

Bcl-2 interacts with ERK to suppress apoptosis. Hydrogen peroxide disrupts the interaction through Bcl-2 cysteine oxidation, which promotes apoptosis. These findings provide a novel redox regulatory mechanism that controls apoptosis via Bcl-2 cysteine oxidation, which could aid in the understanding of pathogenesis under oxidative stress conditions.
Abstract
Hydrogen peroxide is a key mediator of oxidative stress known to be important in various cellular processes, including apoptosis. B-cell lymphoma-2 (Bcl-2) is an oxidative stress–responsive protein and a key regulator of apoptosis; however, the underlying mechanisms of oxidative regulation of Bcl-2 are not well understood. The present study investigates the direct effect of H2O2 on Bcl-2 cysteine oxidation as a potential mechanism of apoptosis regulation. Exposure of human lung epithelial cells to H2O2 induces apoptosis concomitant with cysteine oxidation and down-regulation of Bcl-2. Inhibition of Bcl-2 oxidation by antioxidants or by site-directed mutagenesis of Bcl-2 at Cys-158 and Cys-229 abrogates the effects of H2O2 on Bcl-2 and apoptosis. Immunoprecipitation and confocal microscopic studies show that Bcl-2 interacts with mitogen-activated protein kinase (extracellular signal-regulated kinase 1/2 [ERK1/2]) to suppress apoptosis and that this interaction is modulated by cysteine oxidation of Bcl-2. The H2O2-induced Bcl-2 cysteine oxidation interferes with Bcl-2 and ERK1/2 interaction. Mutation of the cysteine residues inhibits the disruption of Bcl-2–ERK complex, as well as the induction of apoptosis by H2O2. Taken together, these results demonstrate the critical role of Bcl-2 cysteine oxidation in the regulation of apoptosis through ERK signaling. This new finding reveals crucial redox regulatory mechanisms that control the antiapoptotic function of Bcl-2.
INTRODUCTION
Oxidative stress has been implicated in the pathogenesis of several diseases, including cancer and neurodegenerative and cardiovascular diseases (Halliwell, 2007; Valko et al., 2007). Oxidative stress is an imbalance between the production of reactive oxygen species (ROS) and their elimination by the host's defense antioxidant systems. Disturbance in this balance can lead to toxic effects through the accumulation of peroxides and free radicals, which damage the cell and its components. Some ROS can act as messengers through redox regulation of cellular signaling pathways (Forman, 2009).
Apoptosis plays a crucial role in the removal of unwanted or transformed cells. Defects in apoptosis regulatory mechanisms contribute to abnormal cell growth, representing a major causative factor in the development and progression of cancer (Hanahan and Weinberg, 2000; Kasibhatla and Tseng, 2003; Shivapurkar et al., 2003). Apoptosis is a tightly regulated process that operates along two major pathways—the extrinsic and intrinsic pathways (Hancock et al., 2001; Fruehauf and Meyskens, 2007). B-cell lymphoma-2 (Bcl-2) is a key regulator of the intrinsic pathway by interfering with the release of cytochrome c from the mitochondria or its binding to Apaf-1 through the interaction with Bax (Korsmeyer et al., 1995; Gupta, 2003). Bcl-2 also plays a role in the extrinsic pathway, which requires amplification of the apoptosis signal through the intrinsic pathway, that is, via Bid cleavage (Gupta, 2003). Several apoptosis-resistant cell lines and tumor specimens have been shown to overexpress Bcl-2 (Ben-Ezra et al., 1994; Jiang et al., 1995; Osford et al., 2004), suggesting its role in cancer pathogenesis and chemoresistance.
Expression of Bcl-2 is tightly regulated by mechanisms that include transcription, dimerization, and degradation (Willimott and Wagner, 2010). Among them, Bcl-2 degradation is a key mechanism of Bcl-2 regulation during apoptosis (Dimmeler et al., 1999; Chanvorachote et al., 2006). Previous studies showed that Bcl-2 degradation is mediated primarily through the ubiquitin-proteasome pathway and is associated with ROS generation (Dimmeler et al., 1999; Azad et al., 2008; Wang et al., 2008; Luanpitpong et al., 2011). In lung cancer cells, ROS formation is induced by various apoptosis-inducing agents, including cisplatin, chromium, and lipoic acid, and has been shown to be involved in the regulation of Bcl-2 (Moungjaroen et al., 2006; Azad et al., 2008; Wang et al., 2008). However, the underlying mechanisms of Bcl-2 regulation are unclear and are investigated in this study.
Increasing evidence suggests that posttranslational regulation of proteins via redox reactions can serve as a key mechanism for controlling protein function (Reynaert et al., 2006; Michalek et al., 2007; Yang et al., 2012). Key targets for redox regulation of proteins are cysteine residues (Rehder and Borges, 2010; Yang et al., 2012). Bcl-2 protein contains four Bcl-2 homology (BH) domains (BH1–BH4) with two cysteine residues at position 158 in the α5 domain and position 229 in the carboxyl-terminal membrane anchor domain (Cys-158 and Cys-229). The main objective of this study is to investigate the potential redox modification of Bcl-2 and its role in the regulation of apoptosis under oxidative stress conditions. Specifically, we address 1) whether the cysteine residues are targets of oxidative modification by H2O2 in human lung epithelial cells, 2) whether such modification plays a role in apoptosis, and 3) if so, by what mechanisms. To address these questions, we constructed a Bcl-2 mutant plasmid replacing the two existing cysteine residues of Bcl-2 with alanines and expressed them in human lung cells to aid the mechanistic investigations of Bcl-2 cysteine oxidation and its role in apoptosis. Of note, mutation of these Bcl-2 cysteines (to alanines) preserved the overall structure and conformation of protein.
RESULTS
Inhibition of hydrogen peroxide–induced lung cell apoptosis by antioxidants
H2O2 is a signaling molecule generated during oxidative stress and in response to various apoptotic stimuli (Moungjaroen et al., 2006; Wang et al., 2008). To study the direct apoptotic effect of H2O2 in human lung epithelial cells, which are the primary target of inhaled prooxidants, we treated epithelial H460 cells with various pathophysiologically relevant concentrations of H2O2 (Halliwell et al., 2000) and analyzed for apoptosis by Hoechst 33342 and annexin V assays. Figure 1, A and B, shows that treatment of the cells with H2O2 causes a dose-dependent increase in apoptotic cell death, and cotreatment of the cells with antioxidants, including N-acetylcysteine (NAC), reduced glutathione (GSH), and catalase, effectively inhibits the apoptosis. To correlate the apoptosis results with cellular oxidative stress, we analyzed cells for ROS levels using a fluorescent probe, 2′7′-dihydrodichlorofluorescein diacetate (H2DCF-DA). Figure 1C shows that H2O2 induced a dose-dependent increase in cellular fluorescence intensity; the effect can be inhibited by the antioxidant NAC. A similar inhibitory effect was observed with GSH and catalase, although their effect was slightly less efficient (unpublished data). Flow cytometric analysis of apoptotic and necrotic cell death using annexin V and propidium iodide assays shows that apoptosis is the primary mode of cell death induced by H2O2 (Figure 1D) and that this effect is inhibited by the antioxidant NAC (Figure 1E).
FIGURE 1:

Hydrogen peroxide induces apoptosis of human lung epithelial H460 cells and its inhibition by antioxidants. (A) Subconfluent monolayers (80%) of H460 cells were treated with various pathophysiologically relevant concentrations of H2O2 (0–400 μM) for 24 h and analyzed for apoptosis by Hoechst 33342 assay. (B) Cells were similarly treated with H2O2 (300 μM) in the presence or absence of various antioxidants, including NAC (5 mM), GSH (5 mM), and catalase. (5000 U/ml), before analysis for apoptosis by Hoechst assay. (C) Cells were treated with various concentrations of H2O2 in the presence or absence of NAC (2.5 mM) for 2 h, and cellular ROS levels were determined fluorometrically using H2DCF-DA as a fluorescent probe. (D) Cells were treated with H2O2 (300 μM) in the presence or absence of NAC for 24 h and analyzed for apoptosis and necrosis by flow cytometry using annexin V and PI as probes. Histograms show raw data before correcting for background fluorescence, which accounts for ∼21.8% of the signal in the lower right quadrant. The results indicate apoptosis as the primary mode of H2O2-induced cell death. (E) Representative flow cytometric histograms of annexin V comparing cells treated with H2O2 alone and cells treated with H2O2 and NAC (400 μM vs. 400 μM + NAC). A shift to the left indicates the inhibitory effect of NAC on H2O2-induced apoptosis. Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control. #p < 0.05 vs. H2O2-treated control (300 μM).
Hydrogen peroxide induces Bcl-2 down-regulation and extracellular signal-regulated kinase 1/2 activation
Bcl-2 is down-regulated by various apoptotic stimuli through proteasomal degradation (Moungjaroen et al., 2006; Wang et al., 2008). To determine whether H2O2 can induce similar down-regulation of Bcl-2 in H460 cells, we treated the cells with H2O2 and analyzed for Bcl-2 expression by Western blotting. Figure 2A shows that H2O2 treatment substantially down-regulated Bcl-2 in a dose-dependent manner. Because mitogen-activated protein (MAP) kinase activation is linked to oxidant-induced apoptosis (Ueda et al., 2002; Torres and Forman, 2003; Zhuang et al., 2007), we tested the potential involvement of MAP kinase activation in Bcl-2 down-regulation in our cell system. First, cells were treated with H2O2 in the presence or absence of various pharmacological MAP kinase inhibitors, including PD98059 (extracellular signal-regulated kinase 1/2 [ERK1/2] inhibitor), SP600125 (JNK inhibitor), and SB203580 (p38 inhibitor), and their effects on Bcl-2 expression were determined. Figure 2B shows that inhibition of ERK1/2 by PD98059 completely inhibited the effect of H2O2 on Bcl-2 down-regulation, whereas inhibition of JNK and p38 had minimal effect. These results suggest that ERK1/2, but not JNK and p38 kinase, is associated with Bcl-2 expression. Further study revealed that H2O2 did induce activation (phosphorylation) of ERK1/2 in a dose-dependent manner, which is the reverse of its effect on Bcl-2 (Figure 2C). To verify the role of activated ERK1/2 on Bcl-2, we used another ERK1/2 inhibitor, U0216. Figure 2D shows that U0126 dose dependently attenuated the down-regulation of Bcl-2 by H2O2. These results suggest ERK-dependent regulation of Bcl-2, which has not been reported and may be important in oxidant-induced apoptosis.
FIGURE 2:

Effects of H2O2 on Bcl-2 expression and ERK activation. (A) H460 cells were treated with various concentrations of H2O2 (0–400 μM) for 24 h. Cell lysates were prepared and analyzed for Bcl-2 expression by Western blotting. Blots were reprobed with β-actin antibody to confirm equal loading of the samples. Immunoblot signals were quantified by densitometry, and mean data from three independent experiments (one of which is shown here) were normalized to the result obtained in cells without treatment (control). (B) Cells were treated with H2O2 (300 μM) for 12 h in the presence of PD98059 (ERK1/2 inhibitor, 25 μM), SP600125 (JNK inhibitor, 10 μM), and SB203580 (p38 inhibitor, 10 μM), and Bcl-2 expression was determined. (C) Cells were treated with H2O2 (0–400 μM) for 12 h and analyzed for ERK1/2 activation (phosphorylation) by Western blotting. Blots were reprobed for total ERK1/2 to confirm equal loading of the samples. (D) Cells were treated with H2O2 (300 μM) for 24 h in the presence of increasing concentrations of U0216 (0–20 μM), and Bcl-2 expression levels were measured as described. β-Actin was used as a loading control. Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control. #p < 0.05 vs. H2O2-treated control (300 μM).
Hydrogen peroxide induces cysteine oxidation of Bcl-2
To provide a mechanistic insight into the effect of H2O2 on Bcl-2 and its regulation by ERK, we tested the possible modification of Bcl-2 via cysteine oxidation since the cysteine thiol groups are nucleophilic and susceptible to oxidation. We hypothesized that such modification might alter the function of Bcl-2 and its interaction with ERK, which could serve as a key mechanism for controlling apoptotic cell death under oxidative stress conditions. To test cysteine oxidation of Bcl-2, cells were treated with H2O2 and Bcl-2 was isolated and analyzed for cysteine sulfenic acid (Cys-SOH) formation. Bcl-2 isolation was achieved by immunoprecipitation using anti-Bcl-2 antibody, whereas Cys-SOH formation was detected by Western blotting using anti–Cys-SOH antibody. Figure 3A shows that treatment of the cells with H2O2 induced the formation of Bcl-2 Cys-SOH in a dose-dependent manner. Analysis of cysteine oxidation using the specific probe DCP-Bio1 showed a similar dose-dependent pattern of Bcl-2 Cys-SOH formation upon H2O2 treatment (Figure 3B) and thus confirmed the findings. To validate the formation of Bcl-2 Cys-SOH, we also performed analysis of Bcl-2 Cys-SOH in H2O2-treated lung epithelial BEAS-2B cells using the Cys-SOH antibody–based assay. Consistent with the earlier finding in H460 cells, H2O2 was able to dose dependently induce Bcl-2 Cys-SOH formation in BEAS-2B cells (Figure 3C), thus suggesting the generality of the observed Bcl-2 cysteine oxidation. These findings indicate that the cysteine thiol groups of Bcl-2 are key targets for oxidative modification by H2O2. The simultaneous detection of Bcl-2 Cys-SOH formation, Bcl-2 down-regulation, and ERK activation at the same apoptotic doses of H2O2 suggests the possible connection between these molecular events in the regulation of apoptosis.
FIGURE 3:

Effects of H2O2 on Bcl-2 Cys-SOH formation. (A) H460 cells were treated with H2O2 (0–400 μM) for 3 h, and cell lysates were prepared in the presence of dimedone (0.1 mM) and immunoprecipitated with anti–Bcl-2 antibody. The immune complexes were analyzed for cysteine sulfenic acid formation by Western blotting using anti–Cys-SOH antibody. (B) H460 cells were treated with H2O2 (0–400 μM) for 3 h, and cells were labeled with DCP-Bio1 (1 mM) and immunoprecipitated with anti–Bcl-2 antibody. The immune complexes were analyzed for cysteine sulfenic acid formation by Western blotting using anti-biotin antibody. (C) Analysis of Bcl-2 Cys-SOH formation was repeated in lung epithelial BEAS-2B cells using Cys-SOH antibody–based assay. Cells were treated with H2O2 (0–400 μM) for 3 h, and cell lysates were prepared in the presence of dimedone (0.1 mM) and immunoprecipitated with anti–Bcl-2 antibody. Cysteine sulfenic acid formation was analyzed by Western blotting using anti–Cys-SOH antibody (as described in A). Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control.
Mutation of cysteine residues in Bcl-2 suppresses hydrogen peroxide–induced apoptosis
To determine the role of cysteine oxidation in H2O2-induced apoptosis, we transfected cells with Bcl-2 double-mutant plasmid (Bcl-2-DM), replacing Cys-158 and Cys-229 residues with alanines. Cells transfected with wild-type Bcl-2 plasmid or empty vector served as controls. Transfected cells were treated with H2O2, and the effect on apoptosis was examined. Figure 4A shows that the cysteine mutant Bcl-2-DM exhibited a strong antiapoptotic effect as compared with vector control, whereas the wild-type Bcl-2 had considerably lesser effect than the Bcl-2-DM. Morphological analysis of the treated cells by Hoechst nuclear staining showed more shrunken or fragmented nuclei in Bcl-2-DM–than in Bcl-2–transfected cells (Figure 4B). Flow cytometric analysis of apoptosis using annexin V confirmed the Hoechst staining results and indicated an increased antiapoptotic effect of Bcl-2-DM over Bcl-2 (Figure 4C).
FIGURE 4:

Mutation of Bcl-2 cysteine residues suppresses H2O2-induced apoptosis. (A) Cells were transfected with mutant Bcl-2-DM, wild-type Bcl-2, or pcDNA3 control plasmid, as described in Materials and Methods. Transfected cells were treated with H2O2 (400 μM) for 24 h, and apoptosis was determined by Hoechst assay. (B) Fluorescence micrographs of treated cells stained with the Hoechst dye. Apoptotic cells exhibited condensed and/or fragmented nuclei. (C) Representative flow cytometric histograms of annexin V, comparing control and H2O2-treated cells (0 vs. 400 μM) in wild-type Bcl-2– and mutant Bcl-2-DM–expressing cells (top and bottom, respectively). (D) Untreated transfected cells were analyzed for Bcl-2 expression by Western blotting to confirm the overexpression. β-Actin was used as a loading control. Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control. #p < 0.05 vs. H2O2-treated, vector-transfected control. †p < 0.05 vs. H2O2-treated, Bcl-2-transfected control.
The increase in the inhibitory effect of Bcl-2-DM was not due to its antioxidant activity or transgene expression, since the overall expression of Bcl-2 and the ROS levels in Bcl-2-DM– and Bcl-2–transfected cells were comparable (Figures 4D and 5A). Because Bcl-2 is also known to regulate mitochondrial functions, we addressed the possible influence of Bcl-2-DM on mitochondrial ROS by detecting mitochondrial superoxide formation using the mitochondrion-targeted hydroethidium (MitoSOX Red). Figure 5B shows that, although exogenous H2O2 could induce mitochondrial superoxide formation, its effect was not significant among the different transfected cells. Taken together, these results indicate that the cysteine residues are important in the antiapoptotic activity of Bcl-2 against H2O2, probably through cysteine oxidation and ERK signaling.
FIGURE 5:

Effect of H2O2 treatment cellular ROS, mitochondrial superoxide, and Cys-SOH formation. (A) Cells were transfected with mutant Bcl-2-DM, wild-type Bcl-2, or control plasmid as described in Materials and Methods. Transfected cells were treated with H2O2 (400 μM) for 2 h, and cellular ROS levels were determined fluorometrically using H2DCF-DA as a fluorescent probe. (B) Transfected cells were treated with H2O2 (400 μM) for 1 h and analyzed for mitochondrial superoxide generation by fluorescence microscopy using MitoSOX Red as a specific probe. (C) Transfected cells were treated with H2O2 (400 μM) for 3 h, and cell lysates were prepared and immunoprecipitated with anti–Bcl-2 antibody. The immune complexes were analyzed for Cys-SOH by Western blotting. (D) Transfected cells were treated with H2O2 (400 μM) for 3 h, and cells were labeled with DCP-Bio1 (1 mM) and immunoprecipitated with anti–Bcl-2 antibody. The immune complexes were analyzed for Cys-SOH by Western blotting using anti-biotin antibody. Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control. #p < 0.05 vs. vector-transfected control. †p < 0.05 vs. Bcl-2–transfected control.
Mutation of cysteine residues in Bcl-2 attenuates cysteine sulfenic acid formation
To provide a supporting evidence for the role of cysteine oxidation in the antiapoptotic action of Bcl-2, we analyzed Bcl-2 Cys-SOH formation in Bcl-2-DM, Bcl-2, and vector-transfected cells in the presence or absence of H2O2. Figure 5, C and D, shows that H2O2 was able to increase Bcl-2 Cys-SOH formation most prominently in Bcl-2–transfected cells. In Bcl-2-DM–transfected cells, the level of Cys-SOH formation was low and comparable to that of vector-transfected cells (endogenous level), despite the fact that the total expression of Bcl-2 was comparable to that of Bcl-2–transfected cells (Figure 4D).
Cysteine mutation hinders the effects of hydrogen peroxide on Bcl-2 and ERK
Earlier, we showed that H2O2 treatment down-regulated Bcl-2 and induced ERK activation. To test whether cysteine oxidation affects these processes, we treated Bcl-2-DM–, Bcl-2–, and vector-transfected cells with H2O2 and determined the expression levels of Bcl-2 and ERK by Western blotting. Figure 6A shows that Bcl-2 down-regulation by H2O2 was less pronounced in Bcl-2-DM–transfected cells than in Bcl-2– or vector-transfected cells. These results are in good agreement with the apoptosis data and indicate that the overall level of Bcl-2, either wild type or cysteine mutant, determines the cellular apoptotic response to H2O2 treatment. Figure 6B shows that mutation of cysteine residues in Bcl-2 also inhibited ERK activation induced by H2O2, suggesting linkage between Bcl-2 cysteine oxidation and ERK signaling.
FIGURE 6:

Effect of Bcl-2 cysteine residues on Bcl-2 and ERK expression. (A) Cells were transfected with mutant Bcl-2-DM, wild-type Bcl-2, or control plasmid as described in Materials and Methods. Transfected cells were treated with H2O2 (400 μM) for 24 h, and cell lysates were prepared and analyzed for Bcl-2 expression by Western blotting. β-Actin was used as a loading control. (B) Transfected cells were treated with H2O2 (400 μM) for 12 h and analyzed for ERK1/2 activation (phospho-ERK1/2) by Western blotting. Total ERK1/2 was used as a loading control. (C) Cells were treated with the proteasome inhibitors lactacystin (LAC; 10 μM) and MG132 (25 μM) or with the lysosomal inhibitor concanamycin A (CMA; 1 μM) for 1 h and then treated with H2O2 (400 μM) for 24 h. Bcl-2 expression was determined by Western blotting. (D) Cells were transfected with mutant Bcl-2-DM, wild-type Bcl-2, or control plasmid and then treated with H2O2 (400 μM) in the presence of lactacystin (10 μM; to prevent proteasomal degradation of Bcl-2). Cell lysates were immunoprecipitated with Bcl-2 antibody, and the immune complexes were analyzed for ubiquitin by Western blotting. Analysis of ubiquitin was performed at 6 h after treatment, in which ubiquitin was found to be maximal. Equal amounts of protein were loaded in each lane. Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control. #p < 0.05 vs. vector-transfected control. †p < 0.05 vs. Bcl-2-transfected control. §p < 0.05 vs. treated control.
Cysteine mutation blocks Bcl-2 ubiquitination by hydrogen peroxide
Ubiquitination is a major cellular process for rapid down-regulation of Bcl-2 via proteasomal degradation. To verify the role of ubiquitin-proteasomal pathway in Bcl-2 degradation, we treated cells with H2O2 in the presence or absence of lactacystin and MG132, the specific proteasome inhibitors, and determined Bcl-2 expression by Western blotting. Because lysosomal degradation is another possible pathway of protein degradation, cells were also treated with H2O2 in the presence or absence of concanamycin A and analyzed for Bcl-2 expression. Figure 6C shows that lactacystin and MG132, but not concanamycin A, inhibited Bcl-2 down-regulation induced by H2O2, thus confirming proteasomal degradation as the primary mode of Bcl-2 degradation by H2O2. Because cysteine mutation attenuates the effect of H2O2 on Bcl-2, we further examined whether the inhibitory effect of cysteine mutation was regulated by Bcl-2 ubiquitination. We performed immunoprecipitation studies in cells transfected with vector control, wild-type Bcl-2, and Bcl-2-DM plasmids. Transfected cells were treated with H2O2, and cell lysates were prepared and immunoprecipitated using anti–Bcl-2 antibody. The resulting immune complexes were analyzed for ubiquitination by Western blotting. Figure 6D shows that H2O2-induced Bcl-2 ubiquitination was most pronounced in Bcl-2–transfected cells, concomitant with the level of Cys-SOH formation (Figure 4D). These results substantiate the role of cysteine residues in Bcl-2 degradation through the ubiquitin-proteasomal pathway.
Bcl-2 down-regulation by hydrogen peroxide is regulated by ERK interaction
Protein–protein interactions are known to influence protein stability and function (Darnell et al., 2003; Zheng et al., 2011). Because mutation of cysteine residues in Bcl-2 affects ERK activation and Bcl-2 expression, we investigated the possible interaction between Bcl-2 and ERK by generating a correlation plot between Bcl-2 expression and ERK activation in response to H2O2 treatment. Figure 7A shows the correlation plot, which reveals the inverse relationship between the two parameters, with r = 0.93. To experimentally determine the interaction between Bcl-2 and ERK, we used immunoprecipitation and Western blot techniques in combination with immunocytochemistry to aid the analysis. In these experiments, Bcl-2–, Bcl-2-DM–, and vector-transfected cells were treated with H2O2, and cell lysates were prepared and immunoprecipitated with anti-ERK1/2 antibody. The resulting immune complex was then probed for Bcl-2-ERK interaction using anti-Bcl-2 antibody. Figure 7B shows that under a nontreatment condition, a low (basal) level of Bcl-2-ERK complex was observed in the control cells, indicating the direct interaction between Bcl-2 and ERK under these conditions. Treatment of the cells with H2O2 disrupted the interaction in the control as well as Bcl-2–transfected cells but not in Bcl-2-DM–transfected cells (Figure 7B), suggesting the involvement of cysteine oxidation in the disruption. This result, along with the observation that H2O2-induced Bcl-2 down-regulation was substantially less in Bcl-2-DM cells than in Bcl-2 cells (Figure 6A), strongly supports the notion that Bcl-2-ERK interaction helps to stabilize Bcl-2. The disruption of Bcl-2–ERK complex by H2O2 was observed as early as 6 h after the treatment, suggesting that complex disruption was upstream of Bcl-2 down-regulation and ERK activation.
FIGURE 7:
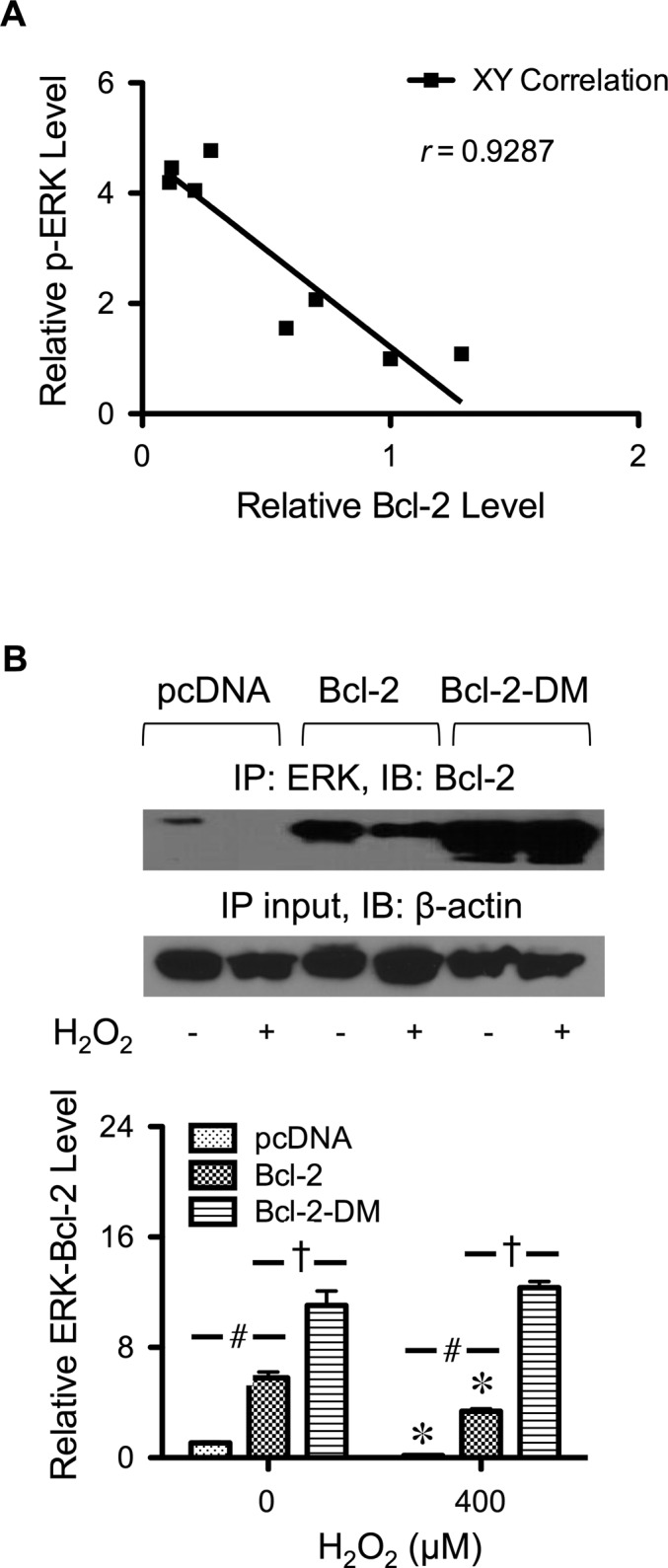
Interaction of Bcl-2 with ERK. (A) Correlation analysis of the expression of Bcl-2 and phospho-ERK in response to H2O2 treatment. (B) Cells were transfected with mutant Bcl-2-DM, wild-type Bcl-2, or control plasmid as described in Materials and Methods. Transfected cells were treated with H2O2 (400 μM) for 6 h, and cell lysates were prepared and immunoprecipitated with anti-ERK1/2 antibody. The immunoblots were probed with anti–Bcl-2 antibody. Equal amounts of protein (60 μg) were loaded in each lane. Plots are mean ± SD (n = 3). *p < 0.05 vs. nontreated control. #p < 0.05 vs. vector-transfected control (400 μM). †p < 0.05 vs. Bcl-2-transfected control.
Immunofluorescence studies were performed to confirm the binding interaction and to evaluate the intracellular localization of Bcl-2and ERK. A high degree of colocalization of Bcl-2 and ERK was observed in the cytosol of untreated cells (Figure 8). After treatment with H2O2, a punctuate pattern of Bcl-2 aggregates was observed in Bcl-2–transfected cells (Figure 8, arrow), thus lowering the degree of colocalization. In contrast, aggregate formation was not observed in Bcl-2-DM–transfected cells after treatment with H2O2. Taken together, these results strongly support the role of cysteine oxidation in Bcl-2–ERK interaction.
FIGURE 8:

Cellular localization of Bcl-2 and ERK. Cells were transfected with mutant Bcl-2-DM, wild-type Bcl-2, or control plasmid as described and seeded onto type I collagen–coated slides. The cells were treated with H2O2 (400 μM) for 6 h and were immunostained and analyzed for protein localization by confocal fluorescence microscopy. Actin was visualized by using phalloidin. Colocalization of ERK and Bcl-2 is shown in the merge display. White arrows indicate aggregations of Bcl-2.
DISCUSSION
Uncontrolled regulation of apoptosis has been implicated in the pathogenesis of various diseases, including cancers and neurodegenerative disorders (Thompson, 1995; Kasibhatla and Tseng, 2003). ROS is a common regulator of apoptosis through the mitochondrial death pathway, which is regulated largely by Bcl-2–family proteins (Hildeman et al., 2003; Juan et al., 2008). The role of ROS in the regulation of Bcl-2 and apoptosis has been well described, but the underlying mechanisms of regulation, particularly posttranslational redox regulation, are not well understood. Increasing evidence has implicated the role of cysteine oxidation in the regulation of protein functions under various oxidative stress conditions (Michalek et al., 2007; Yang et al., 2012); however, detailed investigations of the underlying mechanisms are limited. In this study, we reported several new findings that reveal the mechanisms of Bcl-2 regulation through cysteine oxidation and its effect on apoptosis. First, we demonstrated that H2O2 induced apoptosis through ERK-dependent Bcl-2 down-regulation in human lung epithelial cells (Figures 2 and 4). It was earlier reported that ERK activation resulted in depolarization of mitochondrial membrane potential and cytochrome c release (Kaushal et al., 2004). Because Bcl-2 has long been known to control the mitochondrial membrane permeability transition and cytochrome c release through its interaction with Bax (Korsmeyer et al., 1995; Gupta, 2003), the results of this study therefore suggest that Bcl-2 might be the target where ERK is coupled to the mitochondrial pathway. Our immunoprecipitation and immunofluorescence studies support this notion (Figures 7 and 8).
Overexpression of Bcl-2 in lung epithelial cells inhibited apoptosis induced by H2O2 (Figure 4), indicating the antiapoptotic role of Bcl-2, consistent with the known function of this protein. Bcl-2 cysteine oxidation was observed at the doses of H2O2 that induced apoptosis (Figures 1 and 3), suggesting the potential role of cysteine oxidation in the death-signaling process. Inhibition of Bcl-2 cysteine oxidation by site-specific mutation of cysteine residues (Cys-158 and Cys-229) strongly inhibited the apoptotic effect of H2O2 (Figure 4). A similar result was observed when the cells were treated with antioxidants such as NAC, GSH, and catalase (Figure 1). To our knowledge, this is the first demonstration of cysteine oxidation of Bcl-2 and its effect on cell apoptosis.
We further found that cysteine oxidation of Bcl-2 regulates the apoptotic process by interfering with ERK signaling. Mutation of Bcl-2 cysteine residues abrogated the activation of ERK by H2O2 (Figure 6), suggesting possible interaction between Bcl-2 and ERK. Indeed, our immunoprecipitation and immunofluorescence studies demonstrated for the first time such interaction, which depends on the cellular oxidative stress level (Figures 7 and 8). Treatment of the cells with H2O2 caused a decrease in Bcl-2–ERK interaction (Figures 7 and 8). Because structural modifications of proteins affect protein–protein interactions and bioactivities (Yang et al., 2006; Van Dieck et al., 2009), it is plausible that the observed decrease in Bcl-2-ERK interaction might be a result of cysteine oxidation of Bcl-2. This notion is supported by the observations that 1) H2O2-induced cysteine oxidation of Bcl-2 was concomitant with the disruption of Bcl-2–ERK complex, and 2) prevention of cysteine oxidation by site-specific mutation of cysteine residues inhibited the disruption of Bcl-2–ERK complex by H2O2 (Figures 7 and 8).
The aforementioned results strongly support the causal linkage between Bcl-2 cysteine oxidation and Bcl-2–ERK dissociation. Analysis of the time courses of these two events showed that Bcl-2 cysteine oxidation (Cys-SOH formation) was detected at an earlier time, that is, 3 h after H2O2 treatment, whereas significant Bcl-2–ERK complex disruption was not observed until after 6 h. This result indicates that Bcl-2 cysteine oxidation acts upstream of Bcl-2–ERK complex disruption. Further supporting this notion is the observation that Bcl-2 down-regulation by H2O2 via Bcl-2 ubiquitination was less pronounced in cysteine-mutant Bcl-2-DM cells, which exhibited a higher Bcl-2–ERK level than in wild-type Bcl-2 cells (Figures 6A and 7B). Taken together, these findings indicate cysteine oxidation as a key regulatory event controlling Bcl-2 stability and function through ERK interaction, which is consistent with previous findings showing increased protein stability through protein–protein interactions (Darnell et al., 2003; Zheng et al., 2011).
Bcl-2 has been largely described as a regulator of apoptosis. Increasing evidence suggests that Bcl-2 possesses multiple functions and regulates many cellular processes that are important in carcinogenesis, including cell invasion and migration, anchorage-independent cell growth, and angiogenesis (Medan et al., 2012). Various mutations of Bcl-2 have been reported in cancers, although direct analysis of the function of the mutated Bcl-2 proteins is limited (Reed and Tanaka, 1993; Matolcsy et al., 1996; Packham, 1998). Our study demonstrated the contribution of cysteine residues in Bcl-2 stability and function, which may be important in cancer pathogenesis. Mutation of cysteine residues in Bcl-2 promoted its antiapoptotic activity, rendering cells resistant to oxidative stress–induced apoptosis, which may contribute to cancer development.
In conclusion, evidence presented here demonstrates oxidative modification of Bcl-2 via Cys-SOH formation and establishes its role in the regulation of apoptosis induced by H2O2. Such modification of Bcl-2 causes disruption of the Bcl-2–ERK complex, leading to Bcl-2 down-regulation, ERK activation, and apoptosis. This novel finding on the regulatory mechanism of apoptosis by Bcl-2 could aid in the understanding of disease pathogenesis under oxidative stress conditions.
MATERIALS AND METHODS
Cells and reagents
Human lung epithelial cancer H460 cells and immortalized human normal lung epithelial BEAS-2B cells were obtained from the American Type Culture Collection (Manassas, VA). The cells were cultured in RPMI 1640 and DMEM medium containing 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a 5% CO2 environment at 37°C, respectively. H2O2, NAC, GSH, catalase, and antibody for ubiquitin were obtained from Sigma-Aldrich (St. Louis, MO). Hoechst 33342, annexin V-FITC, H2DCF-DA, and Alexa Fluor secondary antibodies were obtained from Molecular Probes (Grand Island, NY). Lactacystin, concanamycin A, PD98059, and U0216 (MAP kinase ERK1/2 inhibitors), SP600165 (c-Jun N-terminal kinase inhibitor), and SB203580 (p38 kinase inhibitor) were from Calbiochem (La Jolla, CA). Antibodies for Bcl-2, phospho-ERK1/2 (p-p44/p42), ERK1/2 (p44/p42), horseradish peroxidase (HRP)–conjugated secondary antibodies, and HRP-conjugated anti-biotin antibody were obtained from Cell Signaling Technology (Beverly, MA). Antibody for cysteine sulfenic acid was obtained from Millipore (Billerica, MA). DCP-Bio1 was obtained from KeraFAST (Boston, MA).
Plasmids and transfection
Bcl-2 and mutant plasmids were generously provided by C. Stehlik (Northwestern University, Chicago, IL). The authenticity of the plasmid constructs was verified by DNA sequencing. Cells were transfected with hemagglutinin-tagged wild-type Bcl-2, Bcl-2 mutant, or pcDNA3 control plasmid by nucleofection using Nucleofector (Amexa Biosystems, Cologne, Germany), according to the manufacturer's instructions. Briefly, cells were suspended in 100 μl of nucleofection solution with 2 μg of plasmid and nucleofected using the device program T020. The cells were then resuspended in 500 μl of complete medium and seeded in 6-mm cell culture dishes. Cells were allowed to recover for 48 h before each experiment. The efficiency of transfection was determined by using a green fluorescent protein reporter plasmid and was found to be ∼80%.
Apoptosis assays
Apoptosis was determined by Hoechst 33342 assay and by flow cytometric analysis of annexin V and propidium iodide (PI). In the Hoechst assay, cells were incubated with 10 μg/ml Hoechst 33342 for 30 min and analyzed for apoptosis by scoring the percentage of cells having condensed chromatin and/or fragmented nuclei by fluorescence microscopy (Leica Microsystems, Bannockburn, IL). Approximately 1000 nuclei from 10 random fields were analyzed for each sample. The apoptotic index was calculated as the percentage of cells with apoptotic nuclei over total number of cells. For flow cytometric analysis, cells were harvested, washed, stained with annexin V–fluorescein isothiocyanate (FITC) in binding buffer for 30 min at room temperature, and costained with PI (10 μg/ml). Samples were immediately analyzed by FACScan flow cytometer (Becton Dickinson, Rutherford, NJ) using a 488-nm excitation beam and a 530- and 630-nm band-pass filter with CellQuest software (Becton Dickinson).
ROS detection
ROS generation was determined fluorometrically using H2DCF-DA as a fluorescent probe. Briefly, cells were incubated with the probe (10 μM) for 30 min at 37°C, after which they were washed and resuspended in phosphate-buffered saline (PBS), followed by analysis of cellular fluorescence intensity by a fluorescence plate reader at the excitation and emission wavelengths of 485 and 538 nm, respectively.
Western blot analysis
After specific treatments, cells were incubated with lysis buffer containing 20 mM Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM sodium chloride, 10% glycerol, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 100 mM phenylmethylsulfonyl fluoride, and a commercial protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN) at 4°C for 20 min. Cell lysates were collected and determined for protein content using the Bradford method (Bio-Rad Laboratories, Hercules, CA). Proteins (40 μg) were resolved under denaturing conditions by 10% SDS–PAGE and transferred onto nitrocellulose membranes. The transferred membranes were blocked for 1 h in 5% nonfat dry milk in TBST (25 mM Tris-HCl, 125 mM NaCl, 0.05% Tween-20) and incubated with appropriate primary antibodies at 4°C overnight. The membranes were washed twice with TBST for 10 min and incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. The immune complexes were then detected by chemiluminescence detection system (Amersham Biosciences, Piscataway, NJ) and quantified using Analyst/PC densitometry software (Bio-Rad Laboratories).
Immunoprecipitation
After specific treatments, cells were washed with PBS and lysed in lysis buffer at 4°C for 20 min. The lysates were collected and determined for protein content using the Bradford method (Bio-Rad Laboratories). Cell lysates (60 μg protein) were incubated with anti–Bcl-2 or anti-ERK1/2 (p44/p42) antibody at 4°C for 14 h, followed by incubation with protein G–conjugated agarose for 4 h at 4°C. The immune complexes were washed six times with cold lysis buffer and resuspended in 2× Laemmli sample buffer. The immune complexes were separated by 10% SDS–PAGE and analyzed for protein expression by Western blotting.
Cysteine sulfenic acid detection
Cysteine sulfenic acid was determined by Cys-SOH antibody–based and DCP-Bio1 reagent–based assays. In the antibody-based assay, cells were washed with PBS and lysed in lysis buffer containing 0.1 mM dimedone at 4°C for 20 min. Cell lysates (200 μg of protein) were immunoprecipitated using anti–Bcl-2 antibody and separated by 10% SDS–PAGE as described. Cysteine oxidation was detected by Western blotting using 1:1000 anti–cysteine sulfenic acid antibody (Millipore, MA) and anti-rabbit HRP-conjugated secondary antibody. For the reagent-based assay, cells were labeled with 1 mM cysteine oxidation probe DCP-Bio1 (KeraFAST), which is a dimedone analogue containing biotin tag. The DCP-Bio1–labeled proteins were similarly immunoprecipitated using anti–Bcl-2 antibody and separated by 10% SDS–PAGE as described. Subsequently, the samples were detected for cysteine oxidation by Western blotting using 1:1000 anti-biotin (conjugated HRP) antibody. Quantification of the immune signals was performed using Analyst/PC densitometry software.
Immunofluorescence
Cells were seeded on rat type I collagen–coated coverslips (5 μg/cm2), fixed with 3.7% paraformaldehyde for 15 min, incubated in 50 mM glycine for 5 min, permeabilized, and blocked with 0.5% saponin, 1.5% bovine serum albumin, and 1.5% normal goat serum for 30 min. ERK was immunostained with anti-ERK1/2 antibody, and Bcl-2 was immunostained with anti–hemagglutinin tag antibody. Secondary Alexa Fluor 488–, 546–, and 647–conjugated antibodies and phalloidin (Invitrogen, Carlsbad, CA) were used. Cells were washed with PBS containing 0.5% saponin and were mounted on a coverslip using Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL). Cells were visualized with a Zeiss LSM 510 confocal on an AxioImager Z1 microscope using a 63× objective lens (Carl Zeiss, Jena, Germany).
Statistical analysis
The data represent means ± SD from three or more independent experiments. Statistical analysis was performed by Student's t test at a significance level of p < 0.05.
Acknowledgments
This work was supported by the National Institutes of Health Grant R01 HL095579. Imaging experiments were performed in the West Virginia University Microscope Imaging Facility, which is supported in part by the Mary Babb Randolph Cancer Center and National Institutes of Health Grant P20 RR016440. Flow cytometric analysis was performed in the West Virginia University Flow Cytometry Core Facility, which is supported in part by National Institutes of Health Grant P30 GM103488.
Abbreviations used:
- Bcl-2
B-cell lymphoma-2
- Bcl-2-DM
Bcl-2 double mutant
- Cys-SOH
cysteine sulfenic acid
- ERK
extracellular signal-regulated kinase 1/2
- GSH
reduced glutathione
- H2DCF-DA
2′7′-dihydrodichlorofluorescein diacetate
- HRP
horseradish peroxidase
- NAC
N-acetylcysteine
- ROS
reactive oxygen species
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-10-0747) on January 30, 2013.
The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
REFERENCES
- Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y. Superoxide-mediated proteasomal degradation of Bcl-2 determined cell susceptibility to chromium (VI)-induced apoptosis. Carcinogenesis. 2008;29:1538–1545. doi: 10.1093/carcin/bgn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ezra JM, Kornstein MJ, Grimes MM, Krystal G. Small cell carcinomas of the lung express the Bcl-2 protein. Am J Pathol. 1994;145:1036–1040. [PMC free article] [PubMed] [Google Scholar]
- Chanvorachote P, Nimmanit U, Stehlik C, Wang L, Jiang B, Ongpipatanakul B, Rojanasakul Y. Nitric oxide regulates cell sensitivity to cisplatin-induced apoptosis through S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res. 2006;66:6353–6360. doi: 10.1158/0008-5472.CAN-05-4533. [DOI] [PubMed] [Google Scholar]
- Darnell GA, Antalis TM, Johnstone RW, Stringer BW, Ogbourne SM, Harrich D, Suhrbier Inhibition of retinoblastoma protein degradation by interaction with the serpin plasminogen activator inhibitor 2 via a novel consensus motif. Mol Cell Biol. 2003;23:6520–6532. doi: 10.1128/MCB.23.18.6520-6532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmeler S, Breitschopf K, Haendeler J, Zeiher AM. Dephosphorylation targets Bcl-2 for ubiquitin-dependent degradation: a link between the apoptosome and the proteasome pathway. J Exp Med. 1999;189:1815–1822. doi: 10.1084/jem.189.11.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman HJ. Signal transduction and reactive species. Free Radic Biol Med. 2009;47:1237–1238. doi: 10.1016/j.freeradbiomed.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Fruehauf JP, Meyskens JL., Jr Reactive oxygen species: a breath of life or death. Clin Cancer Res. 2007;13:789–794. doi: 10.1158/1078-0432.CCR-06-2082. [DOI] [PubMed] [Google Scholar]
- Gupta S. Molecular signaling in death receptor and mitochondrial pathways of apoptosis. Int J Oncol. 2003;22:15–20. [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1–11. doi: 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Clement MV, Ramalingam J, Long LH. Hydrogen peroxide. Ubiquitous in cell culture and in vivo. IUBMB Life. 2000;50:251–257. doi: 10.1080/713803727. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hancock JT, Desikan R, Neill SJ. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans. 2001;29:345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- Hildeman DA, Mitchell T, Aronow B, Wojciechowski S, Kappler J, Marrack P. Control of Bcl-2 expression by reactive oxygen species. Proc Natl Acad Sci USA. 2003;100:15035–15040. doi: 10.1073/pnas.1936213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang SX, Sato Y, Kuwao S, Kameya T. Expression of bcl-2 oncogene protein is prevalent in small cell lung carcinomas. J Pathol. 1995;177:135–138. doi: 10.1002/path.1711770206. [DOI] [PubMed] [Google Scholar]
- Juan ME, Wenzel U, Daniel H, Planas JM. Resveratrol induces apoptosis through ROS-dependent mitochondrial pathway in HT-29 human colorectal carcinoma cells. J Agric Food Chem. 2008;56:4813–4818. doi: 10.1021/jf800175a. [DOI] [PubMed] [Google Scholar]
- Kasibhatla S, Tseng B. Why target apoptosis in cancer treatment. Mol Cancer Ther. 2003;2:573–580. [PubMed] [Google Scholar]
- Kaushal GP, Liu L, Kaushal V, Hong X, Melnyk O, Seth R, Safirstein R, Shah SV. Regulation of caspase-3 and -9 activation in oxidant stress to RTE by forkhead transcription factors, Bcl-2 proteins, MAP kinases. Am J Physiol Renal Physiol. 2004;287:F1258–F1268. doi: 10.1152/ajprenal.00391.2003. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ, Yin XM, Oltvai ZN, Veis-Novack DJ, Linette GP. Reactive oxygen species and the regulation of cell death by the Bcl-2 gene family. Biochim Biophys Acta. 1995;1271:63–66. doi: 10.1016/0925-4439(95)00011-r. [DOI] [PubMed] [Google Scholar]
- Luanpitpong S, Nimmannit U, Chanvorachote P, Leonard SS, Pongrakhananon V, Wang L, Rojanasakul Y. Hydroxyl radical mediates cisplatin-induced apoptosis in human hair follicle dermal papilla cells and keratinocytes through Bcl-2-dependent mechanism. Apoptosis. 2011;16:769–782. doi: 10.1007/s10495-011-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matolcsy A, Casali P, Warnke RA, Knowles DM. Morphological transformation of follicular lymphoma is associated with somatic mutation of the translocated Bcl-2 gene. Blood. 1996;88:3937–3944. [PubMed] [Google Scholar]
- Medan D, Luanpitpong S, Azad N, Wang L, Jiang BH, Davis ME, Barnett JB, Guo L, Rojanasakul Y. Multifunctional role of Bcl-2 in malignant transformation and tumorigenesis of Cr(VI)-transformed lung cells. PLoS One. 2012;7:e37045. doi: 10.1371/journal.pone.0037045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Nelson KJ, Holbrook BC, Yi JS, Stridiron D, Daniel LW, Fetrow JS, King SB, Poole LB, Grayson JM. The requirement of reversible cysteine sulfenic acid formation for T cell activation and function. J Immunol. 2007;179:6456–6467. doi: 10.4049/jimmunol.179.10.6456. [DOI] [PubMed] [Google Scholar]
- Moungjaroen J, Nimmannit U, Callery PS, Wang L, Azad N, Lipipun V, Chanvorachote P, Rojanasakul Y. Reactive oxygen species mediate caspase activation and apoptosis induced by lipoic acid in human lung epithelial cancer cells through Bcl-2 downregulation. J Pharmacol Exp Ther. 2006;319:1062–1069. doi: 10.1124/jpet.106.110965. [DOI] [PubMed] [Google Scholar]
- Osford SM, Dallman CL, Johnson PW, Ganesan A, Packham G. Current strategies to target the anti-apoptotic Bcl-2 protein in cancer cells. Curr Med Chem. 2004;11:1031–1039. doi: 10.2174/0929867043455486. [DOI] [PubMed] [Google Scholar]
- Packham G. Mutation of Bcl-2 family proteins in cancer. Apoptosis. 1998;3:75–82. doi: 10.1023/a:1009688706783. [DOI] [PubMed] [Google Scholar]
- Reed JC, Tanaka S. Somatic point mutations in the translocated bcl-2 genes of non-Hodgkin's lymphomas and lymphocytic leukemias: implications for mechanisms of tumor progression. Leukemia Lymphoma. 1993;10:157–163. doi: 10.3109/10428199309145877. [DOI] [PubMed] [Google Scholar]
- Rehder DS, Borges CR. Possibilities and pitfalls in quantifying the extent of cysteine sulfenic acid modification of specific proteins with complex biofluids. BMC Biochem. 2010;11:25. doi: 10.1186/1471-2091-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaert NL, et al. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase beta. Proc Natl Acad Sci USA. 2006;103:13086–13091. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivapurkar N, Reddy J, Chaudharry PM, Gazdar AF. Apoptosis and lung cancer: a review. J Cell Biochem. 2003;88:885–898. doi: 10.1002/jcb.10440. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Torres M, Forman HJ. Redox signaling and the MAP kinase pathways. Biofactors. 2003;17:287–296. doi: 10.1002/biof.5520170128. [DOI] [PubMed] [Google Scholar]
- Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxid Redox Signal. 2002;4:405–414. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Van Dieck J, Teufel DP, Jaulent AM, Fernandez-Fernandez MR, Rutherford TJ, Wyslouch-Cieszynska A, Fersht AR. Posttranslational modifications affect the interaction of S100 proteins with tumor suppressor p53. J Mol Biol. 2009;394:922–930. doi: 10.1016/j.jmb.2009.10.002. [DOI] [PubMed] [Google Scholar]
- Wang L, Chanvorachote P, Toledo D, Stehlik C, Mercer RR, Castranova V, Rojanasakul Y. Peroxide is a key mediator of Bcl-2 down-regulation and apoptosis induction by cisplatinin human lung cancer cells. Mol Pharmacol. 2008;73:119–127. doi: 10.1124/mol.107.040873. [DOI] [PubMed] [Google Scholar]
- Willimott S, Wagner SD. Post-transcriptional and post-translational regulation of Bcl-2. Biochem Soc Trans. 2010;38:1571–1575. doi: 10.1042/BST0381571. [DOI] [PubMed] [Google Scholar]
- Yang ML, Doyle HA, Gee RJ, Lowenson JD, Clarke S, Lawson BR, Aswad DW, Mamula MJ. (2006). Intracellular protein modification associated with altered T cell functions in autoimmunity. J Immunol. 177:4541–4549. doi: 10.4049/jimmunol.177.7.4541. [DOI] [PubMed] [Google Scholar]
- Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ. Redox modification of cysteine residues regulates the cytokine activity of HMGB1. Mol Med. 2012;18:250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zheng Y, Wang B, Jayappa KD, Yao X. Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J Biol Chem. 2011;286:17722–17735. doi: 10.1074/jbc.M110.184739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang S, Yan Y, Daubert RA, Han J, Schnellmann RG. ERK promotes hydrogen peroxide-induced apoptosis through caspase-3 activation and inhibition of Akt in renal epithelial cells. Am J Physiol Renal Physiol. 2007;292:F440–F447. doi: 10.1152/ajprenal.00170.2006. [DOI] [PubMed] [Google Scholar]