

Abstract
Heme is an essential molecule for vast majority of organisms serving as a prosthetic group for various hemoproteins. Although most organisms synthesize heme from 5-aminolevulinic acid through a conserved heme biosynthetic pathway composed of seven consecutive enzymatic reactions, nematodes are known to be natural heme auxotrophs. The completely sequenced Caenorhabditis elegans genome, for example, lacks all seven genes for heme biosynthesis. However, genome/transcriptome sequencing of Strongyloides venezuelensis, an important model nematode species for studying human strongyloidiasis, indicated the presence of a gene for ferrochelatase (FeCH), which catalyzes the terminal step of heme biosynthesis, whereas the other six heme biosynthesis genes are apparently missing. Phylogenetic analyses indicated that nematode FeCH genes, including that of S. venezuelensis (SvFeCH) have a fundamentally different evolutionally origin from the FeCH genes of non-nematode metazoa. Although all non-nematode metazoan FeCH genes appear to be inherited vertically from an ancestral opisthokont, nematode FeCH may have been acquired from an alpha-proteobacterium, horizontally. The identified SvFeCH sequence was found to function as FeCH as expected based on both in vitro chelatase assays using recombinant SvFeCH and in vivo complementation experiments using an FeCH-deficient strain of Escherichia coli. Messenger RNA expression levels during the S. venezuelensis lifecycle were examined by real-time RT-PCR. SvFeCH mRNA was expressed at all the stages examined with a marked reduction at the infective third-stage larvae. Our study demonstrates the presence of a bacteria-like FeCH gene in the S. venezuelensis genome. It appeared that S. venezuelensis and some other animal parasitic nematodes reacquired the once-lost FeCH gene. Although the underlying evolutionary pressures that necessitated this reacquisition remain to be investigated, it is interesting that the presence of FeCH genes in the absence of other heme biosynthesis genes has been reported only for animal pathogens, and this finding may be related to nutritional availability in animal hosts.
Introduction
Heme is essential for the vast majority of life serving as a prosthetic group for many hemoproteins such as catalase, cytochrome, hemoglobin, myoglobin, and peroxidase [1]. Although most aerobic organisms possess a complete biosynthetic pathway for this compound [2], certain organisms are deficient in heme biosynthesis, lacking some or all genes for the hemebiosynthetic pathway. Some anaerobic protists, such as Giardia intestinalis, Trichomonas vaginalis, Entamoeba histolytica, Cryptosporidium parvum, Blastocystis hominis, and Encephalitozoon cuniculi do not possess any heme biosynthetic genes [3]. Members of the family Trypanosomatidae lost some or the entire set of heme biosynthesis genes. They acquire heme or heme precursors from their diet [3], [4]. In Trypanosomatidae, members of the genus Trypanosoma lack all the heme biosynthesis genes [3], [5], [6], [7], whereas other members such as Leishmania spp. possess the genes for the last three steps which were horizontally acquired from a gamma–proteobacterium [3]. Insect trypanosomatid species (Blastocrithidia culicis and Crithidia oncopelti) cannot synthesize heme by themselves but harbor bacterial endosymbionts that generate and donate heme or heme precursors to the host (trypanosomatid) cells [4], [8]. More peculiar is the case of Phytomonas serpens, a plant kinetoplastid [9]. This organism lacks most of the known hemoproteins including respiratory cytochromes and does not require heme for viability despite its dependence on oxidative metabolism [9]. The draft genome of P. serpens does not appear to contain heme biosynthesis genes other than ferrochelatase (FeCH, EC 4.99.1.1) [9].
Another important and interesting group of organisms that lack the ability to synthesize heme is the nematodes. Nematodes, or roundworms, are typically small, diverse, and highly abundant metazoan organisms [10]. Although free-living species are found in nearly all habitats (marine, freshwater, and soil), nematodes are also parasites of vertebrate and invertebrate animals as well as plants. Molecular phylogenetics have defined five major nematode clades (I through V), within which parasitism has arisen multiple times [11]. The genome of Caenorhabditis elegans, which was the first metazoan genome to be completely sequenced [12], appears to lack all seven genes necessary to synthesize heme from 5-aminolevulinic acid [13].
Some hemoproteins of animal parasitic nematodes are particularly well studied because of the interests in their roles in low-oxygen environment (host intestine). One such protein is perienteric hemoglobin of Ascaris lumbricoides (parasitic nematode of humans), which has an extraordinary high oxygen affinity, approximately 10,000-fold higher than that of the host’s globin [14]. The proposed functions of this oxygen-avid hemoglobin include oxygen detoxification by a reaction driven by nitric oxide [15] and maintenance of body wall O2 tension by creating an inward-decreasing O2 gradient that is considered important for oxygen unloading from body wall myoglobin, another heme-containing protein [16]. Another example of well-studied nematode hemoproteins is cytochrome b in the mitochondrial respiratory complex II of Ascaris suum (swine parasitic nematode). A. suum larvae utilize classic mammalian-type respiration, expressing a small subunit of larval cytochrome b (CybSL) [17]. In contrast, adult worms live in the host small intestine, where oxygen tension is low and utilize an anaerobic NADH-fumarate reductase system expressing a different small subunit of cytochrome b (CybSA) instead of CybSL [17]. Given the important roles played by the hemoproteins in animal parasitic nematodes, it is interesting to know how heme molecules are synthesized or acquired from the animal hosts.
Strongyloides is a genus of obligate gastrointestinal parasites of vertebrates that belong to nematode clade IV [18]. Among more than 50 documented species, two are known to cause human infections, namely Strongyloides stercoralis and Strongyloides fuelleborni [18]. It is estimated that 30–100 million individuals are infected with Strongyloides worldwide primarily in tropic and subtropic regions [19]. Symptoms are usually absent or mild in immunocompetent hosts. However, in impaired host immunity, severe manifestations can develop, and fatalities may ensue [20].
To study strongyloidiasis, Strongyloides venezuelensis, which is native to rats but can also infect mice, has been widely used as a model [21]. In a transcriptome sequencing project of this Strongyloides species, we identified a partial cDNA sequence that most likely encodes a gene for FeCH [22]. FeCH catalyzes the terminal step of heme biosynthesis [23]. The existence of FeCH sequences was noticed in the genomes of Brugia malayi (another animal parasitic nematode belonging to nematode clade III) and its bacterial endosymbiont, (Wolbachia). However, further analysis was conducted only on the FeCH gene in the endosymbiont genome [24], [25].
In the present study, we cloned the entire cDNA sequence of the FeCH gene from S. venezuelensis (SvFeCH). Our BLAST search on publically available databases revealed that only a fraction of nematode species possesses the FeCH gene. Interestingly, all these species were parasites of mammals. Surprisingly, in our phylogenetic analysis, nematode FeCH formed a distinctive clade, and it was placed distantly from the clade that contains non-nematode metazoan FeCH, suggesting that the origin of nematode FeCH genes are different from those of non-nematode metazoan FeCH. The chelatase activity of the SvFeCH was confirmed by an in vitro assay using recombinant protein and a gene complementation assay using an FeCH-deficient Escherichia. coli.
Nematode genes for heme biosynthesis have not been cloned or characterized to date, essentially because of the nonexistence of these genes in species commonly used in laboratories such as C. elegans. Thus, the present study represents the first report of a cloned active FeCH from organisms in the phylum Nematoda.
Although the biological significance of carrying only the FeCH gene among other heme biosynthesis genes is unclear, the presence of this gene only in animal parasites suggests a possible role for this gene in nutritional adaptation to the animal host environment.
Materials and Methods
Ethics Statement
S. venezuelensis has been maintained over serial passages in male Wistar rats purchased from Kyudo Co. Ltd. (Kumamoto, Japan). The animals were housed and handled in the Division of Parasitology, Department of Infectious Diseases, University of Miyazaki [26]. All animal studies were conducted under the applicable laws and guidelines for the care and use of laboratory animals in the University of Miyazaki and approved by the Animal Experiment Committee of the University, as specified in the Fundamental Guidelines for Proper Conduct of Animal Experiment and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology, Japan, 2006.
5′- and 3′-rapid Amplification of cDNA Ends (RACE)
To determine the sequences of the 3′- and 5′-ends of FeCH cDNA, RACE experiments were performed [27], [28]. The priming sites used for these experiments were based on a contig sequence obtained from our S. venezuelensis transcriptome sequencing project [22]. For 3′-RACE, a PrimeScript RT-PCR kit (Takara, Japan) was used with oligo(dT) adaptor primers to synthesize cDNA from total RNA prepared from parasitic adult worms. Using this 3′-RACE-ready cDNA as a template, hemi-nested PCR was performed first with primer pairs ENM059/ENM008, followed by ENM060/ENM008. The primer sequences used in this study are summarized in Table S1. The resultant PCR products were cloned into pCR2.1 TOPO (Invitrogen, Carlsbad, CA, USA) for DNA sequencing.
For 5′-RACE, a gene specific-primer (reverse) ENM070 was used to synthesize cDNA from total RNA prepared from adult worms. The addition of a homopolymeric A-tail to the 3′-end of the synthesized first-strand cDNA was performed using dATP and terminal transferase. The dA-tailed cDNA was used as a template for hemi-nested PCR first with primers ENM5_6_7, and ENM008/ENM071, then with primers ENM008/ENM072. The resultant PCR products were cloned into pCR2.1 TOPO for DNA sequencing.
Based on the sequence information obtained from the 5′- and 3′-RACE experiments, a PCR primer pair (ENM073/ENM074) was designed to amplify the entire ORF of the SvFeCH gene. The PCR products obtained using an adult-stage cDNA sample as a template were cloned into pCR2.1 TOPO vectors to determine the sequence. The resultant full-length ORF sequence was deposited into DNA Data Bank of Japan under the accession number AB710465, which can be accessed through GenBank (http://www.ncbi.nlm.nih.gov/genbank/), and used to deduce the amino acid sequence of the SvFeCH.
BLAST Homology Search
To search for heme biosynthesis genes, BLAST homology searches [29] were performed against predicted protein sequence data from published nematode genome projects (Caenorhabditis briggsae (nematode clade V) [30], C. elegans (V) [12], Pristionchus pacificus (V) [31], Meloidogyne incognita (IV) [31], Meloidogyne hapla (IV) [32], Bursaphelenchus xylophilus (IV) [33], B. malayi (III) [25], Ascaris suum (III) [34], and Trichinella spiralis (I) [35]) and nematode expressed sequence tags (ESTs) from NEMBASE4 [36], and S. venezuelensis genome (obtained by the Roche-454 pyrosequencing platform [37] with an estimated coverage of more than 20, unpublished), and transcriptome [22] datasets, using human sequences as queries with cutoff value of 1×10−4. For the FeCH gene, the S. venezuelensis protein sequence, deduced from the cDNA sequence, was also used as a query to search for potential orthologs against the aforementioned set of nematode genome and EST datasets, as well as the NCBI nonredundant protein database. Similarly, nematode heme biosynthesis gene sequences identified during these database searches were used as queries, instead of the human sequences, to search for potential orthologs in our S. venezuelensis genome and transcriptome datasets.
Phylogenetic Analyses
We retrieved the gene sequences encoding FeCH of 71 bacterial and 65 eukaryotic species from the GENBANK nonredundant protein database (note that some eukaryotes possess more than two FeCH homologs). These amino acid sequences and those of the S. venezuelensis homolog were firstly aligned using MAFFT [38], and the resultant alignment was edited manually. After the exclusion of ambiguously aligned positions, the final FeCH alignment containing 71 eukaryotic and 71 bacterial homologs with 177 amino acid positions was subjected to phylogenetic analyses, as described below. Taxonomic affiliation and accession numbers for the sequences considered in our FeCH alignment are listed in Table S2.
Maximum likelihood (ML) phylogenetic analyses were performed using RAxML 7.2.8 [39]. The substitution model used was the LG model incorporating the among-site rate variation approximated with a discrete gamma distribution with four categories (LG+Γ). This particular substitution model was selected as the most appropriate model for the FeCH alignment using Aminosan [40]. The ML tree was selected from heuristic tree search initiated from 20 distinctive parsimonious trees. In ML bootstrap analysis (with 100 replicates), a single tree search was performed per replicate.
Bayesian analysis based on the LG+Γ model was also conducted using MrBayes 3.2.1 [41]. Four parallel Metropolis-coupled Markov chain Monte Carlo runs, each consisting of one cold and seven heated chains with a chain temperature of 0.1, were run for 5,000,000 generations. Log-likelihood scores and trees with branch lengths were sampled every 1000 generations. The first 1,250,000 generations were excluded as burn-in, and the remaining trees were summarized to obtain Bayesian posterior probabilities.
Bacterial Expression of Recombinant SvFeCH and Measurement of Porphyrin-metal Chelatase Activity
A cDNA sequence corresponding to the entire catalytic core region of SvFeCH (amino acid positions 29–373) was obtained by PCR using the primer pair TKT001/TKT002. The PCR product was cloned into pET-21a (+), an E. coli expression vector (Merck, Darmstadt, Germany), and the plasmid obtained was transferred to E. coli BL21. The bacteria were grown in LB medium for 16 h, and then the culture medium was diluted by 10-fold in fresh LB medium. The enzyme was expressed with 0.3 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 30°C for 2 h.
The cells were harvested by centrifugation and suspended in 20 mM Tris-HCl (pH 8.0), 10% glycerol, 1 mM DTT, 0.1% Tween 20, and 0.3 M NaCl. Cells were disrupted by sonication and centrifuged at 5000×g at 4°C for 10 min. The supernatants were used for the enzyme assay.
The FeCH activity was determined by measuring the insertion of zinc ions into mesoporphyrin, as described previously [42]. After incubation at 30°C for 30 min, the protoporphyrin or zinc-protoporphyrin formed was measured fluorophotometrically.
Genetic Complementation Assay of hemH (Bacterial FeCH) Deficient E. coli
E. coli strain VS200 (ΔhemH), a deletion mutant for hemH gene [43] was provided by the National Bioresource Project of MEXT, Japan.
The entire ORF of SvFeCH, obtained by RT-PCR with the primer pair ENM089/ENM098, was cloned into the XhoI/BglII restriction site of pFLAG-CTC plasmid, an E. coli expression vector containing a tac promoter (Sigma-Aldrich, St. Louis, MO, USA). The resultant plasmid pFLAG-CTC-SvFeCH was tested as a gene complementation vector. The original pFLAG-CTC plasmid served as a control.
ΔhemH was transformed with pFLAG-CTC-SvFeCH or with pFLAG-CTC. The transformed and untransformed E. coli ΔhemH strains were cultured overnight in LB medium supplemented with hemin (10 µg/ml).
For the culture of the transformed ΔhemH, ampicillin was also added at a concentration of 50 µg/ml. The bacteria from the overnight culture were pelleted by centrifugation and washed thrice with LB medium. After washing, the bacteria pellets were resuspended to give an OD600 of 0.1 in hemin-containing (10 µg/ml) or hemin-free LB medium with (for the transformed ΔhemH) or without (for the untransformed ΔhemH) ampicillin, and incubated at 37°C with rocking. O.D. 600 of each culture was measured every hour up to 20 h.
Real-time RT-PCR Analysis
Total RNA samples were prepared from eggs, a mixture of first- and second-stage larvae (L1/L2), third-stage infective larvae (L3i), lung third-stage larvae (LL3), mucosal larvae (ML) and parasitic adult stages. Eggs were obtained by the floatation method with saturated salt solution from rat feces. L1/L2 and L3i were prepared from fecal culture. LL3 and ML were collected from infected male ICR mice 72 and 85 h after infection, respectively. Parasitic adults were collected from the small intestine of rats 10 days after infection. Eggs and worms were washed extensively with PBS, pelleted by centrifugation and stored at −80°C until used.
Frozen eggs or worms were crushed with a crushing device (SK-200) purchased from Tokken, Japan. Trizol (Invitrogen) was used for total RNA preparation following the manufacturer’s instructions. After DNase I treatment, cDNA was synthesized using PrimeScript RT-PCR kit. Real-time RT-PCR was performed by the GoTaq qPCR system (Promega, Madison, WI, USA) using specific primer pairs (ENM056/ENM057 for SvFeCH and 377F/501R for 18S ribosomal RNA genes). The real-time RT-PCR analyses were performed using biological triplicate samples.
Results
Initially, we identified an EST contig that appeared to represent a transcript from SvFeCH gene [22]. The entire cDNA sequence was determined by 3′- and 5′- RACE experiments. This sequence could be mapped to the genomic DNA sequence of this organism obtained from our genome sequencing project, the details of which will be published elsewhere. The genomic and cDNA sequences of the SvFeCH gene are presented in Figure 1. The length of the coding region was 1122 bp including the stop codon. There was one short (49 bp) intron. The deduced amino acid sequence had a length of 373 residues and an expected molecular mass of 43.3 kDa.
Figure 1. Genomic DNA and cDNA sequences of the Strongyloides venezuelensis FeCH gene.

Both sequences were identical excluding the intronic region, which existed only in the genomic DNA, and a nucleotide at the 54th codon (single-underlined), which was cytosine in the cDNA sequence but was thymidine in the genomic DNA sequences (silent mutation). The deduced amino acid sequence is shown below the nucleotide sequence. In-frame stop codons are indicated by asterisks. 5′- and 3′-splice junction sites that obey the GT-AG rule of eukaryotic introns are indicated by double lines.
In our search for the presence of other heme biosynthesis genes, BLAST homology searches were performed against nematode genome and EST databases, using human heme biosynthesis gene sequences as queries (Tables S3 and S4). Overall, many nematodes appeared to lack all the heme biosynthesis genes, as reported for C. elegans [13]. However, some exceptions were also noticed, including the presence of the aminolevulinic acid dehydrogenase (ALAD) gene in several species and the uroporphyrinogen decarboxylase (UROD) gene in Meloidogyne paranaensis, the coproporphyrinogen oxidase (CPOX) gene in Ancylostoma caninum, and the FeCH gene in B. malayi and Strongyloides ratti. No heme biosynthesis gene other than FeCH was found in our S. venezuelensis genome and transcriptome data using the human sequences as queries. We did not obtain any significant hit from the BLAST analyses for S. venezuelensis genome and transcriptome datasets using the ALAD, UROD, and CPOX gene sequences identified in the nematode genome/EST datasets (see above) as queries. When the SvFeCH protein sequence was used as a query for the BLAST analysis, two additional species were found to carry FeCH gene (Table S5), namely Litomosoides sigmodontis and Onchocerca volvulus. The S. venezuelensis sequence was also used for BLAST searches against NCBI non-redundant protein database, which led to the identification of two more nematode species that carry FeCH, namely Dirofilaria immitis and Acanthocheilonema viteae. These results are interesting because all the species found to carry the FeCH gene were animal parasites (filarial nematodes in clade III and Strongyloides in clade IV).
A multiple sequence alignment of FeCH protein sequences from selected organisms is presented in Figure 2. Amino acid residues in the catalytic core (boxed by a red dotted line) displayed moderate similarity. Key residues for FeCH activity, such as H263 (human sequence numbering), which was proposed to be involved in metal substrate binding [23], [44], were well conserved. Characteristically, nematode (S. venezuelensis and B. malayi) FeCH lacked a protein region called the “C-terminal extension,” a short (approximately 30–50 amino acid residues) stretch of sequences at the C-terminus of the protein that is commonly present in the FeCH of non-nematode opisthokonts [23], [45] (boxed by a green dotted line in Figure 2). To measure the similarities of these selected sequences, BLAST scores and amino acid identities were retrieved by the BLASTP program (Table S6). All the nematode FeCH sequences had higher BLAST scores and percent similarity values to the E. coli sequence (BLAST score: 202–221; similarity: 33.6%–36.8%) than to human (92–106 and 26.8%–27.5%, respectively), Drosophila (93–102 and 24.0%–25.9%, respectively) and Saccharomyces sequences (74–96 and 23.0%–27.2%, respectively). When BLAST homology searches were conducted using the SvFeCH protein sequence as a query against the NCBI non-redundant protein as described above, virtually all the top hits were bacterial sequences excluding the sequences of filarial nematodes (data not shown). These findings prompted us to conduct a phylogenetic analysis to better clarify the evolutionary origin of nematode FeCH genes.
Figure 2. Multiple sequence alignment of FeCH sequences.

The FeCH sequences were taken from the NCBI protein database together with the S. venezuelensis sequence (this study); Homo sapiens (CAB65962), Drosophila melanogaster (AAC26225), Saccharomyces cerevisiae (EDV10759), Brugia malayi 1 (ADI33748), Brugia malayi 2 (ADI33749), Acanthocheilonema viteae (ADI33750), Dirofilaria immitis (ADI33752), Onchocerca volvulus (ADI33751), Escherichia coli (AP_001124), and Bacillus subtilis (NP_388894). The sequences were computationally aligned by the ClustalX program [55]. The catalytic core and the C-terminal extension are boxed by red and green dotted lines, respectively. A histidine residue reported to be critical for metal substrate binding (H263, human sequence numbering) is indicated by an inverted triangle. A cluster of three acidic residues are marked with asterisks. Two residues at the active site that were reported to be identical in all known FeCH sequences (Y276 and W310) [56] are indicated by number (#) marks.
Phylogenetic Analysis
The amino acid alignment of FeCH sampled from 71 eukaryotic and 71 bacterial species was phylogenetically analyzed by ML and Bayesian methods (Figure 3). Overall, the FeCH trees inferred by the ML and Bayesian methods were concordant with each other as well as the results of previously published FeCH phylogenies [7], [46], [47], [48]. Four major clades including the FeCH homologues sampled from eukaryotes were reconstructed with ML bootstrap support values (MLBPs) of 86%–96% and a Bayesian posterior probability (BPP) of 1.00 (shaded in blue, green, pink, and orange; Figure 3): (1) a ‘blue’ clade comprising a single bacterial homolog (Gemmatimonas aurantiaca) and those of eukaryotes–non-nematode metazoans, fungi, Capsaspora owczarzaki, oomycetes, amoebozoans, and ciliates; (2) a ‘green’ clade of the homolog of cyanobacteria including an obligate endosymbiont in the testate amoeba, Paulinella chromatophora [49], and putative plastid homolog in photosynthetic eukaryotes; (3) a ‘pink’ clade comprising the homolog of insect trypanosomatids [3]; (4) an ‘orange’ clade comprising the homolog of parasitic nematodes including S. venezuelensis. Other homologs sampled from eukaryotes were scattered amongst the bacterial homologues, and they exhibited no specific evolutionary affinity to other homologs.
Figure 3. Phylogenetic analysis of FeCH sequences.

The ML phylogeny inferred from FeCH amino acid sequences from 71 bacteria and 65 eukaryotes. Numerical values at the nodes represent MLBPs and BPPs. Only MLBPs greater than 60% are shown. The majority of the FeCH homologs sampled from eukaryotes (colored in red) were separated into four clades shaded in blue, green, pink, and orange. The homologs from Strongyloides venezuelensis identified in this study formed the ‘orange’ clade with those of other nematodes. We compared the ML tree and the alternative hypotheses for the origin of the nematode FeCH genes by pruning and regrafting the entire nematode clade (shaded in orange) to the branches marked by diamonds in the ‘blue’ clade.
The FeCH phylogeny suggested that the homologs from non-nematode metazoans nested in the ‘blue’ clade and those of nematodes forming the ‘orange’ clade were distantly related to each other. Although they received little support from the ML bootstrap and Bayesian analyses, the homologs from non-nematode metazoans, Capsaspora, and fungi were grouped together, corresponding to members of Opisthokonta, a well-established monophyletic assemblage [50]. Curiously, the nematode FeCH homologs formed a robust clade with an MLBP of 96% and BPP of 1.00, being distinct from other metazoan homologs. This tree topology can be rationalized by the vertical inheritance of FeCH genes from the ancestral opisthokont species to non-nematode metazoans and horizontal transfer of a FeCH gene between the ancestral nematodes and a non-metazoan organism. This conjecture was further supported by a topology test comparing the ML tree shown in Figure 3 with three alternative trees, in which the nematode homologs were enforced to branch at the base of (1) the non-nematode metazoan clade, (2) the clade of the opisthokont homologues (excluding that of Schizosaccharomyces pombe), and (3) the ‘blue’ clade composed of the eukaryotic and Gemmatimonas homologs (highlighted by diamonds in Figure 3). Importantly, all the alternative trees were successfully rejected with very small p values (2.0×10−78–2.0×10−36).
Chelatase Assay using Recombinant SvFeCH
To determine whether the FeCH gene of S. venezuelensis identified in the present study encodes an active enzyme, we conducted a chelatase assay using a bacterially expressed recombinant SvFeCH. We constructed an expression plasmid, pET-SvFeCH, which was used to transform E. coli strain BL21. Protein expression was induced by incubation with 0.3 mM IPTG at 30°C for 2 h. The enzyme activity was measured using the cell extracts of untransformed and transformed bacteria. The FeCH activity in transformed bacteria, which was derived from overexpressed SvFeCH and endogenous E. Coli FeCH, was much higher than that in the untransformed control, which originated solely from endogenous FeCH, indicating that the enzyme was active (Figure. 4).
Figure 4. Chelatase assay using bacterially expressed recombinant SvFeCH.

The cell extracts were incubated with 20 mM Tris-HCl, pH 8.0, 0.1% Tween 20, 15 µM mesoporphyrin IX, and 40 µM zinc acetate in a final volume of 200 µl at 30°C for 60 min. The formation of zinc mesoporphyrin was measured. Data are expressed as the mean ± SD of triplicate experiments.
Genetic Complementation Assay of hemH Deficient E. coli
The VS200 strain of E. coli K12, a hemH null-mutant, was used for the gene complementation assay, and the results are shown in Figure 5. VS200 could not grow in LB medium, unless hemin (10 µg/ml) was supplemented. The expression of SvFeCH by pFLAG-CTC-SvFeCH made the bacteria capable of growing in the LB medium in the absence of hemin. Transforming the bacteria with the control vector (pFLAG-CTC) did not have such an effect. Therefore, it was concluded that SvFeCH is an active enzyme that can function as FeCH.
Figure 5. Genetic complementation assay of ΔhemH E. coli.

An untransformed ΔhemH strain of E. coli was grown in the absence (diamond) or presence 10 µg/ml hemin (square). In the same experiment, a transformed ΔhemH strain of E. coli either with SvFeCH gene expression vector (triangle) or with empty vector (x-mark) was cultured in the absence of hemin. OD600 was measured every hour up to 20 h to monitor bacterial growth.
Expression of FeCH during the Life Cycle of S. venezuelensis
The relative expression levels of SvFeCH mRNA were assessed by real-time RT-PCR analysis using RNA samples prepared from the six major developmental stages of S. venezuelensis (Figure 6). It was observed that although SvFeCH mRNA expression was present throughout the stages, it was relatively low in L3i.
Figure 6. Expression analysis of SvFeCH gene by real-time RT-PCR.
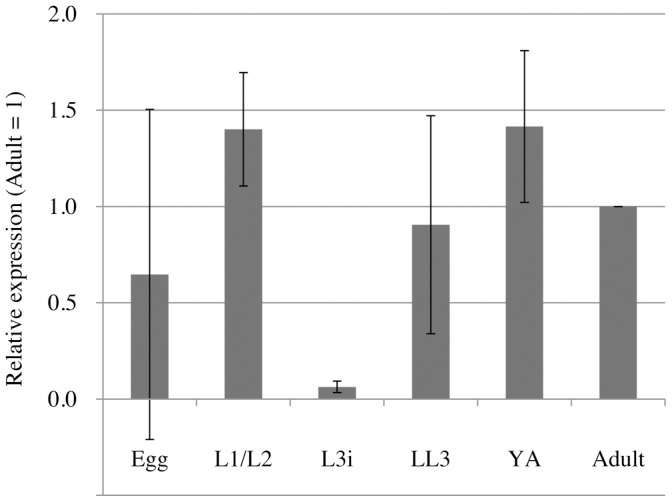
mRNA abundance is shown relative to the expression level at the adult stage, after normalizing to 18S rRNA expression levels. The bars represent the means and standard deviations (±) of biological triplicates. Real-time RT-PCR was performed in triplicate wells for each biological replicate.
Discussion
We demonstrated that a gene for FeCH exists in the S. venezuelensis genome. Although the presence of the FeCH gene in the draft genome of B. malayi was reported previously [24], [25], no further characterization was reported. The present study represents the first cloning and characterization of nematode FeCH, particularly in an evolutionary context.
Phylogenetic analyses revealed that nematode FeCH forms a distinct clade from that of non-nematode metazoans, indicating that the evolutionary origin of nematode FeCH is fundamentally different from that of the FeCH genes of other metazoan organisms. In the ML phylogeny, the nematode clade was placed within the homologs from a subset of alpha-proteobacteria, although the statistical support for this hypothesis is inconclusive. If the affinity between the nematode and alpha-proteobacterial FeCH homologs is genuine, then an as-yet-unknown alpha-proteobacterium was the source of the FeCH homologs working in the extant nematodes. This hypothesis is intriguing because replacement of the eukaryotic FeCH gene by a bacterial FeCH gene had been suggested only for unicellular eukaryotes, such as apicomplexan parasites (Plasmodium falciparum, P. chabaudi, P. berghei, Eimeria tenella, Toxoplasma gondii, and Neospora caninum) [47], [48], [51], the chromerid Chromera velia [47], rhodophytes (Cyanidioschyzon merolae, Porphyra yezoensis, and Galdieria sulphuraria) [48], and the euglenid Euglena gracilis [46].
BLAST analysis of the sequenced nematode genomes and transcriptomes revealed that the FeCH gene is present only in Strongyloides (clade IV) and filarial parasites (clade III). It is still not clear at which point of nematode evolution the proposed horizontal gene transfer event occurred. Regarding B. malayi and related filarial nematodes, horizontal gene transfer from Wolbachia, a bacterial symbiont, is known to have occurred [52]. However, the FeCH sequences present in nematode genomes do not appear to originate from Wolbachia based on the positions of the Wolbachia species in the phylogenetic tree (Figure 3).
We hypothesize two possible scenarios concerning the evolutionary histories of FeCH genes in nematodes, using a current view of the phylogenetic relationship of nematode clades [10]. Because no nematode species possesses the ‘blue clade’ FeCH commonly found in opisthokonts, it can be speculated that this type of FeCH was lost early in nematode evolution (Figure. 7). Strongyloides and the filarias may have acquired FeCH genes from alpha-proteobacteria independently. Alternatively, a common ancestral lineage leading to clades III, IV, and V may have received such an alpha-proteobacterial FeCH gene (scenarios 1 and 2, respectively: Fig. 7a and 7b). For scenario 1 to be true, the hypothetical alpha-proteobacterial species that provided FeCH genes to Strongyloides and filarias, need to be closely related to each other, because the nematode homologs were robustly grouped together in the FeCH phylogeny (Figure. 3). In scenario 2, the lateral transfer of a bacterial FeCH gene occurred through an ancestor leading to species that belong to clades III, IV, and V, and again, the FeCH gene disappeared in some species in clades III and IV such as Ascaris and Meloidogyne and in the branch leading to clade V (Figure. 7b).
Figure 7. Proposed hypotheses for the loss of the original (common opisthokontal) FeCH gene and the re-acquisition of alpha-proteobacterial FeCH in the evolution of the phylum Nematoda.

The initial loss of the common opisthokontal FeCH gene may have occurred at the common ancestor level (red arrows). (a) Scenario 1: The first scenario hypothesizes that alpha-proteobacterial FeCH was acquired independently by some species in clades III and IV (green arrows). b) Scenario 2: Reacquisition of FeCH from an alpha-proteobacterium may have occurred at the common ancestor level of clades III, IV and V (blue arrow) followed by a secondary loss in some species in clade III and IV and in the branch leading to clade V (pink arrows). The phylogenetic relationships of the nematode clades are based on Sommer and Streit [10].
Among the parasitic nematodes, the reason why only Strongyloides and filarias needed to reacquire (scenario 1) or retain (scenario 2) FeCH gene is unclear, particularly when the other six heme biosynthesis genes are still absent. This situation (the presence of FeCH gene in the absence of other heme biosynthesis genes) has been documented for a limited number of organisms, such as Haemophilus influenzae [53] and P. serpens [9]. As was suggested for H. influenzae [9], [54], there may be a possibility that FeCH is used to obtain Fe2+ through its reverse activity rather than obtain heme from protoporphyrin IX using its forward activity.
Supporting Information
List of primers used in this study.
(PDF)
Taxonomic affiliation and accession numbers for the sequences considered in the phylogenetic analysis.
(PDF)
BLAST homology search against predicted proteins from nematode genome projects.
(PDF)
BLAST homology search against nematode EST database (NEMBASE 4).
(PDF)
BLAST homology search against nematode EST database (NEMBASE 4) using Strongyloides venezuelensis ferrochelatase sequence as a query.
(PDF)
Sequence similarities of FeCH proteins assessed by BLAST program.
(PDF)
Acknowledgments
SI is a research fellow supported by the Japan Society for Promotion of Sciences for Young Scientists (No. 00007). We thank Dr. Tadasuke Ooka (University of Miyazaki, Japan) for helpful discussions and Amy Hombu (University of Miyazaki) for critical reading of the manuscript.
Funding Statement
This study was supported by Grant-in-Aid for Young Scientists (B) (23790461) from Japan Society for the Promotion of Science (JSPS), Grant-in-Aid for Scientific Research C (21590466), Grant-in-aid for Scientific Research on Priority Areas ‘Matrix of Infection Phenomena’ (21022041), Health Labor Sciences Research Grant (H23-Shinko-Ippan-014, H21-kokui-shitei-004), and Grant-in-Aid for Scientific Research on Innovative Areas “Genome Science” from MEXT. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Furuyama K, Kaneko K, Vagras P (1997) Heme as a magnificent molecule with multiple missions: Heme determines its own fate and governs cellular homeostasis. Tohoku J Exp Med 213: 1–116. [DOI] [PubMed] [Google Scholar]
- 2. Dailey HA (1997) Enzymes of heme biosynthesis. JBIC 2: 411–417. [Google Scholar]
- 3. Koreny L, Lukes J, Obornik M (2010) Evolution of the haem synthetic pathway in kinetoplastid flagellates: an essential pathway that is not essential after all? Int J Parasitol 40: 149–156. [DOI] [PubMed] [Google Scholar]
- 4. Chang KP, Chang CS, Sassa S (1975) Heme biosynthesis in bacterium-protozoon symbioses: enzymic defects in host hemoflagellates and complemental role of their intracellular symbiotes. Proc Natl Acad Sci U S A 72: 2979–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, et al. (2005) The genome of the African trypanosome Trypanosoma brucei . Science 309: 416–422. [DOI] [PubMed] [Google Scholar]
- 6. El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, et al. (2005) The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 309: 409–415. [DOI] [PubMed] [Google Scholar]
- 7. Alves JM, Voegtly L, Matveyev AV, Lara AM, da Silva FM, et al. (2011) Identification and phylogenetic analysis of heme synthesis genes in trypanosomatids and their bacterial endosymbionts. PLoS One 6: e23518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang KP, Trager W (1974) Nutritional significance of symbiotic bacteria in two species of hemoflagellates. Science 183: 531–532. [DOI] [PubMed] [Google Scholar]
- 9. Koreny L, Sobotka R, Kovarova J, Gnipova A, Flegontov P, et al. (2012) Aerobic kinetoplastid flagellate Phytomonas does not require heme for viability. Proc Natl Acad Sci U S A 109: 3808–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sommer RJ, Streit A (2011) Comparative genetics and genomics of nematodes: genome structure, development, and lifestyle. Annu Rev Genet 45: 1–20. [DOI] [PubMed] [Google Scholar]
- 11. Blaxter ML, De Ley P, Garey JR, Liu LX, Scheldeman P, et al. (1998) A molecular evolutionary framework for the phylum Nematoda. Nature 392: 71–75. [DOI] [PubMed] [Google Scholar]
- 12. The C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018. [DOI] [PubMed] [Google Scholar]
- 13. Rao AU, Carta LK, Lesuisse E, Hamza I (2005) Lack of heme synthesis in a free-living eukaryote. Proc Natl Acad Sci U S A 102: 4270–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Blaxter ML (1993) Nemoglobins: divergent nematode globins. Parasitol Today 9: 353–360. [DOI] [PubMed] [Google Scholar]
- 15. Minning DM, Gow AJ, Bonaventura J, Braun R, Dewhirst M, et al. (1999) Ascaris haemoglobin is a nitric oxide-activated ‘deoxygenase’. Nature 401: 497–502. [DOI] [PubMed] [Google Scholar]
- 16. Takamiya S, Hashimoto M, Kazuno S, Kikkawa M, Yamakura F (2009) Ascaris suum NADH-methemo(myo)globin reductase systems recovering differential functions of hemoglobin and myoglobin, adapting to environmental hypoxia. Parasitol Int 58: 278–284. [DOI] [PubMed] [Google Scholar]
- 17. Amino H, Osanai A, Miyadera H, Shinjyo N, Tomitsuka E, et al. (2003) Isolation and characterization of the stage-specific cytochrome b small subunit (CybS) of Ascaris suum complex II from the aerobic respiratory chain of larval mitochondria. Mol Biochem Parasitol 128: 175–186. [DOI] [PubMed] [Google Scholar]
- 18.Viney ME, Lok JB (2007) Strongyloides spp. WormBook: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bethony J, Brooker S, Albonico M, Geiger SM, Loukas A, et al. (2006) Soil-transmitted helminth infections: ascariasis, trichuriasis, and hookworm. The Lancet 367: 1521–1532. [DOI] [PubMed] [Google Scholar]
- 20. Montes M, Sawhney C, Barros N (2010) Strongyloides stercoralis: there but not seen. Curr Opin Infect Dis 23: 500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sato Y, Toma H (1990) Strongyloides venezuelensis infections in mice. Int J Parasitol 20: 57–62. [DOI] [PubMed] [Google Scholar]
- 22. Nagayasu E, Ogura Y, Itoh T, Yoshida A, Chakraborty G, et al. (2013) Transcriptomic analysis of four developmental stages of Strongyloides venezuelensis . Parasitol Int 62: 57–65. [DOI] [PubMed] [Google Scholar]
- 23. Ferreira GC (1999) Ferrochelatase. Int J Biochem Cell Biol 31: 995–1000. [DOI] [PubMed] [Google Scholar]
- 24. Wu B, Novelli J, Foster J, Vaisvila R, Conway L, et al. (2009) The heme biosynthetic pathway of the obligate Wolbachia endosymbiont of Brugia malayi as a potential anti-filarial drug target. PLoS Negl Trop Dis 3: e475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, et al. (2007) Draft genome of the filarial nematode parasite Brugia malayi . Science 317: 1756–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maruyama H, El-Malky M, Kumagai T, Ohta N (2003) Secreted adhesion molecules of Strongyloides venezuelensis are produced by oesophageal glands and are components of the wall of tunnels constructed by adult worms in the host intestinal mucosa. Parasitology 126: 165–171. [DOI] [PubMed] [Google Scholar]
- 27. Scotto-Lavino E, Du G, Frohman MA (2006) 3′ end cDNA amplification using classic RACE. Nat Protoc 1: 2742–2745. [DOI] [PubMed] [Google Scholar]
- 28. Scotto-Lavino E, Du G, Frohman MA (2006) 5′ end cDNA amplification using classic RACE. Nat Protoc 1: 2555–2562. [DOI] [PubMed] [Google Scholar]
- 29. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 30. Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, et al. (2003) The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1: E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dieterich C, Clifton SW, Schuster LN, Chinwalla A, Delehaunty K, et al. (2008) The Pristionchus pacificus genome provides a unique perspective on nematode lifestyle and parasitism. Nat Genet 40: 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Opperman CH, Bird DM, Williamson VM, Rokhsar DS, Burke M, et al. (2008) Sequence and genetic map of Meloidogyne hapla: A compact nematode genome for plant parasitism. Proc Natl Acad Sci U S A 105: 14802–14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kikuchi T, Cotton JA, Dalzell JJ, Hasegawa K, Kanzaki N, et al. (2011) Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus . PLoS Pathog 7: e1002219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jex AR, Liu S, Li B, Young ND, Hall RS, et al. (2011) Ascaris suum draft genome. Nature 479: 529–533. [DOI] [PubMed] [Google Scholar]
- 35. Mitreva M, Jasmer DP, Zarlenga DS, Wang Z, Abubucker S, et al. (2011) The draft genome of the parasitic nematode Trichinella spiralis . Nat Genet 43: 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Elsworth B, Wasmuth J, Blaxter M (2011) NEMBASE4: the nematode transcriptome resource. Int J Parasitol 41: 881–894. [DOI] [PubMed] [Google Scholar]
- 37. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, et al. (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Katoh K, Kuma K, Toh H, Miyata T (2005) MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57: 758–771. [DOI] [PubMed] [Google Scholar]
- 40. Tanabe AS (2011) Kakusan4 and Aminosan: two programs for comparing nonpartitioned, proportional and separate models for combined molecular phylogenetic analyses of multilocus sequence data. Mol Ecol Resour 11: 914–921. [DOI] [PubMed] [Google Scholar]
- 41. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taketani S, Ishigaki M, Mizutani A, Uebayashi M, Numata M, et al. (2007) Heme synthase (ferrochelatase) catalyzes the removal of iron from heme and demetalation of metalloporphyrins. Biochemistry 46: 15054–15061. [DOI] [PubMed] [Google Scholar]
- 43. Nakahigashi K, Nishimura K, Miyamoto K, Inokuchi H (1991) Photosensitivity of a protoporphyrin-accumulating, light-sensitive mutant (visA) of Escherichia coli K-12. Proc Natl Acad Sci U S A 88: 10520–10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kohno H, Okuda M, Furukawa T, Tokunaga R, Taketani S (1994) Site-directed mutagenesis of human ferrochelatase: identification of histidine-263 as a binding site for metal ions. Biochim Biophys Acta 1209: 95–100. [DOI] [PubMed] [Google Scholar]
- 45. Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, et al. (2000) Ferrochelatase at the millennium: structures, mechanisms and [2Fe-2S] clusters. Cell Mol Life Sci 57: 1909–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koreny L, Obornik M (2011) Sequence evidence for the presence of two tetrapyrrole pathways in Euglena gracilis . Genome Biol Evol 3: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koreny L, Sobotka R, Janouskovec J, Keeling PJ, Obornik M (2011) Tetrapyrrole synthesis of photosynthetic chromerids is likely homologous to the unusual pathway of apicomplexan parasites. Plant Cell 23: 3454–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Obornik M, Green BR (2005) Mosaic origin of the heme biosynthesis pathway in photosynthetic eukaryotes. Mol Biol Evol 22: 2343–2353. [DOI] [PubMed] [Google Scholar]
- 49. Nakayama T, Ishida K (2009) Another acquisition of a primary photosynthetic organelle is underway in Paulinella chromatophora . Curr Biol 19: R284–285. [DOI] [PubMed] [Google Scholar]
- 50. Wainright PO, Hinkle G, Sogin ML, Stickel SK (1993) Monophyletic origins of the metazoa: an evolutionary link with fungi. Science 260: 340–342. [DOI] [PubMed] [Google Scholar]
- 51. Sato S, Wilson RJ (2003) Proteobacteria-like ferrochelatase in the malaria parasite. Curr Genet 42: 292–300. [DOI] [PubMed] [Google Scholar]
- 52. Dunning Hotopp JC, Clark ME, Oliveira DC, Foster JM, Fischer P, et al. (2007) Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317: 1753–1756. [DOI] [PubMed] [Google Scholar]
- 53. Schlor S, Herbert M, Rodenburg M, Blass J, Reidl J (2000) Characterization of ferrochelatase (hemH) mutations in Haemophilus influenzae . Infect Immun 68: 3007–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Loeb MR (1995) Ferrochelatase activity and protoporphyrin IX utilization in Haemophilus influenzae . J Bacteriol 177: 3613–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 56. Medlock AE, Dailey TA, Ross TA, Dailey HA, Lanzilotta WN (2007) A pi-helix switch selective for porphyrin deprotonation and product release in human ferrochelatase. J Mol Biol 373: 1006–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of primers used in this study.
(PDF)
Taxonomic affiliation and accession numbers for the sequences considered in the phylogenetic analysis.
(PDF)
BLAST homology search against predicted proteins from nematode genome projects.
(PDF)
BLAST homology search against nematode EST database (NEMBASE 4).
(PDF)
BLAST homology search against nematode EST database (NEMBASE 4) using Strongyloides venezuelensis ferrochelatase sequence as a query.
(PDF)
Sequence similarities of FeCH proteins assessed by BLAST program.
(PDF)