH1 (corresponds to human T759A). For sumoylation studies, cells were transfected with wild-type or T559A pEGFP-mDia and pSG5-His-SUMO1 (generated in the laboratory of Stephen Goff, Columbia University, New York, NY; Yueh et al., 2006; purchased from Addgene [Cambridge, MA; plasmid 17271]) and harvested in RIPA containing 20 mM N-ethylmaleimide. Five percent of the lysate was retained (input), and the remaining volume was diluted 10-fold with RIPA. Lysates were precleared with 1 μg of rabbit IgG, 10 mg/ml bovine serum albumin, and 25 μl of protein A/G agarose (Santa Cruz Biotechnology) for 30 min at 4ºC with rotation. Samples were centrifuged for 2 min at 4000 × g and the supernatants incubated with 5 μg of rabbit anti-GFP (A11122; Invitrogen) and 50 μl of protein A/G agarose for 16 h at 4ºC with rotation. The immobilized proteins were washed three times in RIPA and twice in PBS (5 min each wash), and the eluted proteins were separated by SDS–PAGE (10% acrylamide). Output proteins were transferred to polyvinylidene fluoride (PVDF) membranes, incubated with anti–dynamin 1 (1:1000; sc-12724; Santa Cruz Biotechnology), anti-kinesin, heavy chain (1:1000; MAB1613; Millipore), anti–β-tubulin (1:1000; sc-23949; Santa Cruz Biotechnology), anti–β-actin (1:1000; sc-81178; Santa Cruz Biotechnology), anti-vimentin (1:7500; 574P; Cell Signaling), anti-ORP2 (1:1000; sc-66567; Santa Cruz Biotechnology), anti-ORP4 (1:1000; sc-376601, Santa Cruz Biotechnology), anti-ORP10 (1:1000; GeneTex, Irvine, CA), anti-RhoA (1:1000; sc-418; Santa Cruz Biotechnology), anti-SUMO1 (1:500; sc-9060; Santa Cruz Biotechnology), and alkaline phosphate–conjugated secondary antibodies (1:5000; ECF Western Blotting Kit; GE Biosciences, Piscataway, NJ), and imaged using a VersaDoc 4000 Imager (Bio-Rad, Hercules, CA). Input samples were subjected to SDS–PAGE and Western blotting using an anti-GFP antibody (Invitrogen).
Site-directed mutagenesis and DIAPH1 phosphomutant screening
PCR was used to generate alanine-substituted variants of the pEGFP-mDia expression plasmid using the Stratagene QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). All mutants were confirmed by sequencing (Retrogen, San Diego, CA). Putative phosphomutants were tested by transfecting cells with wild-type or mutant pEGFP-mDia and then immunoprecipitating the isolated lysates with anti-GFP (Invitrogen). The purified proteins were separated by SDS–PAGE, transferred to PVDF membranes, and incubated with anti–phospho-Ser/Thr-Pro antibody (1:1000; MAB1613; Millipore). Ten percent of the lysates were (input samples) subjected to SDS–PAGE and Western blotting using an anti-GFP antibody (Invitrogen).
Generation of phosphospecific DIAPH1 antibody
An affinity-purified phosphospecific antibody was produced to recognize DIAPH1 phosphorylated at T759 by Pacific Immunology (Ramona, CA). Two splice variants of DIAPH1 have been identified in humans, isoforms 1 and 2. Isoform 1 is the longer form (1272 amino acids), and isoform 2 (1263 amino acids) lacks an in-frame exon in the 5′-coding region. However, the amino acid sequence of the antigenic region is identical. The phosphopeptide antigen, LPFGLT[PO3]PKKLYKPEVQLRR-C, corresponds to the region between amino acid region 754–772 of isoform 2 and regions 763 and 781 of isoform 1. A cysteine residue was added to the C-terminus of the peptide to facilitate chemical cross-linking. Sera from immunized rabbits were tested for antibody titer, and specificity for the phosphorylated peptide was tested by enzyme-linked immunosorbent assay and subjected to affinity purification. A nonphosphorylated antibody raised against the same peptide sequence was also produced.
Western blotting
H295R cells were treated with 10 μM H89 or 10 μM U0126 for 15 min and treated with Bt2cAMP (0.4 mM) for 30 min, and whole-cell extracts were prepared in RIPA. Lysates were separated by SDS–PAGE and transferred to PVDF membranes. In some experiments, cells were transfected with wild-type or T751A pEGFP-mDia and then treated with 0.4 mM Bt2cAMP for 30 min or with 20 μM MG132 for 6 h. In other experiments, cells were treated with 1 μM S1P for 15–60 min or with 50 μg/ml CHX for 3 or 6 h. Some lysates were incubated with λ-phosphatase (EMD Biosciences) before SDS–PAGE. Whole-cell lysates were immunoprecipitated with anti-GFP antibodies and protein A/G plus-agarose and washed, and the purified protein was separated by SDS–PAGE. Blots were incubated with anti-DIAPH1 (1:2000; AB3888; Millipore) or anti–phospho Thr-759 DIAPH1 antibody (1:250; Pacific Immunology) and alkaline phosphate–conjugated secondary antibody (1:5000; ECF Western Blotting Kit; GE Healthcare, Piscataway, NJ) and imaged on a VersaDoc 4000 (Bio-Rad). Densitometric analysis was carried out using Quantity One software (Bio-Rad).
Confocal microscopy and time-lapse video imaging
Cells were plated onto coverslips and transfected with 50 ng of wild-type or T759A pEGFP-DIAPH1 (transfection efficiency ranged from 70 to 85%) using Gene Juice (EMD Biosciences) and Opti-MEM (Invitrogen) for 48 h. Confocal images were collected in control and treated cells using an LSM510 confocal microscope (Carl Zeiss, Thornwood, NY) equipped with a helium–neon coherent laser, as previously described (Kulik et al., 2002; Li and Sewer, 2010). Emissions were collected with a C-Apochromat 40/1.3 numerical aperture oil immersion objective (Carl Zeiss) using a 560-nm long-pass filter. The Zeiss Imaging Physiology platform was used to determine the velocity of movement of individual mitochondria from taped recordings. To quantify the rate of movement, we set the time of exposure of each frame to 1 s, with a 3-s interval between successive frames. The track of mitochondria was obtained by subtracting the change in position after each frame interval. Only mitochondria that changed position during a given time interval were calculated. For these mobile mitochondria, the translocation between neighbor frames was measured, and the mean rate of movement was calculated. Each experiment was performed at least three times, and the movement of at least 20 mitochondria was analyzed in each experimental condition.
Statistical analysis
One-way analysis of variance, Tukey–Kramer multiple comparison, and unpaired Student's t tests were performed using InStat software (GraphPad Software, San Diego, CA). Significant differences from a compared value were defined as p < 0.05.
Acknowledgments
This work was supported by National Institutes of Health Grant DK094151.
Abbreviations used:
- ACTH
adrenocorticotropin
- Bt2cAMP
dibutyryl cAMP
- ERK
extracellular signal-regulated kinase
- DIAPH1
diaphanous homologue 1
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-08-0597) on January 16, 2013.
REFERENCES
- Alberts AS. Identification of a carboxyl-terminal diaphanous-related formin homology protein autoregulatory domain. J Biol Chem. 2001;276:2824–2830. doi: 10.1074/jbc.M006205200. [DOI] [PubMed] [Google Scholar]
- Alberts AS, Bouquin N, Johnston LH, Treisman R. Analysis of RhoA-binding proteins reveals an interaction domain conserved in heterotrimeric G protein beta subunits and the yeast response regulator protein Skn7. J Biol Chem. 1998;273:8616–8622. doi: 10.1074/jbc.273.15.8616. [DOI] [PubMed] [Google Scholar]
- Allan VJ, Schroer TA. Membrane motors. Curr Opin Cell Biol. 1999;11:476–482. doi: 10.1016/s0955-0674(99)80068-4. [DOI] [PubMed] [Google Scholar]
- Bartolini F, Gundersen GG. Formins and microtubules. Biochim Biophys Acta. 2010;1803:164–173. doi: 10.1016/j.bbamcr.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolini F, Moseley JB, Schmoranzer J, Cassimeris L, Goode BL, Gundersen GG. The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J Cell Biol. 2008;181:523–536. doi: 10.1083/jcb.200709029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci USA. 2008;105:1442–1447. doi: 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook TA, Nagasaki T, Gundersen GG. Rho guanosine triphosphatase mediates the selective stabilization of microtubules induced by lysophosphatidic acid. J Cell Biol. 1998;141:175–185. doi: 10.1083/jcb.141.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland JW, Treisman R. The diaphanous-related formin mDia1 controls serum response factor activity through its effects on actin polymerization. Mol Biol Cell. 2002;13:4088–4099. doi: 10.1091/mbc.02-06-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWard AD, Alberts AS. Ubiquitin-mediated degradation of the formin mDia2 upon completion of cell division. J Biol Chem. 2009;284:20061–20069. doi: 10.1074/jbc.M109.000885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWard AD, Eisenmann KM, Matheson SF, Alberts AS. The role of formins in human disease. Biochim Biophys Acta. 2010;1803:226–233. doi: 10.1016/j.bbamcr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- Diviani D, Abuin L, Cotecchia S, Pansier L. Anchoring of both PKA and 14–3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004;23:2811–2820. doi: 10.1038/sj.emboj.7600287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diviani D, Soderling J, Scott JD. AKAP-Lbc anchors protein kinase A and nucleates Galpha12-selective Rho-mediated stress fiber formation. J Biol Chem. 2001;276:44247–44257. doi: 10.1074/jbc.M106629200. [DOI] [PubMed] [Google Scholar]
- Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol. 2011;192:121–135. doi: 10.1083/jcb.201004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CH, Huckaba TM, Gundersen GG. The formin mDia regulates GSK3beta through novel PKCs to promote microtubule stabilization but not MTOC reorientation in migrating fibroblasts. Mol Biol Cell. 2006;17:5004–5016. doi: 10.1091/mbc.E05-10-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairn GD, McMaster CR. Emerging roles of the oxysterol-binding protein family in metabolism, transport, and signaling. Cell Mol Sci. 2008;65:228–236. doi: 10.1007/s00018-007-7325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8:1668–1675. doi: 10.1111/j.1600-0854.2007.00644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geneste O, Copeland JW, Treisman R. LIM kinase and diaphanous cooperate to regulate serum response factor and actin dynamics. J Cell Biol. 2002;157:831–838. doi: 10.1083/jcb.200203126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath SD, Narumiya S, Dhawan J. The RhoA effector mDiaphanous regulates MyoD expression and cell cycle progression via SRF-dependent and SRF-independent pathways. J Cell Sci. 2007;120:3086–3098. doi: 10.1242/jcs.006619. [DOI] [PubMed] [Google Scholar]
- Gross SP. Hither and yon: a review of bi-directional microtubule-based transport. Phys Biol. 2004;1:R1–R11. doi: 10.1088/1478-3967/1/2/R01. [DOI] [PubMed] [Google Scholar]
- Hall PF. The roles of calmodulin, actin, and vimentin in steroid synthesis by adrenal cells. Steroids. 1997;62:185–189. doi: 10.1016/s0039-128x(96)00179-1. [DOI] [PubMed] [Google Scholar]
- Higashi T, et al. Biochemical characterization of the Rho GTPase-regulated actin assembly by diaphanous-related formins, mDia1 and Daam1, in platelets. J Biol Chem. 2008;283:8746–8755. doi: 10.1074/jbc.M707839200. [DOI] [PubMed] [Google Scholar]
- Ishizaki T, Morishima Y, Okamoto M, Furuyashiki T, Kato T, Narumiya S. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat Cell Biol. 2001;3:8–14. doi: 10.1038/35050598. [DOI] [PubMed] [Google Scholar]
- Ivaska J, Pallari HM, Nevo J, Eriksson JE. Novel functions of vimentin in cell adhesion. Exp Cell Res. 2007;313:2050–2062. doi: 10.1016/j.yexcr.2007.03.040. [DOI] [PubMed] [Google Scholar]
- Janes ME, Chu KM, Clark AJ, King PJ. Mechanisms of adrenocorticotropin-induced activation of extracellularly regulated kinase 1/2 mitogen-activated protein kinase in the human H295R adrenal cell line. Endocrinology. 2008;149:1898–1905. doi: 10.1210/en.2007-0949. [DOI] [PubMed] [Google Scholar]
- Kamasani U, Duhadaway JB, Alberts AS, Prendergast GC. mDia function is critical for the cell suicide program triggered by farnesyl transferase inhibition. Cancer Biol Ther. 2007;6:1422–1427. doi: 10.4161/cbt.6.9.4546. [DOI] [PubMed] [Google Scholar]
- Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2001;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, et al. Rho family guanine nucleotide exchange factor Brx couples extracellular signals to the glucocorticoid signaling system. J Biol Chem. 2006;281:9118–9126. doi: 10.1074/jbc.M509339200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klussmann E, Edemir B, Pepperle B, Tamma G, Henn V, Klauschenz E, Hundsrucker C, Maric K, Rosenthal W. Ht31: the first protein kinase A anchoring protein to integrate protein kinase A and Rho signaling. FEBS Lett. 2001;507:264–268. doi: 10.1016/s0014-5793(01)02995-7. [DOI] [PubMed] [Google Scholar]
- Kulik AV, Gioeva FK, Minin AA. Videomicroscopic studies of the movement of mitochondria. Russ J Dev Biol. 2002;33:299–305. [Google Scholar]
- Lammers M, Rose R, Scrima A, Wittinghofer A. The regulation of mDia1 by autoinhibition and its release by Rho*GTP. EMBO J. 2005;24:4176–4187. doi: 10.1038/sj.emboj.7600879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le T, Schimmer BP. The regulation of MAPKs in Y1 mouse adrenocortical tumor cells. Endocrinology. 2001;142:4282–4287. doi: 10.1210/endo.142.10.8441. [DOI] [PubMed] [Google Scholar]
- Lehto M, Laitinen S, Chinetti G, HJohansson M, Ehnholm C, Staels B, Ikonen, Olkkonen VM. The OSBP-related protein family in humans. J Lipid Res. 2001;42:1203–1213. [PubMed] [Google Scholar]
- Li D, Sewer MB. RhoA and DIAPH1 mediate adrenocorticotropin-stimulated cortisol biosynthesis by regulating mitochondrial trafficking. Endocrinology. 2010;151:4313–4323. doi: 10.1210/en.2010-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi CF, Costa ET, Schwindt TT, Armelin HA. Role of ERK/MAP kinase in mitogenic interaction between ACTH and FGF2 in mouse Y1 adrenocortical tumor cells. Endocr Res. 2000;26:873–877. doi: 10.3109/07435800009048611. [DOI] [PubMed] [Google Scholar]
- Nagasaki T, Gundersen GG. Depletion of lysophosphatidic acid triggers a loss of oriented detyrosinated microtubules in motile fibroblasts. J Cell Sci. 1996;109:2461–2469. doi: 10.1242/jcs.109.10.2461. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis, and invasion. Cancer Metastasis Rev. 2009;28:65–76. doi: 10.1007/s10555-008-9170-7. [DOI] [PubMed] [Google Scholar]
- Olkkonen VM, Levine TP. Oxysterol binding proteins: in more than one place at one time. Biochem Cell Biol. 2004;82:87–98. doi: 10.1139/o03-088. [DOI] [PubMed] [Google Scholar]
- Otomo T, Otomo C, Tomchick DR, Machius M, Rosen MK. Structural basis of Rho GTPase-mediated activation of the formin mDia1. Mol Cell. 2005;18:273–281. doi: 10.1016/j.molcel.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Cook TA, Alberts AS, Gundersen GG. mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat Cell Biol. 2001a;3:723–729. doi: 10.1038/35087035. [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Joseph HL, Chen YJ, Dujardin DL, Alberts AS, Pfister KK, Vallee RB, Gundersen GG. Cdc42, dynein, and dynactin regulate MTOC reorientation independent of Rho-regulated microtubule stabilization. Curr Biol. 2001b;11:1536–1541. doi: 10.1016/s0960-9822(01)00475-4. [DOI] [PubMed] [Google Scholar]
- Ridgway N. Oxysterol-binding proteins. Subcell Biochem. 2010;51:159–182. doi: 10.1007/978-90-481-8622-8_6. [DOI] [PubMed] [Google Scholar]
- Schonichen A, Geyer M. Fifteen formins for an actin filament: a molecular view on the regulation of human formins. Biochim Biophys Acta. 2010;1803:152–163. doi: 10.1016/j.bbamcr.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Schweitzer SC, Evans RM. Vimentin and lipid metabolism. Subcell Biochem. 1998;31:437–462. [PubMed] [Google Scholar]
- Shen WJ, Patel S, Eriksson JE, Kraemer FB. Vimentin is a functional partner of hormone sensitive lipase and facilitates lipolysis. J Proteome Res. 2010;9:1786–1794. doi: 10.1021/pr900909t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WJ, Zaidi SK, Patel S, Cortez Y, Ueno M, Azhar R, Azhar S, Kraemer FB. Ablation of vimentin results in defective steroidogenesis. Endocrinology. 2012;153:3249–3257. doi: 10.1210/en.2012-1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A, Nyitrai M, Vetter IR, Kuhlmann D, Bugyi B, Narumiya S, Geeves MA, Wittinghofer A. The core FH2 domain of diaphanous-related formins is an elongated actin binding protein that inhibits polymerization. Mol Cell. 2004;13:511–522. doi: 10.1016/s1097-2765(04)00059-0. [DOI] [PubMed] [Google Scholar]
- Stefan CJ, Manford AG, Baird D, Yamada-Hanff Mao Y, Emr SD. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 2011;144:389–401. doi: 10.1016/j.cell.2010.12.034. [DOI] [PubMed] [Google Scholar]
- Tan YC, Wu H, Wang WN, Zheng Y, Wang ZX. Characterization of the interactions between the small GTPase RhoA and its guanine nucleotide exchange factors. Anal Biochem. 2002;310:156–162. doi: 10.1016/s0003-2697(02)00382-2. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell. 1998;93:1147–1158. doi: 10.1016/s0092-8674(00)81459-2. [DOI] [PubMed] [Google Scholar]
- Tanizaki H, et al. Rho-mDia1 pathway is required for adhesion, migration, and T-cell stimulation in dendritic cells. Blood. 2010;116:5875–5884. doi: 10.1182/blood-2010-01-264150. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Wang C, JeBailey L, Ridgway N. Oxytsterol-binding-protein (OSBP)-related protein 4 binds 25-hydroxycholesterol and interacts with vimentin intermediate filaments. Biochem J. 2002;361:461–472. doi: 10.1042/0264-6021:3610461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Watanabe NMP, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Eng CH, Schmoranzer J, Cabrera-Poch N, Morris EJS, Chen M, Wallar BJ, Alberts AS, Gundersen GG. EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat Cell Biol. 2004;6:820–830. doi: 10.1038/ncb1160. [DOI] [PubMed] [Google Scholar]
- Xie Y, Tan EJ, Wee S, Manser E, Lim L, Koh CG. Functional interactions between phosphatase POPX2 and mDia modulate RhoA pathways. J Cell Sci. 2008;121:514–521. doi: 10.1242/jcs.013557. [DOI] [PubMed] [Google Scholar]
- Xu Y, Liu Y, Ridgway ND, McMaster CR. Novel members of the human oxysterol-binding protein family bind phospholipids and regulate vesicular transport. J Biol Chem. 2001;276:18407–18414. doi: 10.1074/jbc.M101204200. [DOI] [PubMed] [Google Scholar]
- Yamana N, et al. The Rho-mDia1 pathway regulates cell polarity and focal adhesion turnover in migrating cells through mobilizing Apc and c-Src. Mol Cell Biol. 2006;26:6844–6858. doi: 10.1128/MCB.00283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young KG, Copeland JW. Formins in cell signaling. Biochim Biophys Acta. 2010;1803:183–190. doi: 10.1016/j.bbamcr.2008.09.017. [DOI] [PubMed] [Google Scholar]
- Yueh A, Leung J, Bhattacharyya S, Perrone LA, de los Santos K, Pu SY, Goff SP. Interaction of Moloney murine leukemia virus capsid with Ubc9 and PIASy mediates SUMO-1 addition required early in infection. J Virol. 2006;80:342–352. doi: 10.1128/JVI.80.1.342-352.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaoui K, Honore S, Isnardon D, Braguer D, Badache A. Memo-RhoA-mDia1 signaling controls microtubules, the actin network, and adhesion site formation in migrating cells. J Cell Biol. 2008;183:401–408. doi: 10.1083/jcb.200805107. [DOI] [PMC free article] [PubMed] [Google Scholar]