

Abstract
Background
The emergence of drug-resistant pathogen strains and new infectious agents pose major challenges to public health. A promising approach to combat these problems is to target the host’s genes or proteins, especially to discover targets that are effective against multiple pathogens, i.e., host-oriented broad-spectrum (HOBS) drug targets. An important first step in the discovery of such drug targets is the identification of host responses that are commonly perturbed by multiple pathogens.
Results
In this paper, we present a methodology to identify common host responses elicited by multiple pathogens. First, we identified host responses perturbed by each pathogen using a gene set enrichment analysis of publicly available genome-wide transcriptional datasets. Then, we used biclustering to identify groups of host pathways and biological processes that were perturbed only by a subset of the analyzed pathogens. Finally, we tested the enrichment of each bicluster in human genes that are known drug targets, on the basis of which we elicited putative HOBS targets for specific groups of bacterial pathogens. We identified 84 up-regulated and three down-regulated statistically significant biclusters. Each bicluster contained a group of pathogens that commonly dysregulated a group of biological processes. We validated our approach by checking whether these biclusters correspond to known hallmarks of bacterial infection. Indeed, these biclusters contained biological process such as inflammation, activation of dendritic cells, pro- and anti- apoptotic responses and other innate immune responses. Next, we identified biclusters containing pathogens that infected the same tissue. After a literature-based analysis of the drug targets contained in these biclusters, we suggested new uses of the drugs Anakinra, Etanercept, and Infliximab for gastrointestinal pathogens Yersinia enterocolitica, Helicobacter pylori kx2 strain, and enterohemorrhagic Escherichia coli and the drug Simvastatin for hematopoietic pathogen Ehrlichia chaffeensis.
Conclusions
Using a combination of automated analysis of host-response gene expression data and manual study of the literature, we have been able to suggest host-oriented treatments for specific bacterial infections. The analyses and suggestions made in this study may be utilized to generate concrete hypothesis on which gene sets to probe further in the quest for HOBS drug targets for bacterial infections. All our results are available at the following supplementary website: http://bioinformatics.cs.vt.edu/ murali/supplements/2013-kidane-plos-one
Introduction
Infectious diseases are the second leading cause of death worldwide, next to cardiovascular diseases [1]. Bacterial infections such as tuberculosis and food- and water- borne infections from Salmonella enterica and Escherichia coli still present many challenges to biomedical researchers. Foremost among these challenges is that infectious agents rapidly mutate and become resistant to drugs [2]. The conventional approach of targeting pathogen proteins has accelerated the spread of resistance, resulting in the re-emergence of once-contained infectious diseases, such as those caused by multidrug-resistant strains of Mycobacterium tuberculosis, Staphylococcus aureus, and Salmonella enterica [3]. In an effort to combat the issue of drug resistance, anti-infective drug discovery is shifting to a new approach that targets the host instead of pathogens [3], [4]. “Host-oriented” drug discovery focuses on manipulating or subverting biological processes in the host that pathogens utilize [5]. Another problem facing the treatment of infectious diseases is the increasing number of pathogenic agents [6]. Furthermore, new pathogens are appearing regularly, e.g., the pandemic swine flu H1N1 virus recognized in 2009. The expanding range of infectious agents coupled with the high cost associated with drug discovery have made it economically infeasible and practically impossible to tackle each pathogen individually [6], [7]. These factors have necessitated treatment regimens that are effective against a wide variety of infectious agents.
These factors have encouraged efforts in host-oriented broad-spectrum (HOBS) drug discovery, i.e., finding targets in the host that can simultaneously cure multiple infections [3], [8]. Examples of HOBS drugs currently available in the market include Statins and Isoprinosine. Statins are used in the treatment of Leishmania, Staphylococcus aureus, and HIV infections [9]–[11]. Statins lower the cholesterol level in human body. They are effective against pathogens that utilize cholesterol in binding and internalization to the host cell. Isoprinosine, which stimulates the proliferation of T-cells, is used in the treatment of Herpes simplex, Hepatitis, and Epstein-Barr virus infections [12].
A first and important step in HOBS drug discovery is the development of computational tools to discover common physiological processes and cellular pathways that different pathogens utilize to infect, proliferate, and spread in the host. We hypothesized that comprehensive molecular datasets of host responses to diverse varieties of pathogens might form a powerful resource to discover such pathways. Transcriptional datasets that correspond to different infectious diseases, cell/tissue types, and organisms are the most abundantly available. Meta-analysis of transcriptional datasets have been performed for a wide range of diseases. For instance, Rhodes et al. [13] analyzed 40 cancer related microarray datasets to identify common signatures of cancer. English and Butte [14] integrated 49 obesity-related genome-wide experiments obtained from human, mouse, rat, and worm to predict new genes that may be associated with obesity. Magalhaes et al. [15] performed meta-analysis of 27 age-related gene expression profile datasets from human, mouse, and rat to reveal several common signatures of aging. Jenner et al. [16] used hierarchical clustering of gene expression profiles of 77 pathogens in order to find genes that exhibited similar expression profiles across several disease types.
Recent approaches have taken meta-analysis of DNA microarray datasets one step further by incorporating drug targets into the analysis and inferring new uses for existing drugs on the basis of disease similarities. The premise underlying these approaches is that diseases with a high degree of transcriptional similarity might be treated with similar drugs [17]. Hu et al. [18] discovered disease-disease links by using correlation-based methods and gene set enrichment analysis to measure the similarities between gene expression profiles of diseases. They also integrated gene expression profiles that pertain to responses of cell lines to drugs derived from the Connectivity Map [19] to create a drug-disease network where clusters of drugs and diseases suggested shared drug mechanisms and molecular disease pathology. Suthram et al. [20] performed integrative analysis of 54 disease-related mRNA expression datasets. They measured the perturbation of predefined protein functional modules using the mean normalized transcriptional activity of each module's component genes in the disease's transcriptional profile. Furthermore, they identified known drug targets in the modules that were perturbed by multiple disease types, which they proposed as pluripotent/broad-spectrum drug targets .
The goal of our work is similar to that of Jenner et al. , Hu et al. , and Suthram et al. : to discover transcriptional responses common to many diseases, specifically those caused by bacterial pathogens, and to discover existing drug targets within those transcriptional signatures. The previous authors have used global correlation measures to detect disease associations, which may obscure relationships that exist over only a subset of the diseases or genes. In contrast, we use a combination of gene set level enrichment and biclustering. As we demonstrate in this work, this approach enables us to group sets of host genes that are dysregulated only by a subset of the pathogens, facilitating the capture of pathway-specific relationships among groups of pathogens.
Results
We start with an overview of the method (Figure 1). We obtained genome-wide transcriptional data sets of host responses after infection by bacterial pathogens from the NCBI's Gene Expression Omnibus (GEO) (Figure 1A). After data filtering (see Methods), we retained 29 gene expression profiling studies which represent 213 host samples and 38 bacterial pathogens or pathogen strains. We sub-divided the datasets into four major kinds of infection: gastrointestinal, oral cavity, hematopoietic, and respiratory. A complete description of these datasets and their GEO accession numbers is provided in Table S1.
Figure 1. Overview of our system.

Overview of our computational system to compute host-oriented broad-spectrum drug targets. (A) Obtaining relevant collection of taxonomic names for human bacterial pathogens. Querying the GEO metadatabase in search of relevant transcriptional datasets. (B) Gene Set Enrichment Analysis of the transcriptional datasets collected in Step A. (C) Identification of pathogen-gene set biclusters and estimation of statistical significance of biclusters (D) Testing bicluster enrichment for known drug targets. (E) Literature analysis of putative HOBS drug targets contained in biclusters.
Since these datasets were generated by different research groups with different objectives in mind, they tended to be very diverse, e.g., in the microarray platform used, the infected host, and the tissue or cell type from which the gene expression measurements were taken. Such variations made the direct comparison of the datasets difficult. To alleviate this problem, we computed gene sets perturbed by each pathogen using Gene Set Enrichment Analysis (GSEA) (Figure 1B), thereby enabling comparison across pathogens at the level of perturbed gene sets. We recorded all pathogens and the gene sets they perturbed in a matrix. Next, we biclustered this matrix in order to identify all subsets of the gene sets that were co-perturbed across a subset of the pathogens (Figure 1C). We assessed the statistical significance of the biclusters by comparing their sizes to biclusters found in randomized matrices. This process yielded 84 up-regulated and three down-regulated significant biclusters at a 0.05 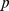 -value cutoff, after adjusting for multiple-hypothesis testing [21] (Tables S2 and S3). In this paper, we focus our discussion on up-regulated biclusters as (a) they are far greater in number than down-regulated biclusters and (b) up-regulated genes and pathways may be controlled, in general, by drugs that prevent function of their targets. We used Fisher's exact test to estimate the enrichment of a bicluster in known drug targets (Figure 1D). We acknowledge that even a bicluster with a single drug target may be worthy of study. We computed bicluster enrichment in drug targets in order to prioritize biclusters for examination since we had a large number of biclusters. Finally, we searched the literature for biologically meaningful connections among the gene sets, pathogens, and drug targets in a bicluster in order to find support for the hypothesis that modulating the activity of the drug targets may control the infection caused by the pathogens (Figure 1E).
-value cutoff, after adjusting for multiple-hypothesis testing [21] (Tables S2 and S3). In this paper, we focus our discussion on up-regulated biclusters as (a) they are far greater in number than down-regulated biclusters and (b) up-regulated genes and pathways may be controlled, in general, by drugs that prevent function of their targets. We used Fisher's exact test to estimate the enrichment of a bicluster in known drug targets (Figure 1D). We acknowledge that even a bicluster with a single drug target may be worthy of study. We computed bicluster enrichment in drug targets in order to prioritize biclusters for examination since we had a large number of biclusters. Finally, we searched the literature for biologically meaningful connections among the gene sets, pathogens, and drug targets in a bicluster in order to find support for the hypothesis that modulating the activity of the drug targets may control the infection caused by the pathogens (Figure 1E).
We have organized the results from our study into two major sections. First, we asked if the biclusters we computed could reveal well-known immunological responses in the host to bacterial infection. To this end, we identified host gene sets that were contained in those biclusters that were also perturbed by many pathogens. Our analysis revealed that biological functions pertaining to the up-regulation of inflammatory gene sets, Lipopolysaccharide (LPS)-inducible gene sets, innate immunity response, induction and inhibition of apoptosis, and maturation of dendritic cells are host responses that are triggered by most of the bacterial pathogens. Rediscovering well known host responses to infection established the validity of our approach in detecting common host signatures. Second, we analyzed the biclusters for putative HOBS targets. Out of the 84 significantly up-regulated biclusters, 47 of them were enriched in known drug targets at the 0.05 significance level (Table S2). We identified seven biclusters where all the pathogens contained in each of these biclusters infected a single tissue or organ in the human body. For instance, in bicluster 38, we found four gastrointestinal pathogens, namely, Yersinia enterocolitica wap and p60 strains, Helicobacter pylori kx2 strain, and Enterohemorrhagic Escherichia coli. From this bicluster, we suggested the potential use of chronic inflammation suppressors such as Anakinra, Etanercept and Infliximab in treating infection caused by these four pathogens.
Gene Sets Perturbed in Response to Bacterial Infection
There are several stages and outcomes that are hallmarks of generalized infection. On one hand, pathogens try to enter, multiply, and spread in the host, causing disease. On the other hand, hosts attempt to defend the attack from pathogens using processes conferring innate and adaptive immunity, leading to the elimination of pathogens. There are different strategies that are utilized by pathogens and by hosts to achieve these objectives. Among other things, pathogens induce or inhibit apoptosis, import their genetic material into the host, and replicate their genome [22], [23]. Hosts utilize various arms of the immune system such as inflammation, response to stimulus, maturation of dendritic cells and activation of various components of the innate immunity to lessen pathogenicity.
The 84 statistically significant up-regulated biclusters contained 1,364 distinct gene sets and 34 pathogens. To determine if our biclusters capture the hallmarks of infection mentioned above, we asked which gene sets belonged to the largest number of biclusters. Upon ranking the gene sets in decreasing order of number of biclusters they were perturbed in, we observed that the number of biclusters that a gene set was contained in had a high positive correlation (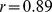 ,
, 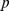 -value
-value 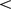
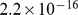 ) with the number of pathogens that perturb the gene set (Figure S1). Table 1 shows the top ten gene sets in this ranked list. Then, for each gene set, we assigned Gene Ontology (GO) biological processes for intuitive interpretation (Table 2) using the procedure described in Methods. We now proceed to discuss these highly-ranked gene sets and correlate them to well-known hallmarks of infection.
) with the number of pathogens that perturb the gene set (Figure S1). Table 1 shows the top ten gene sets in this ranked list. Then, for each gene set, we assigned Gene Ontology (GO) biological processes for intuitive interpretation (Table 2) using the procedure described in Methods. We now proceed to discuss these highly-ranked gene sets and correlate them to well-known hallmarks of infection.
Table 1. Gene sets perturbed in many pathogens.
| Gene Set | # Pathogens | # Biclusters |
| Zhang Response to IKK Inhibitor and TNF up | 33 | 83 |
| Seki Inflammatory Response LPS up | 33 | 83 |
| Dirmeier LMP1 Response Early | 32 | 76 |
| Dauer STAT3 Targets up | 31 | 75 |
| Hinata NFKB Targets Keratinocyte up | 31 | 74 |
| Tian TNF Signaling via NFKB | 32 | 73 |
| Lindstedt Dendritic Cell Maturation B | 30 | 67 |
| Uzonyi Response to Leukotriene and Thrombin | 31 | 63 |
| Netpath IL 4 Pathway Down | 30 | 59 |
| Mahadevan Response to MP470 up | 30 | 53 |
For each gene set, the table shows the number of pathogens that perturb it and the number of biclusters it appears in.
Table 2. Mapping of Gene Sets to GO Biological Processes.
| Gene Set | GO Enriched Processes (Top Three) |
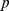 -value -value |
| Zhang Response to IKK Inhibitor and TNF up | Inflammatory Response |
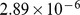
|
| Response to Wounding |
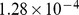
|
|
| Defense Response |
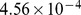
|
|
| Seki Inflammatory Response LPS up | Locomotory Behavior |
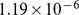
|
| Response to External Stimulus |
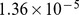
|
|
| Defense Response |
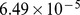
|
|
| Dirmeier LMP1 Response Early | Apoptosis GO |
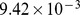
|
| Programmed Cell Death |
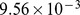
|
|
| Viral Genome Replication |
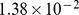
|
|
| Dauer STAT3 Targets up | Cyclic Nucleotide Metabolic Process |
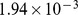
|
| Protein Import into Nucleus Translocation |
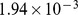
|
|
| DNA Damage Response Signal Transduction Resulting in Induction of Apoptosis |
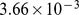
|
|
| Hinata NFKB Targets Keratinocyte up | Response to Wounding |
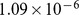
|
| Inflammatory Response |
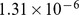
|
|
| Response to Stress |
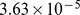
|
|
| Tian TNF Signaling via NFKB | Defense Response |
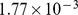
|
| Regulation of I KAPPAB Kinase NF KAPPAB Cascade |
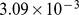
|
|
| Response to Wounding |
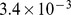
|
|
| Lindstedt Dendritic Cell Maturation B | Apoptosis GO |
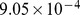
|
| Programmed Cell Death |
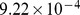
|
|
| Cell Development |
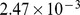
|
|
| Uzonyi Response to Leukotriene and Thrombin | Heart Development |
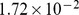
|
| Inflammatory Response |

|
|
| Regulation of Transcription |
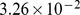
|
|
| Netpath IL 4 Pathway Down | Activation of Innate Immune Response |
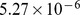
|
| Pattern Recognition Receptor Signaling Pathway |
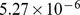
|
|
| Toll-like Receptor Signaling Pathway |

|
|
| Mahadevan Response to MP470 up | Locomotory Behavior |
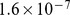
|
| Defense Response |
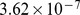
|
|
| Inflammatory Response |
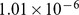
|
The table shows top three GO biological processes that have the highest overlap with each of the ten most frequently perturbed gene sets (in Table 1). The  -value indicates the statistical significance of the overlap, based on Fisher's exact test.
-value indicates the statistical significance of the overlap, based on Fisher's exact test.
Inflammatory Response
Inflammation is one of the immediate reactions by the host against pathogenic infections. Of the top ten gene sets, four gene sets have a high overlap with genes annotated with GO's inflammatory response process (GO:0006954; “Zhang Response to IKK Inhibitor and TNF up”, “Uzonyi Response to Leukotriene and Thrombin”, “Hinata NFKB Targets Keratinocyte up”, and “Mahadevan Response to MP470 up”). For each of these gene sets, we describe the experiment that generated it. We note that these experiments were conducted in diverse tissues and were not directly related to pathogen infection. Nevertheless, by examining the connection between each of these gene sets and inflammation, we demonstrate that inflammation is a non-specific response triggered by many of the pathogens irrespective of the type of cell being infected. The gene set “Zhang Response to IKK Inhibitor and TNF up” is perturbed in 83 biclusters spanning 33 different bacterial pathogens. This gene set contains 219 genes that are up-regulated in BxPC3 pancreatic cancer cells after treatment with tumor necrosis factor (TNF)- , a pro-inflammatory cytokine [24]. This gene set consists of genes encoding for pro-inflammatory mediators such as IL1A, IL1B, TNFSF10 and a number of other chemokines including CCL20, CCL5, CXCL1, CXCL10, CXCL11, CXCL16, CXCL2, and CXCL3. The next set in the list is “Hinata NFKB Targets Keratinocyte up”, which was perturbed by 31 pathogens and appeared in 74 biclusters. This gene set contains 71 genes that were up-regulated in primary keratinocyte cells after transduction with NF-kappa B [25]. The majority of the genes in this gene set are cytokines and growth factor genes including chemokines ( CCL20, CCL5, CXCL10, CXCL11, CXCL3, CXCL6); interleukins ( IL15, IL1B, IL1RN, IL6, IL8); and growth factor genes (TNC, VEGFA, ESM1, MP2). The “Uzonyi Response to Leukotriene and Thrombin” gene set is perturbed by the same number of pathogens as “Hinata NFKB Targets Keratinocyte up”. It contains 37 genes that were up-regulated in Human Umbilical Vein Endothelial Cells (HUVEC) after stimulation with leukotriene LTD4, a leukocyte produced at sites of inflammation [26]. The fourth gene set is “Mahadevan Response to MP470 up”, which is perturbed by 30 pathogens and appeared in 53 biclusters. This gene set contains 19 genes that were up-regulated in gastrointestinal stromal tumor cell-line after treatment with protein-kinase inhibitor drug (MP470) [27]. This gene set also contains chemokines and proinflammatory cytokines such as CCL5, CXCL1, CXCL10, CXCL3, CXCL5, CXCL6, IL8, and IL6.
, a pro-inflammatory cytokine [24]. This gene set consists of genes encoding for pro-inflammatory mediators such as IL1A, IL1B, TNFSF10 and a number of other chemokines including CCL20, CCL5, CXCL1, CXCL10, CXCL11, CXCL16, CXCL2, and CXCL3. The next set in the list is “Hinata NFKB Targets Keratinocyte up”, which was perturbed by 31 pathogens and appeared in 74 biclusters. This gene set contains 71 genes that were up-regulated in primary keratinocyte cells after transduction with NF-kappa B [25]. The majority of the genes in this gene set are cytokines and growth factor genes including chemokines ( CCL20, CCL5, CXCL10, CXCL11, CXCL3, CXCL6); interleukins ( IL15, IL1B, IL1RN, IL6, IL8); and growth factor genes (TNC, VEGFA, ESM1, MP2). The “Uzonyi Response to Leukotriene and Thrombin” gene set is perturbed by the same number of pathogens as “Hinata NFKB Targets Keratinocyte up”. It contains 37 genes that were up-regulated in Human Umbilical Vein Endothelial Cells (HUVEC) after stimulation with leukotriene LTD4, a leukocyte produced at sites of inflammation [26]. The fourth gene set is “Mahadevan Response to MP470 up”, which is perturbed by 30 pathogens and appeared in 53 biclusters. This gene set contains 19 genes that were up-regulated in gastrointestinal stromal tumor cell-line after treatment with protein-kinase inhibitor drug (MP470) [27]. This gene set also contains chemokines and proinflammatory cytokines such as CCL5, CXCL1, CXCL10, CXCL3, CXCL5, CXCL6, IL8, and IL6.
Activation of Innate Immunity
In addition to inflammation, innate immunity also involves the activation of anatomical barriers, mechanical removal of antigens, pattern-recognition receptors, complement pathways, and phagocytosis. The “Netpath IL 4 Pathway down” gene set (which contains 90 genes that are supposed to be transcriptionally down-regulated by the activation of IL4 pathway) is among the top ten most perturbed gene sets. It is perturbed by 30 pathogens and is implicated in 59 biclusters. This gene set has a high overlap with three GO biological process namely “Activation of Innate Immune Response”, “Pattern Recognition Receptor Signaling Pathway”, and “Toll-like Receptor Signaling Pathway”. The perturbation of this gene set indicated that in addition to inflammation, other components of the innate immunity process are also perturbed by multiple bacterial pathogens.
Maturation of Dendritic Cells
Dendritic cells have the ability to develop from immature antigen-capturing cells to more specialized antigen-presenting cells. The maturation of dendritic cells is a very important aspect of the host response to bacterial infection. This step indicates the stimulation of various cytokines, chemokines, and other co-stimulatory molecules that are necessary for the onset of adaptive immunity [28]. A number of factors drive the maturation of dendritic cells including the type of antigen (e.g., lipopolysaccharide) and the presence of inflammatory cytokines (e.g., IL-1 and TNF-alpha). In our study, we found that the “Lindstedt Dendritic Cell Maturation A” gene set was perturbed by 30 pathogens and implicated in 67 biclusters. This gene set contains 54 genes that were up-regulated in a transcriptional study involving stimulation of human monocyte-derived dendritic cells with inflammatory stimuli, consisting of tumor necrosis factor (TNF)- and IL-1
and IL-1 [29].
[29].
Induction and Inhibition of Apoptosis
Induction and inhibition of apoptosis are important mechanisms of bacterial pathogenesis [22]. The “Dirmeier LMP1 Response Early” gene set, which has a high overlap with GO's “Apoptosis” (GO:0006915) and “Programmed Cell Death” (GO:0012501) biological processes is the second most highly perturbed gene set across the significant biclusters. It is perturbed by 32 pathogens spanning 76 biclusters. This gene set contains 54 genes that are dysregulated in B lymphocyte cells after induction of LMP1, an oncogene. This gene set contains both pro- and antiapoptotic genes whose balance permitted survival of B lymphocyte cells [30]. Perturbation of the “Dirmeier LMP1 Response Early” gene set by most of the pathogens we analyzed indicated that genes with opposing activities involved in cell survival were up-regulated during bacterial infection. This gene set contains tumor suppressors (KLF6, TNFAIP3), oncogenes (BIRC3, CXCR7, HERPUD1, HSP90AB1, LCP1, MYC, NFKB2), cell differentiation markers (CD69, CD83, ICAM1, SLAMF1), and growth markers (LTA, NPPB, TNFSF9).
Response to Lipopolysaccharide Stimulation
The host responds in a variety of ways against internal or external stimuli. An example of an external stimulus is a lipopolysaccharide (LPS). LPS is a molecule found on the outer membrane of Gram-negative bacteria. It triggers the expression of a number of signaling molecules, pro-inflammatory cytokines, and antibacterial genes when interacting with the Toll-like receptor of the host cell [31]. The “Seki Inflammatory Response LPS up” gene set [32], [33] contains genes that were up regulated in hepatic stellate cells of the mouse after stimulation with bacterial LPS. This gene set is up-regulated in as many as 83 biclusters (similar to “Zhang Response to IKK Inhibitor and TNF up” gene set) indicating that, genes related to LPS stimulation are predominantly perturbed across a significant number of Gram-negative pathogens. Previous studies have shown that LPS and Gram-negative bacteria such as Salmonella elicit identical patterns of gene regulation in macrophages [34], [35].
We expected this gene set would be perturbed only by Gram-negative bacteria, as LPS is a characteristic of these bacteria [31]. However, we observed that 30% of the pathogens that up-regulated this gene set were Gram-positive. Figure 2 shows 20 distinct pathogens (without counting strains of the same pathogen) that up-regulated the “Seki Inflammatory Response LPS up” gene set. Six of these pathogens are Gram-positive, namely Streptococcus pneumoniae, Listeria monocytogenes, Bifidobacterium bifidum, Streptococcus pyogenes, Lactobacillus acidophilus, and Bacillus anthracis. We noted that this gene set has a significant overlap with genes annotated with the biological process “Response to External Stimulus” (GO:0009605). This biological process represents the cells's response to external stimuli. Of the 83 genes annotated to this GO term, 14 genes also belong to “Seki Inflammatory Response LPS up” gene set (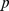 -value
-value 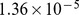 ). This high degree of overlap suggests that many genes that respond to LPS may belong to a broader class of genes that are perturbed by any external stimulus, including a pathogenic bacterium. This possibility may explain our finding that many Gram-positive bacteria perturb the gene set “Seki Inflammatory Response LPS up”.
). This high degree of overlap suggests that many genes that respond to LPS may belong to a broader class of genes that are perturbed by any external stimulus, including a pathogenic bacterium. This possibility may explain our finding that many Gram-positive bacteria perturb the gene set “Seki Inflammatory Response LPS up”.
Figure 2. Pathogens that perturb the “Seki Inflammatory Response LPS up” gene set.

Pathogens that perturb the “Seki Inflammatory Response LPS up” gene set. The second column contains the 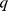 -values as well as a color indicating the magnitude of the
-values as well as a color indicating the magnitude of the 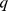 -value. Figure 3 contains the legend mapping
-value. Figure 3 contains the legend mapping 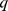 -values to colors. All pathogens up-regulate this gene set, except Streptococcus gordonii, which down-regulates it.
-values to colors. All pathogens up-regulate this gene set, except Streptococcus gordonii, which down-regulates it.
Putative HOBS Drug Targets
We now turn our attention to discovering potential HOBS drug targets in our biclusters. To this end, we further filtered the 84 significant biclusters based on the type of infection caused by the pathogens they contained. Table 3 shows biclusters that contained pathogens that cause an infection in a single type of tissue. We identified seven such biclusters: five gastrointestinal, one respiratory, and one hematopoietic. We selected the most statistically significant bicluster from each category for discussion in this paper.
Table 3. Biclusters divided by kind of infection.
| Pathogens | Bicluster 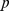 -value -value |
# Gene Sets | # Targets | Target Enrich. (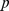 -value) -value) |
| Gastrointestinal | ||||
| Yersinia Enterocolitica wap and p60 strains, Helicobacter Pylori, and Escherichia Coli |
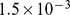
|
227 | 18 |
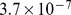
|
| Yersinia Enterocolitica, Lactobacillus Acidophilus, Listeria Monocytogenes, and Helicobacter Pylori |
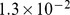
|
173 | 11 |
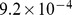
|
| Yersinia Enterocolitica and Helicobacter Pylori |
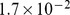
|
272 | 21 |
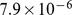
|
| Yersinia Enterocolitica, Listeria Monocytogenes, and Bifidobacterium Bifidum |
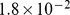
|
269 | 17 |
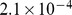
|
| Yersinia Enterocolitica, Bifidobacterium Bifidum, Streptococcus Pyogenes, and Helicobacter Pylori |
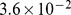
|
101 | 6 |
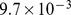
|
| Respiratory | ||||
| Pseudomonas Aeruginosa, and Mycobacterium Tuberculosis |
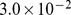
|
245 | 16 |
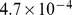
|
| Hematopoietic | ||||
| Ehrlichia Chaffeensis; Strains: arkansa and wakulla |
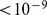
|
979 | 186 |
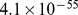
|
The table shows the biclusters that contained pathogens that cause an infection in a single type of tissue. The columns from left to right are: (i) list of pathogens contained in a bicluster, (ii) a 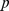 -value indicating the statistical significance of the bicluster, (iii) the number of gene sets in the bicluster, (iv) the number of known human drug target genes/proteins in the bicluster, and (v)
-value indicating the statistical significance of the bicluster, (iii) the number of gene sets in the bicluster, (iv) the number of known human drug target genes/proteins in the bicluster, and (v) 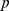 -value indicating the enrichment of the bicluster in know human drug-target genes/proteins.
-value indicating the enrichment of the bicluster in know human drug-target genes/proteins.
Gastrointestinal Pathogens
Bicluster 38 consisting of the Gram-negative pathogens Yersinia enterocolitica wap and p60 strains, Helicobacter pylori kx2 strain, and enterohemorrhagic Escherichia coli is the bicluster most enriched with gastrointestinal pathogens (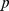 -value
-value  ). Yersinia enterocolitica causes a broad range of gastrointestinal syndromes ranging from acute diarrhea, terminal ileitis, mesenteric lymphadenitis, and pseudoappendicitis [36]. Helicobacter pylori kx2 strain is responsible for causing gastric adenocarcinoma [37]. Enterohemorrhagic Escherichia coli causes diarrhea or hemorrhagic colitis in humans [38]. The four pathogens jointly up-regulate 227 gene sets (Figure 3A shows the gene sets in this bicluster that contain drug targets). There are 18 known drug targets in this bicluster (
). Yersinia enterocolitica causes a broad range of gastrointestinal syndromes ranging from acute diarrhea, terminal ileitis, mesenteric lymphadenitis, and pseudoappendicitis [36]. Helicobacter pylori kx2 strain is responsible for causing gastric adenocarcinoma [37]. Enterohemorrhagic Escherichia coli causes diarrhea or hemorrhagic colitis in humans [38]. The four pathogens jointly up-regulate 227 gene sets (Figure 3A shows the gene sets in this bicluster that contain drug targets). There are 18 known drug targets in this bicluster (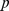 -value
-value 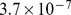 ). Below we will discuss the drug targets IL1R1 and TNF, which are both primary pro-inflammatory cytokines.
). Below we will discuss the drug targets IL1R1 and TNF, which are both primary pro-inflammatory cytokines.
Figure 3. Dendrogram of hierarchical clustering of gene sets for three tissue-specific biclusters.

Dendrogram of hierarchical clustering of gene sets for three tissue-specific biclusters. (A) Yersinia enterocolitica wap and p60 strains, Helicobacter pylori kx2 strain, and enterohemorrhagic Escherichia coli. (B) Pseudomonas aeruginosa and Mycobacterium tuberculosis. (C) E.chaffeensis Arkansa and Wakulla strains. The figure only shows gene sets that contain one or more known human drug targets.
Interleukin-1 type 1 receptor (IL-1R1) is a target molecule for the drug Anakinra (DrugBank ID DB00026). Anakinra is designed to treat rheumatoid arthritis by competitively binding to IL-1R1 thereby inhibiting the action of elevated levels of the pro-inflammatory cytokine IL-1. Previous studies have shown that Yersinia enterocolitica, Helicobacter pylori kx2 strain, and Enterohemorrhagic Escherichia coli induce chronic inflammation [37], [39], [40]. These observations suggest the potential use of drugs that suppress elevated levels of IL-1, such as Anakinra, in the treatment of gastrointestinal infections caused by these four pathogens. Another pro-inflammatory molecule produced by cells infected with bacteria is TNF- , which can cause TNF-
, which can cause TNF- -induced apoptosis. TNF-
-induced apoptosis. TNF- has been implicated as a target molecule for a number of FDA-approved drugs. Etanercept (DrugBank ID: DB00005) and Infliximab (DrugBank ID: DB00065) are TNF-
has been implicated as a target molecule for a number of FDA-approved drugs. Etanercept (DrugBank ID: DB00005) and Infliximab (DrugBank ID: DB00065) are TNF- blockers. Anti-TNF therapies have shown to be effective in the treatment of Crohn's disease and ulcerative colitis, which are both disease of the gastrointestinal tract that are characterized by inflammation [41], [42]. Although we did not find supporting evidence on the use of these drugs in the treatment of infections caused by Yersinia enterocolitica, Helicobacter pylori kx2 strain, and Enterohemorrhagic Escherichia coli, the potential use of TNF-
blockers. Anti-TNF therapies have shown to be effective in the treatment of Crohn's disease and ulcerative colitis, which are both disease of the gastrointestinal tract that are characterized by inflammation [41], [42]. Although we did not find supporting evidence on the use of these drugs in the treatment of infections caused by Yersinia enterocolitica, Helicobacter pylori kx2 strain, and Enterohemorrhagic Escherichia coli, the potential use of TNF- blockers such as Etanercept and Infliximab in the treatment of infection caused by these four pathogens may be worth investigating.
blockers such as Etanercept and Infliximab in the treatment of infection caused by these four pathogens may be worth investigating.
Respiratory Pathogens
Bicluster 72 is enriched with respiratory pathogens (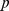 -value
-value 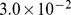 ). It contains the pathogens Pseudomonas aeruginosa and Mycobacterium tuberculosis. Pseudomonas aeruginosa causes major infections in immunocompromised patients. It is also a leading cause of hospital-acquired infections such as pneumonia [43]. Mycobacterium tuberculosis is a causative agent of tuberculosis. The two pathogens jointly perturb 245 gene sets including the IL-12 and IL-23 pathways (Figure 3B shows the gene sets in this bicluster that contain drug targets). The role of IL-12 induction in the treatment of M. tuberculosis has been reported in previous studies. For instance, Lowrie et al. have shown that up-regulation of IL-12 suppressed proliferation of M.tuberculosis in mice [44]. They further suggested the inclusion of this cytokine in tuberculosis vaccines. IL-12 plays a significant role in the host response against P.aeruginosa. It is an important molecule in the generation of IFN-
). It contains the pathogens Pseudomonas aeruginosa and Mycobacterium tuberculosis. Pseudomonas aeruginosa causes major infections in immunocompromised patients. It is also a leading cause of hospital-acquired infections such as pneumonia [43]. Mycobacterium tuberculosis is a causative agent of tuberculosis. The two pathogens jointly perturb 245 gene sets including the IL-12 and IL-23 pathways (Figure 3B shows the gene sets in this bicluster that contain drug targets). The role of IL-12 induction in the treatment of M. tuberculosis has been reported in previous studies. For instance, Lowrie et al. have shown that up-regulation of IL-12 suppressed proliferation of M.tuberculosis in mice [44]. They further suggested the inclusion of this cytokine in tuberculosis vaccines. IL-12 plays a significant role in the host response against P.aeruginosa. It is an important molecule in the generation of IFN- and TNF-
and TNF- , which are essential to promote bacterial clearance. Up-regulation of IL-12 by the host cell is a common strategy used by the host to fight infections caused by these two pathogens. Boosting the level of this molecule when needed, e.g., in immunocompromised patients, might be a viable strategy to treat infection caused by Pseudomonas aeruginosa and Mycobacterium tuberculosis. Studies suggest that Pseudomonas aeruginosa up-regulates IL-23 thereby creating airway inflammation in the host. Dubin et al.
[45] suggested the suppression of IL-23 as a potential avenue for immunotherapy to infection with this pathogen. Another study indicated that IL-23 is not required by the host to control Mycobacterium tuberculosis infection [46] indicating that the down-regulation of IL-23 may not disrupt the host defense mechanism during M.tuberculosis infection. Therefore, we suggest that down-regulating IL-23 might be a common strategy to treat infection caused by Pseudomonas aeruginosa and Mycobacterium tuberculosis.
, which are essential to promote bacterial clearance. Up-regulation of IL-12 by the host cell is a common strategy used by the host to fight infections caused by these two pathogens. Boosting the level of this molecule when needed, e.g., in immunocompromised patients, might be a viable strategy to treat infection caused by Pseudomonas aeruginosa and Mycobacterium tuberculosis. Studies suggest that Pseudomonas aeruginosa up-regulates IL-23 thereby creating airway inflammation in the host. Dubin et al.
[45] suggested the suppression of IL-23 as a potential avenue for immunotherapy to infection with this pathogen. Another study indicated that IL-23 is not required by the host to control Mycobacterium tuberculosis infection [46] indicating that the down-regulation of IL-23 may not disrupt the host defense mechanism during M.tuberculosis infection. Therefore, we suggest that down-regulating IL-23 might be a common strategy to treat infection caused by Pseudomonas aeruginosa and Mycobacterium tuberculosis.
Hematopoietic Pathogens
Bicluster 0 contains two E.chaffeensis species, Arkansa and Wakulla. Infection with Ehrlichia chaffeensis causes ehrlichiosis, which is characterized by an influenza-like illness, elevation of transaminase levels and sepsis [47]. These two strains commonly up-regulated as many as 979 gene sets, which is not surprising considering the fact that they are different strains of the same bacterial pathogen. However, what is interesting is that the E.chaffeensis Liberty strain, which is a part of our study, is not part of this bicluster. This result indicates E.chaffeensis Arkansa and E.chaffeensis wakulla elicit similar host responses that are different from those perturbed by the Liberty strain. Considering the similarity in the host transcriptional responses, it is tempting to speculate that a common treatment regimen may exist for infection caused by the strains Arkansa and Wakulla.
Among the commonly up-regulated gene sets, “Hsiao Liver Specific Genes” contains the highest number of known drug targets (Figure 3C shows the gene sets in this bicluster that contain drug targets). There are 49 known drug-target proteins in this gene set alone. The “Hsiao Liver Specific Genes” gene set determined by Hsiao et al. [48] contains 255 genes that are selectively expressed in the human liver in a gene expression profiling study that involved 59 human samples of 19 different tissue types. The genes in “Hsiao Liver Specific Genes” genes are annotated with liver-specific function including blood coagulation (GO:0007596) and homeostasis (GO:0007599). The up-regulation of the “Hsiao Liver Specific Genes” gene sets by by Wakulla and Arkansas (but not by Liberty) might indicate that E.chaffeensis Liberty is inactive in the liver.
The liver is an important organ in cholesterol synthesis, regulation, and export to the other cells. The “Hsiao Liver Specific Genes” gene set contains the protein F2, coagulation factor II (thrombin), which is linked to the cholesterol lowering drug Simvastatin (DrugBank ID: DB00641). Simvastatin reduces total and LDL-cholesterol as well as plasma triglycerides and apolipoprotein B. Previous studies have indicated that E.chaffeensis requires cholesterol for survival and growth. However, E.chaffeensis does not have the genes for synthesizing cholesterol. Instead, it depends on the host cell to acquire this molecule [49]. In another study, treatment of E.chaffeensis with cholesterol extraction reagent methyl-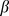 -cyclodextrin hampred the ability of this pathogen to infect leukocytes [50]. With this observation in mind, we reasoned that cholesterol lowering drugs such as Simvastatin can be used in the treatment of E.chaffeensis infection.
-cyclodextrin hampred the ability of this pathogen to infect leukocytes [50]. With this observation in mind, we reasoned that cholesterol lowering drugs such as Simvastatin can be used in the treatment of E.chaffeensis infection.
Known Anti-infective Drug-targets in Biclusters
In the previous section, we attempted to predict HOBS drug targets for three biclusters where the pathogens contained in each bicluster are known to infect similar organs of the human host. In this section, we ranked all statistically significant biclusters based on the number of known anti-infective drug targets that they contain. Identification of such biclusters may be useful to predict other HOBS drug targets in the same bicluster.
To this end, we used the Anatomical Therapeutic Chemical (ATC) Classification from DrugBank and categorized drug targets that are found in statistically significant biclusters as anti-infective or non-anti-infective targets (Table S4). Out of 479 drug targets that are contained in these biclusters, 73 of them are known to be targeted by one or more anti-infective drugs. A functional enrichment analysis of these drug-target genes using DAVID [51] revealed that “response to wounding” (GO:0009611), “inflammatory response” (GO:0006954), “defense response” (GO:0006952), and the KEGG complement and coagulation cascades pathway are among the top five highly enriched biological processes (Table S5). These results shed light on which biological processes in the host are commonly targeted by existing anti-infective drugs.
Bicluster 0 and Bicluster 72 that we discussed in the previous section are the two biclusters that contain the highest number of anti-infective drug targets. Bicluster 0 that contains two strains of Ehrlichia Chaffeensis, arkansa and wakulla, has 58 anti-infective drug targets. Bicluster 72 that contains two respiratory pathogens Pseudomonas aeruginosa and Mycobacterium tuberculosis, has 12 anti-infective targets. It appears that biclusters that had the highest number of anti-infective targets also contained pathogens that are related to one another. This result provided support to our approach of focusing on biclusters that contained pathogens infecting similar organs/tissue of the host.
Conclusions
In this paper, we have presented a computational approach to identify potential host-oriented broad-spectrum drug targets. Gene set enrichment and biclustering were key ingredients of our method. We combined these two techniques to compute subsets of pathogens that commonly up- or down- regulated sets of biological pathways, gene sets, or protein complexes. We applied this approach on a compendium of gene expression data that represented 38 bacterial pathogens and pathogen strains, from which we identified 84 up-regulated and three down-regulated statistically significant biclusters. Using this approach we were successful in detecting common host responses that are hallmarks of bacterial infections.
Motivated by the premise that diseases that have high degree of transcriptional similarity may be treated with similar drugs [17], we integrated drug target information into our analysis to predict HOBS targets for bacterial infections. Focusing on biclusters that contained pathogens that infected same tissue, we predicted new uses of the drugs Anakinra, Etanercept, and Infliximab for gastrointestinal pathogens Yersinia enterocolitica, Helicobacter pylori kx2 strain, and enterohemorrhagic Escherichia coli and the drug Simvastatin for hematopoietic pathogen Ehrlichia chaffeensis.
Broadly, the approach we presented in this paper falls in the realm of integrative DNA microarray data analysis. It can be viewed as an alternative approach to the existing methods developed to discover transcriptional responses common to many diseases [16], [18], [20]. Unlike previous approaches, our method leverages biclustering to detect pathway-specific relationships only among subsets of pathogens.
Our computational approach depends on the identification and targeting of genes whose expression is modulated during host-pathogen interactions. A potential concern with this approach is that it may not distinguish between beneficial host responses and those that may worsen the pathogenecity of the microbe. Dysregulation of a particular biological pathway may not have the same effect on the host under all kinds of infections. For instance, inflammation is often an important host defensive mechanism that may be harmful if uncontrolled.
We computed biclusters that contained groups of biological pathways that are commonly dysregulated by group of pathogens. We acknowledged that a pathway may not be appropriate to target by HOBS drugs simply because a group of pathogens dysregulated that pathway. Accordingly, we used biclustering as a filtering step that would provide potential candidates for HOBS drug targets. In our analysis, we subjected each commonly dysregulated pathway to additional examination, wherein we studied the literature on these pathways and the genes they contained in the context of the pathogens that perturbed them. We used this additional manual step in order to prevent us from proposing an intervention mechanism that would inadvertently block beneficial host responses.
Another difficulty that may arise with our approach is that the number of pathways in a bicluster can sometimes be overwhelming for subsequent analysis. A rational extension to our work is to design methods to prioritize non-redundant biclusters and biological processes based on the similarity of their perturbation. Recent techniques for functional enrichment [52] may be appropriate for this task.
The perturbation of a group of gene sets by a group of pathogens indicates by itself that there might be some underlying similarities in the mechanisms used by the pathogens to infect the host. Therefore, we would ideally like to examine each statistically significant bicluster regardless of whether it contains a drug target or not. The large number of biclusters we computed precluded this detailed analysis. Hence, we chose the strategy of prioritizing biclusters based on drug-target enrichment. The other statistically significant biclusters presented in our supplementary results may also be worthy of further study in the future.
In this study, we analyzed host response data from bacterial infections. In the future, we plan to apply the approach developed here to fungal and viral data sets as well. The results from our studies and related approaches [20] may serve as powerful resources for researchers engaged in host-oriented broad-spectrum drug target discovery.
Methods
Gene Expression Datasets
We retrieved 808 distinct taxonomic names of bacterial pathogens from the American Biological Safety Association database of human pathogens. We downloaded the GEO meta database [53] that contains metadata associated with the NCBI's Gene Expression Omnibus (GEO) [54] samples, platforms, and datasets. Next, we queried the meta database using the taxonomic names as keywords. We obtained gene expression datasets for 105 of the 808 bacterial pathogens. Next, we pruned the datasets using the following criteria: (i) We removed time-course data to avoid complications that could arise due to temporal variation of cellular responses to the various pathogens. (ii) We excluded datasets that have less than six samples (infected and healthy samples combined) so that our datasets conform to the recommended sample size for conducting t-tests. (iii) We considered DNA microarray data collected from three hosts, namely, Homo sapiens, Mus musclus, and Rattus norvegicus. (iv) We considered experiments that involved the comparison of normal and infected samples. After this process, we retained 29 GEO datasets for subsequent analysis. Details on these datasets are given in Table S1.
Gene Set Compendium
We built comprehensive functional annotation data sets encompassing biological pathways and functionally associated genes. We integrated data from four sources:
1.National Cancer Institute-Nature Pathway Interaction Database (NCI-PID): The NCI-PID contains a collection of curated and peer-reviewed pathways of molecular signaling, regulatory events, and cellular processes [55].
2.NetPath: The NetPath database contains cancer and immune signaling pathways, such as the T- and B- cell receptor signaling pathways [56].
3.CORUM: The CORUM database houses protein complexes mainly from human, rat, and mouse. A protein complex contains multiple gene products annotated by the same function or localization e.g., respiratory chain protein complex mitochondrial [57].
4.The Molecular Signature Database (MsigDB): MsigDB contains genes that are biologically related. This relatedness can be defined by participation in the same biological pathway, chromosomal location, or response to some treatment as evidenced by high-throughput experiments such as gene expression profiling. MsigDB houses four categories of gene sets namely, positional gene sets, curated gene sets, motif gene sets, and computational gene sets. In our analyses we used only curated gene sets.
We collected 449 curated pathways from NCI-PID, 20 curated pathways from the NetPath database, 1,765 protein complexes from the CORUM database, and 3,272 curated gene sets from MsigDB.
Drugs and Drug Targets Data
We collected 1,652 human drug target proteins from DrugBank [58]. These drug targets were linked to 6,796 therapeutically-validated and experimental drugs.
Computation of Gene Sets Perturbed in the Host by a Pathogen
We downloaded the raw gene expression profiles (CEL files) from the NCBI's Gene Expression Omnibus (GEO) [54] for the 29 GEO accessions identified above. We normalized the datasets with the Microarray Analysis Suite (MAS5) [59] using the ExpressionFileCreator Module of the GenePattern genomic analysis platform [60]. We ran Gene Set Enrichment Analysis (GSEA) [61] on each gene expression dataset using the compendium of gene sets collected above. We collected the resulting q-values (False Discovery Rate or FDR values) into a matrix that indicates the significance of perturbation of each gene set by each pathogen. A q-value is the expected probability that GSEA's assessment that a pathogen perturbs a gene set represents a false positive finding. We use a cutoff of 0.2 on q-value, which implies that four out of five gene sets that we consider to be perturbed by a pathogen are likely to be true discoveries. As we describe below, we further reduce the possibility of false discoveries in three steps: (i) we compute pathogen-gene set biclusters, (ii) we estimate the statistical significance of each bicluster, and (iii) we compute the enrichment of biclusters in known drug targets. A bicluster associates multiple pathogens with multiple gene sets. Therefore, each gene set in a bicluster is perturbed by more than one pathogen, decreasing the possibility that the perturbation of this gene set is a random occurrence. Furthermore, by estimating the statistical significance of each bicluster, we discard biclusters (and the pathogen-gene set associations that they represent) that could have arisen by random chance. Finally, we filter-out biclusters that are not significantly enriched in known drug targets. This process enabled us to focus on drug-target enriched, non-random, pathogen-gene set associations.
Biclustering the q-value Matrix
Then, we created two binary matrices representing up-regulated and down-regulated biclusters, respectively. In each matrix, each row corresponded to a gene set and each column to a pathogen. An entry in one of these matrices had a value of  if and only if the GSEA q-value for that gene set-pathogen pair was at least 0.2. We applied the BiMax algorithm [62] implemented in the BicAT biclustering analysis toolbox [63] on these matrices to obtain two sets of biclusters, one for up-regulated gene sets and another for down-regulated gene sets.
if and only if the GSEA q-value for that gene set-pathogen pair was at least 0.2. We applied the BiMax algorithm [62] implemented in the BicAT biclustering analysis toolbox [63] on these matrices to obtain two sets of biclusters, one for up-regulated gene sets and another for down-regulated gene sets.
Computing the Statistical Significance of Biclusters
We generated 10,000 randomized binary matrices using the swap randomization algorithm [64]. Given a binary matrix 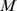 with values
with values  and
and  , the swap randomization algorithm creates a random matrix
, the swap randomization algorithm creates a random matrix 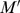 such that each row (respectively, column) of
such that each row (respectively, column) of 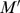 has the same number of
has the same number of 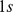 as the corresponding row (respectively, column) of
as the corresponding row (respectively, column) of 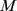 . The algorithm achieves this goal through a series of steps that swap row-column pairs. We used our own Perl implementation of this algorithm. We computed biclusters in each of these matrices. We built two sets of distributions reflecting the number of pathogens and the number of genes sets in random biclusters. First, for every integer
. The algorithm achieves this goal through a series of steps that swap row-column pairs. We used our own Perl implementation of this algorithm. We computed biclusters in each of these matrices. We built two sets of distributions reflecting the number of pathogens and the number of genes sets in random biclusters. First, for every integer 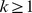 , we recorded the number of biclusters that contained
, we recorded the number of biclusters that contained 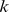 pathogens and at least
pathogens and at least 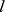 gene sets, for different values of
gene sets, for different values of 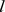 . Next, we repeated this process for each integer
. Next, we repeated this process for each integer 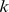 , considering the number of gene sets in a bicluster. Now, given a bicluster in the original data containing
, considering the number of gene sets in a bicluster. Now, given a bicluster in the original data containing 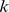 pathogens and
pathogens and 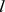 gene sets, we computed two
gene sets, we computed two 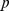 -values. One
-values. One 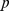 -value was the fraction of random biclusters that contained
-value was the fraction of random biclusters that contained 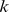 pathogens and at least
pathogens and at least 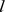 gene sets. The second
gene sets. The second 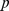 -value was the fraction of random biclusters that contained
-value was the fraction of random biclusters that contained 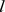 gene sets and at least
gene sets and at least 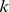 pathogens. These
pathogens. These 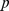 -values indicate the probability of observing a bicluster that contains at least a certain number of pathogens or gene sets in the original dataset by chance. We adjusted the
-values indicate the probability of observing a bicluster that contains at least a certain number of pathogens or gene sets in the original dataset by chance. We adjusted the 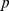 -values for multiple hypothesis testing using the method of Benjamini-Hochberg [21]. Finally, we chose the greater of the two
-values for multiple hypothesis testing using the method of Benjamini-Hochberg [21]. Finally, we chose the greater of the two 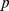 -values as a
-values as a 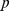 -value for each bicluster. We further considered only biclusters with
-value for each bicluster. We further considered only biclusters with 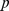 -value of at most 0.05.
-value of at most 0.05.
Computation of Bicluster Enrichment
We computed the enrichment of each bicluster in various attributes such as the number of known drug targets, host type (human, mouse, and rat), infected cell type (epithelial, dendritic, and macrophage), Gram stain of the pathogen (positive and negative), and infection kind (gastrointestinal, respiratory, oral cavity, and hematopoietic). We used Fisher's exact test for testing the significance of enrichment of a bicluster in each of these attributes.
Translating Gene Identifiers
Different data sources use different naming schemes for identifying genes . For instance, the molecular signature database uses HUGO symbols while DrugBank uses UniProt namespaces. We used HUGO gene symbols as the common gene identifier in our study. We used the Synergizer service for translating gene/protein's identifiers from other namespaces to HUGO [65].
Assigning Gene Ontology Biological Processes to a Gene Set
Some of the gene set names in the MsigBD are not self-explanatory, affecting intuitive interpretation of results. In order alleviate this problem, we considered the Gene Ontology biological processes that have the highest overlaps with each respective gene set. To this end, we used the pre-computed overlap/hypergeometric 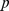 -values between a gene set and GO processes that are provided on the MsigDB website. For the “Netpath IL 4 Pathway Down” gene set, we obtained the corresponding GO biological processes using GOrilla [66].
-values between a gene set and GO processes that are provided on the MsigDB website. For the “Netpath IL 4 Pathway Down” gene set, we obtained the corresponding GO biological processes using GOrilla [66].
Supporting Information
Scatter plot of number of pathogens vs. biclusters. Plot indicates that number of pathogens perturbing a gene set are positively correlated with the number of biclusters a particular gene set appeared in. Supporting information can also be accessed from our supplementary website: http://bioinformatics.cs.vt.edu/ murali/supplements/2013-kidane-plos-one.
(PDF)
Details of DNA microarray dataset used in the study. It contains GEO accession numbers, microarray platform used, infected host, and tissue or cell type from which the gene expression measurements were taken.
(HTML)
Up-regulated biclusters. It contains detail information on all up-regulated biclusters. This include: bicluster ID, list of pathogens and gene sets in bicluster, 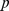 -values indicating statistical significance of bicluster and enrichment of these biclusters in various attributes such as drug targets and host type.
-values indicating statistical significance of bicluster and enrichment of these biclusters in various attributes such as drug targets and host type.
(HTML)
Down-regulated biclusters. It contains detail information on all down-regulated biclusters. This include: bicluster ID, list of pathogens and gene sets in bicluster, 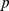 -values indicating statistical significance of bicluster and enrichment of these biclusters in various attributes such as drug targets and host type.
-values indicating statistical significance of bicluster and enrichment of these biclusters in various attributes such as drug targets and host type.
(HTML)
Known anti-infective targets in biclusters. It contains bicluster ID, list of all drug targets, and anti-infective targets in bicluster.
(XLS)
Functional annotations of anti-infective targets. It contains 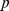 -values indicating enrichment of anti-infective drug targets in GO biological processes.
-values indicating enrichment of anti-infective drug targets in GO biological processes.
(XLS)
Acknowledgments
We thank Oswald Crasta, Josep Bassaganya-Riera and Stephen Melville for useful discussions.
Funding Statement
The work presented in this paper was supported by Initiative to Maximize Students Development program, Genetics, Bioinformatics and Computational Biology program at Virginia Polytechnic Institute and State University, and Southern Regional Education Board dissertation fellowship program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Fauci AS, Touchette NA, Folkers GK (2005) Emerging infectious diseases: A 10-year perspective from the national institute of allergy and infectious diseases. The International Journal of Risk and Safety in Medicine 17: 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Walsh C (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406: 775–781. [DOI] [PubMed] [Google Scholar]
- 3.Schwegmann A, Brombacher F (2008) Host-directed drug targeting of factors hijacked by pathogens. Sci Signal 1 : re8+. [DOI] [PubMed] [Google Scholar]
- 4. Schneider DS, Ayres JS (2008) Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol 8: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan SL, Ganji G, Paeper B, Proll S, Katze MG (2007) Systems biology and the host response to viral infection. Nature Biotechnology 25: 1383–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woolhouse ME, Gowtage-Sequeria S (2005) Host range and emerging and reemerging pathogens. Emerging Infectious Diseases 11: 1842–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rawlins MD (2004) Cutting the cost of drug development? Nature reviews Drug discovery 3: 360–364. [DOI] [PubMed] [Google Scholar]
- 8. Finlay BB, Hancock RE (2004) Can innate immunity be enhanced to treat microbial infections? Nature reviews Microbiology 2: 497–504. [DOI] [PubMed] [Google Scholar]
- 9. Del Real G, Jiménez-Baranda S, Mira E, Lacalle RAA, Lucas P, et al. (2004) Statins inhibit HIV-1 infection by down-regulating rho activity. The Journal of Experimental Medicine 200: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu CII, Liu GY, Song Y, Yin F, Hensler ME, et al. (2008) A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 319: 1391–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pucadyil TJ, Chattopadhyay A (2007) Cholesterol: a potential therapeutic target in Leishmania infection? Trends in Parasitology 23: 49–53. [DOI] [PubMed] [Google Scholar]
- 12. Hamill P, Brown K, Jenssen H, Hancock R (2008) Novel anti-infectives: is host defence the answer? Current Opinion in Biotechnology 19: 628–636. [DOI] [PubMed] [Google Scholar]
- 13. Rhodes D, Yu J, Shanker K, Deshpande N, Varambally R, et al. (2004) Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A 101: 9309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perusse L, Rankinen T, Zuberi A, Chagnon YC, Weisnagel SJ, et al. (2005) The human obesity gene map: The 2004 update. Obesity 13: 381–490. [DOI] [PubMed] [Google Scholar]
- 15. De Magalhães JaP, Curado Ja, Church GM (2009) Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 25: 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jenner RG, Young RA (2005) Insights into host responses against pathogens from transcriptional profiling. Nature Reviews Microbiology 3: 281–294. [DOI] [PubMed] [Google Scholar]
- 17. Dudley JT, Deshpande T, Butte AJ (2011) Exploiting drug-disease relationships for computational drug repositioning. Briefings in Bioinformatics 12: 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu G, Agarwal P (2009) Human disease-drug network based on genomic expression profiles. PLoS ONE 4 : e6536+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, et al. (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313: 1929–1935. [DOI] [PubMed] [Google Scholar]
- 20.Suthram S, Dudley JT, Chiang AP, Chen R, Hastie TJ, et al. (2010) Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput Biol 6 : e1000662+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society 57: 289–300. [Google Scholar]
- 22. Boya P, Roques B, Kroemer G (2001) Viral and bacterial proteins regulating apoptosis at the mitochondrial level. The EMBO Journal 20: 4325–4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hayden MS, West AP, Ghosh S (2006) NF-κB and the immune response. Oncogene 25: 6758–6780. [DOI] [PubMed] [Google Scholar]
- 24. Zhang Y, Gavriil M, Lucas J, Mandiyan S, Follettie M, et al. (2008) IκBα kinase inhibitor IKI-1 conferred tumor necrosis factor α sensitivity to pancreatic cancer cells and a xenograft tumor model. Cancer Research 68: 9519–9524. [DOI] [PubMed] [Google Scholar]
- 25. Hinata K, Gervin AM, Jennifer Zhang Y, Khavari PA (2003) Divergent gene regulation and growth effects by NF-κB in epithelial and mesenchymal cells of human skin. Oncogene 22: 1955–1964. [DOI] [PubMed] [Google Scholar]
- 26. Uzonyi B, Lotzer K, Jahn S, Kramer C, Hildner M, et al. (2006) Cysteinyl leukotriene 2 receptor and protease-activated receptor 1 activate strongly correlated early genes in human endothelial cells. Proc Natl Acad Sci U S A 103: 6326–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mahadevan D, Cooke L, Riley C, Swart R, Simons B, et al. (2007) A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 26: 3909–3919. [DOI] [PubMed] [Google Scholar]
- 28. Théry C (2001) The cell biology of antigen presentation in dendritic cells. Current Opinion in Immunology 13: 45–51. [DOI] [PubMed] [Google Scholar]
- 29. Lindstedt M, Johansson-Lindbom B, Borrebaeck CAK (2002) Global reprogramming of dendritic cells in response to a concerted action of inammatory mediators. International Immunology 14: 1203–1213. [DOI] [PubMed] [Google Scholar]
- 30. Dirmeier U, Hoffmann R, Kilger E, Schultheiss U, Briseño C, et al. (2005) Latent membrane protein 1 of Epstein-Barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 24: 1711–1717. [DOI] [PubMed] [Google Scholar]
- 31. Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annual Review of Immunology 21: 335–376. [DOI] [PubMed] [Google Scholar]
- 32. Foster SL, Hargreaves DC, Medzhitov R (2007) Gene-specific control of inammation by TLRinduced chromatin modifications. Nature 447: 972–978. [DOI] [PubMed] [Google Scholar]
- 33. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, et al. (2007) TLR4 enhances TGF-β signaling and hepatic fibrosis. Nature Medicine 13: 1324–1332. [DOI] [PubMed] [Google Scholar]
- 34.McDermott JE, Archuleta M, Thrall BD, Adkins JN, Waters KM (2011) Controlling the response: predictive modeling of a highly central, pathogen-targeted core response module in macrophage activation. PloS one 6 : e14673+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramsey SA, Klemm SL, Zak DE, Kennedy KA, Thorsson V, et al. (2008) Uncovering a macrophage transcriptional program by integrating evidence from motif scanning and expression dynamics. PLoS computational biology 4 : e1000021+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bercovier H, Brenner D, Ursing J, Steigerwalt A, Fanning G, et al. (1980) Characterization of Yersinia enterocolitica sensu stricto. Current Microbiology 4: 201–206. [Google Scholar]
- 37. Giannakis M, Chen SL, Karam SM, Engstrand L, Gordon JI (2008) Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc Natl Acad Sci U S A 105: 4358–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Campos LC, Whittam TS, Gomes TA, Andrade JR, Trabulsi LR (1994) Escherichia coli serogroup O111 includes several clones of diarrheagenic strains with different virulence properties. Infection and Immunity 62: 3282–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saebø A, Lassen J (1994) Yersinia enterocolitica: an inducer of chronic inammation. International Journal of Tissue Reactions 16: 51–57. [PubMed] [Google Scholar]
- 40. Ritchie JM, Thorpe CM, Rogers AB, Waldor MK (2003) Critical roles for stx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inammation in infant rabbits. Infection and Immunity 71: 7129–7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Teshima CW, Thompson A, Dhanoa L, Dieleman LA, Fedorak RN (2009) Long-term response rates to iniximab therapy for crohn's disease in an outpatient cohort. Canadian Journal of Gastroenterology 23: 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, et al. (2005) Iniximab for induction and maintenance therapy for ulcerative colitis. The New England Journal of Medicine 353: 2462–2476. [DOI] [PubMed] [Google Scholar]
- 43.Bodey GP, Bolivar R, Fainstein V, Jadeja L (1983) Infections caused by Pseudomonas aeruginosa. Reviews of Infectious Diseases 5.. [DOI] [PubMed] [Google Scholar]
- 44. Lowrie DB, Tascon RE, Bonato VLD, Lima VMF, Faccioli LH, et al. (1999) Therapy of tuberculosis in mice by DNA vaccination. Nature 400: 269–271. [DOI] [PubMed] [Google Scholar]
- 45. Dubin PJ, Kolls JK (2007) IL-23 mediates inammatory responses to mucoid Pseudomonas aerug- inosa lung infection in mice. American Journal of Physiology - Lung Cellular and Molecular Physiology 292: L519–L528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, et al. (2005) IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. Journal of Immunology 175: 788–795. [DOI] [PubMed] [Google Scholar]
- 47. Paddock CD, Childs JE (2003) Ehrlichia chaffeensis: a prototypical emerging pathogen. Clinical Microbiology Reviews 16: 37–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hsiao LL, Dangond F, Yoshida T, Hong R, Jensen RV, et al. (2001) A compendium of gene expression in normal human tissues. Physiological Genomics 7: 97–104. [DOI] [PubMed] [Google Scholar]
- 49. Rikihisa Y (2010) Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nature Reviews Microbiology 8: 328–339. [DOI] [PubMed] [Google Scholar]
- 50. Lin M, Rikihisa Y (2003) Ehrlichia chaffeensis and Anaplasma phagocytophilum lack genes for lipid biosynthesis and incorporate cholesterol for their survival. Infect Immun 71: 5324–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Huang DWaW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 52. Bauer S, Gagneur J, Robinson PN (2010) GOing bayesian: model-based gene set analysis of genome-scale data. Nucleic Acids Research 38: 3523–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhu Y, Davis S, Stephens R, Meltzer PS, Chen Y (2008) GEOmetadb: powerful alternative search engine for the gene expression omnibus. Bioinformatics 24: 2798–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Barrett T, Suzek T, Troup D, Wilhite S, Ngau W, et al. (2005) NCBI GEO: mining millions of expression profiles–database and tools. Nucleic Acids Research 33: D562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, et al. (2009) PID: the Pathway Interaction Database. Nucleic Acids Research 37: D674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kandasamy K, Mohan SS, Raju R, Keerthikumar S, Kumar GS, et al. (2010) NetPath: a public resource of curated signal transduction pathways. Genome Biol 11: R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ruepp A, Brauner B, Dunger-Kaltenbach I, Frishman G, Montrone C, et al. (2008) CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Research 36: D646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wishart DS, Knox C, Guo ACC, Cheng D, Shrivastava S, et al. (2008) DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Research 36: D901–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.MAS5. Available: http://media.affymetrix.com/support/technical/whitepapers/saddwhitepaper.pdf.
- 60. Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, et al. (2006) GenePattern 2.0. Nature Genetics 38: 500–501. [DOI] [PubMed] [Google Scholar]
- 61.Subramanian A, Tamayo P, Mootha V, Mukherjee S, Ebert B, et al.. (2005) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed]
- 62. Prelić A, Bleuler S, Zimmermann P, Wille A, Bühlmann P, et al. (2006) A systematic comparison and evaluation of biclustering methods for gene expression data. Bioinformatics 22: 1122–1129. [DOI] [PubMed] [Google Scholar]
- 63. Barkow S, Bleuler S, PreliÄ A, Zimmermann P, Zitzler E (2006) BicAT: a biclustering analysis toolbox. Bioinformatics 22: 1282–1283. [DOI] [PubMed] [Google Scholar]
- 64.Gionis A, Mannila H, Mielikäinen T, Tsaparas P (2007) Assessing data mining results via swap randomization. ACM Trans Knowl Discov Data 1: : 14+. [Google Scholar]
- 65. Berriz GF, Roth FP (2008) The synergizer service for translating gene, protein and other biological identifiers. Bioinformatics 24: 2272–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10: : 48+. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scatter plot of number of pathogens vs. biclusters. Plot indicates that number of pathogens perturbing a gene set are positively correlated with the number of biclusters a particular gene set appeared in. Supporting information can also be accessed from our supplementary website: http://bioinformatics.cs.vt.edu/ murali/supplements/2013-kidane-plos-one.
(PDF)
Details of DNA microarray dataset used in the study. It contains GEO accession numbers, microarray platform used, infected host, and tissue or cell type from which the gene expression measurements were taken.
(HTML)
Up-regulated biclusters. It contains detail information on all up-regulated biclusters. This include: bicluster ID, list of pathogens and gene sets in bicluster, -values indicating statistical significance of bicluster and enrichment of these biclusters in various attributes such as drug targets and host type.
(HTML)
Down-regulated biclusters. It contains detail information on all down-regulated biclusters. This include: bicluster ID, list of pathogens and gene sets in bicluster, -values indicating statistical significance of bicluster and enrichment of these biclusters in various attributes such as drug targets and host type.
(HTML)
Known anti-infective targets in biclusters. It contains bicluster ID, list of all drug targets, and anti-infective targets in bicluster.
(XLS)
Functional annotations of anti-infective targets. It contains -values indicating enrichment of anti-infective drug targets in GO biological processes.
(XLS)